
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel-catalyzed C(sp2)–C(sp3) coupling via photoactive electron donor–acceptor complexes†
Salman
Alsharif‡
a,
Chen
Zhu‡
 ac,
Xiushan
Liu
b,
Shao-Chi
Lee
a,
Huifeng
Yue
*b and
Magnus
Rueping
*a
ac,
Xiushan
Liu
b,
Shao-Chi
Lee
a,
Huifeng
Yue
*b and
Magnus
Rueping
*a
aKAUST Catalysis Center, KCC, King Abdullah University of Science and Technology, KAUST, Thuwal 23955-6900, Saudi Arabia. E-mail: magnus.rueping@kaust.edu.sa
bKey Laboratory of Molecule Synthesis and Function Discovery (Fujian Province University), College of Chemistry, Fuzhou University, Fuzhou, 350108, China. E-mail: yuehuifeng@fzu.edu.cn
cEastern Institute for Advanced Study, Eastern Institute of Technology, Ningbo, 315200, China
First published on 19th April 2024
Abstract
We have developed a novel Ni-catalyzed reductive cross-coupling reaction of aryl bromides and alkyl iodides via a photoactive electron donor–acceptor (EDA) complex. This photo-induced process enables the efficient construction of C(sp2)–C(sp3) bonds in the absence of an external photocatalyst. Electronically and structurally diverse aryl bromides, as well as secondary and primary alkyl iodides could undergo this transformation smoothly. Natural product derivatives were employed successfully, and UV-vis spectroscopy was utilized to gain mechanistic insight.
The formation of C–C bonds is a fundamental process in organic synthesis, facilitating the creation of complex molecules from simpler precursors. Over the past few decades, transition-metal-catalyzed cross-coupling reactions have emerged as powerful tools for C–C bond formation, finding widespread application in both academic and industrial settings.1,2 Notably, reductive cross-coupling, which utilizes two electrophiles as coupling partners, provides a robust catalytic approach to constructing diverse chemical bonds. This method stands out from traditional cross-coupling strategies, which typically involve organometallic reagents and electrophiles, due to its milder reaction conditions, enhanced functional group compatibility, and broader substrate range.3–5 Recent years have seen the advent of Ni- or Co-catalyzed procedures for challenging C(sp2)–C(sp3) bond formations, with notable contributions from research groups including those of Weix,6 Gong,7 Gosmini,8 Molander,9 and others (Scheme 1a). However, the reliance on substantial amounts of metal reductants like Zn or Mn, coupled with inevitable waste production, has raised questions about the scalability and sustainability of these methods. To address such concerns, the Lei group and Barham group introduced photoredox and nickel dual-catalyzed C(sp2)–C(sp3) reductive cross-coupling of organic halides, employing Et3N as the external reductant or XAT reagent.10 Electrochemical strategies for this transformation have also been put forth by the Sevov11 and Weix12 groups. Despite these significant advances, the utilization of precious metal complexes as photocatalysts, metal sacrificial anodes, and costly ligands continues to pose challenges (Scheme 1b). As a result, there is an ongoing demand for more versatile, efficient, and sustainable methods for C(sp2)–C(sp3) reductive cross-coupling.
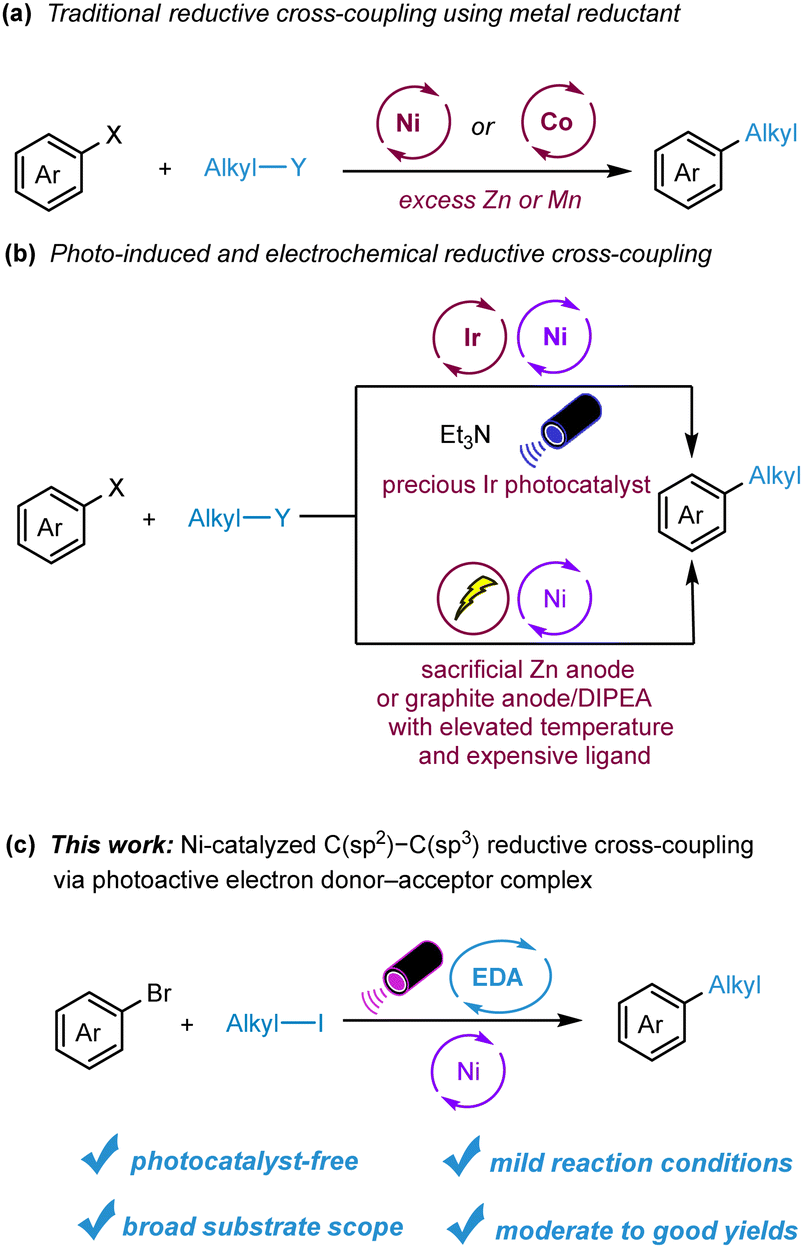 | ||
| Scheme 1 Currently available C(sp2)–C(sp3) reductive cross-coupling processes and the newly developed method via EDA complex. | ||
In recent years, photoredox catalysis has risen as a potent and versatile tool in organic synthesis, facilitating a myriad of novel and challenging transformations.13,14 However, many of these processes depend on either precious metal complexes or specially designed organic photocatalysts. An intriguing alternative is the electron donor–acceptor (EDA) complex.15–17 Formed between an electron-rich substrate and an electron-deficient counterpart, the EDA complex can undergo intermolecular single-electron transfer (SET) when exposed to light, yielding coupling products or radical intermediates suitable for further functionalization. Due to its ability to bypass the need for external photocatalysts, the EDA complex approach is garnering interest for photo-induced transformations. For instance, the Melchiorre group pioneered a range of photochemical radical reactions, encompassing both intermolecular and asymmetric intramolecular EDA complexes.18,19 Other groundbreaking contributions in this field have been made by several researcher including the Leonori,20 Chen,21 Miyake,22 Köenig,23 Aggarwal,24 and Chu,25 groups achieving imporanant transformations such as hydroimination cyclization, allylation/alkenylation, thiolation, amination, borylation, and alkene carbopyridylation. Furthermore, the efforts of Fu/Shang team led to the development of an EDA complex involving a catalytic amount of sodium iodide/triphenylphosphine and stoichiometric redox-active esters.26 Expanding on this concept, the Molander group integrated the EDA complex concept with nickel catalysis, realizing photo-induced C(sp2)–C(sp3) bond formations using aliphatic redox-active esters as acceptor and Hantzsch ester (HE) as both the donor and robust reductant.27,28 Motivated by these achievements and aligning with our ongoing exploration of the EDA complex methodology, we hypothesized that an EDA complex could form between alkyl iodide and Hantzsch ester (HE), paving the way for reductive cross-coupling between alkyl iodides and aryl bromides (Scheme 1c).
We started to explore the Ni-catalyzed C(sp2)–C(sp3) reductive cross-coupling by choosing 4-bromo-1,1′-biphenyl 1a as a model substrate in reaction with iodocyclohexane 2a (Table 1). The initial reaction conditions using NiBr2·d(OMe)-bpy as nickel source, HE as donor, nBu3N as additive, DMA as the solvent, and 390 nm purple LED as light source gave the desired C(sp2)–C(sp3) product in 75% yield (entry 1). Further optimization showed that the application of 440 nm blue LED decreased the yield significantly (entry 2). Other nickel and ligand combinations including NiBr2·dme/d-(OMe)-bpy, NiBr2·3H2O/d-(OMe)-bpy, and NiI2/d-(OMe)-bpy gave slightly lower yields (entries 3–5). The adjustment of reaction concentration led to no improvement (entries 6 and 7). Utilizing Cy2NH instead of nBu3N improved the yield to 83% (entry 8). Moreover, comparable yield was observed when lowing the amount of Cy2NH to two equivalents (entry 9). Other solvents, such as CH3CN and DCE, diminished the yield (entries 10 and 11). The absence of amine decreased the yield dramatically (entry 12). The control experiments shows that both nickel catalyst and HE are essential for the success of this transformation (entries 13 and 14). With the optimized reaction conditions in hand, the scope with respect to the aryl bromides was first investigated (Table 2). A series of aryl bromides bearing electron-withdrawing and electron-donating groups could undergo this reaction smoothly, delivering the corresponding products in good to high yields. π-extended substrates coupled with alkyl iodide with good efficiency (3a and 3b). The good chemoselectivity of this newly developed protocol was well illustrated by the tolerance of various functionalities such as ketone, cyano, trifluoromethoxy, trifluoromethylthio, fluoro, and sulfone (3c–3h).
|
|
||
|---|---|---|
| Entry | Variables | Yieldb (%) |
| a Reaction conditions: 1a (0.20 mmol), 2a (0.40 mmol), NiBr2·d(OMe)-bpy (0.02 mmol), HE (2 equiv.), nBu3N (5 equiv.) in DMA (2 mL) was irradiated under 390 nm purple LED at rt. for 16 h. b GC Yields using dodecane as internal standard. c 2 Equivanlents of Cy2NH was used. d Isolated yield. | ||
| 1 | None | 75 |
| 2 | 440 nm blue LED | 26 |
| 3 | NiBr2·dme, d-(OMe)-bpy | 71 |
| 4 | NiBr2·3H2O, d-(OMe)-bpy | 70 |
| 5 | NiI2, d-(OMe)-bpy | 60 |
| 6 | 4 mL DMA | 66 |
| 7 | 1 mL DMA | 65 |
| 8 | Cy2NH instead of nBu3N | 83 |
| 9c | Cy2NH instead of nBu3N | 83(80d) |
| 10c | CH3CN instead of DMA | 45 |
| 11c | DCE instead of DMA | 21 |
| 12 | w/o Amine | 45 |
| 13 | w/o Nickel catalyst | 0 |
| 14 | w/o HE | 0 |
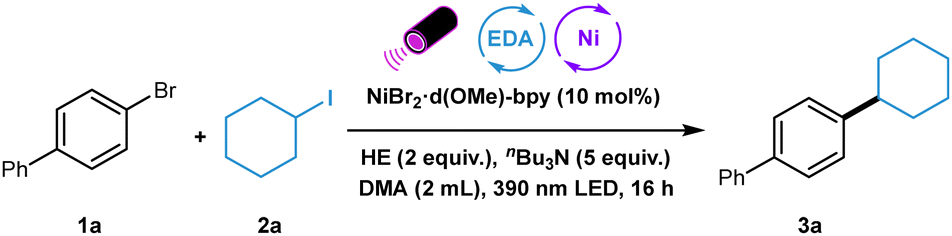
| a Reaction conditions: 1 (0.20 mmol), 2a (0.40 mmol), NiBr2·d(OMe)-bpy (0.02 mmol), HE (2 equiv.), Cy2NH (2 equiv.) in DMA (2 mL) was irradiated under 390 nm purple LED at rt. for 16 h. b Yield after purification. c 1a (1.0 mmol), 2a (2.0 mmol), NiBr2·d(OMe)-bpy (0.05 mmol), HE (2 equiv.), Cy2NH (2 equiv.) in DMA (10 mL) was irradiated under two 390 nm purple LED at rt. for 24 h. d 2a (0.80 mmol), NiBr2·d(OMe)-bpy (0.04 mmol), HE (4 equiv.) and Cy2NH (4 equiv.) were used. |
|---|
|
Di-substituented aryl bromide was also suitable for this transformation, albeit with moderate yield (3i). Significantly, electron-rich aryl bromides bearing alkyl, methoxy, methylthio, and even dimethylamino groups could also participated in this photoactive protocol effectively (3k–3o). Notably, cyclopropyl nitrile, one versatile synthon, was also tolerated here (3p). The reactions proceeded smoothly with heteoaryl bromides containing imide, lactone, and benzothiophene, affording the corresponding products in good to excellent yields (3q–3s). When dibromo-aryl substrate was employed, dialkylation occurred in good yield (3t). 1-Bromo-2-methylbenzene could also undergo this reaction in moderate yield. (3u).
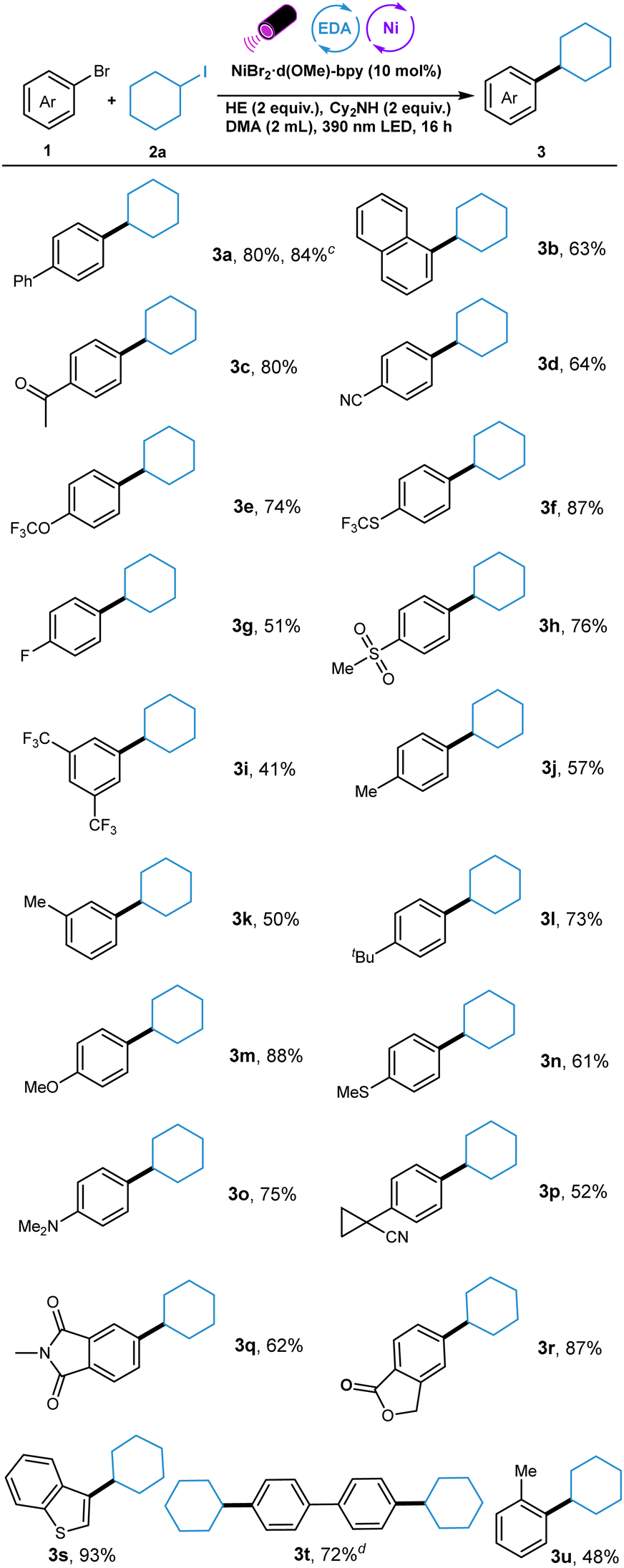
Next, the scope of alkyl iodides was explored (Table 3). A wide range of structurally diverse alkyl iodides including cyclic and linear ones were suitable substrates for this transformation. For example, cyclopentanyl- and O-containing heterocyclic alkyl iodides underwent this reductive cross-coupling in moderate to high yields (4a–4c). Also, linear secondary alkyl iodides with different chain length worked well in this system, furnishing the coupling products in high yields (4d and 4e). In addition, primary alkyl iodides with diverse electronical nature also showed good reactivity in this protocol (4f–4i). Notably, trimethyl silanyl group was tolerated in this reaction, providing possibility for the further functionalization (4j).
Significantly, the effectiveness of this photoactive methodology is illustrated by the efficient transformation of a series of complex molecules. The substrates derived from natural products such as cholestanol, cholestanol, and probenecid were alkylated smoothly (5a–5c), demonstrating the practicability of this protocol (Scheme 2).
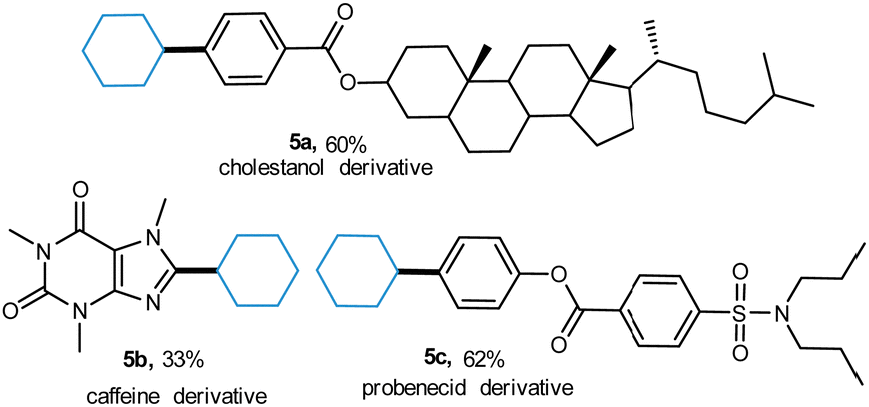 | ||
| Scheme 2 Alkylation of the nature-derived substrates. | ||
To further understand the mechanism of this reaction, the UV-vis absorption spectroscopy of mixtures HE, amine, cyclohexyl iodide, and nickel source in DMA (path length = 1 cm) were measured at standard reaction concentrations (Scheme 3a). Individually, the amine, alkyl iodide, and nickel catalyst exhibited no absorption in the visible spectrum; however, HE showed significant absorption. The addition of amine, alkyl iodide, or nickel catalyst to the HE solution resulted in a decrease in absorption intensity. Conversely, introducing amine/cyclohexyl iodide or amine/nickel catalyst to the mixture increased absorption, indicative of EDA complex formation with HE. Upon the visible light irradiation, the intermolecular charge transfer event occurred between the EDA complex from amine, and HE complex to the alkyl iodide, affording alkyl radical, radical cation II, and iodide ions. The alkyl radical is then trapped by the Ar–NiII–Br intermediate III generated by the oxidative addition of aryl bromide to Ni0Ln species. The resulting NiIII intermediate IV undergoes reductive elimination, delivering the desired alkylation product and the NiI intermediate V that subsequently forms another EDA complex VI with amine and HE. This EDA complex VI is primed to undergo an intermolecular charge transfer to generate the active Ni0Ln species and the radical cation II, thus completing the catalytic cycle.
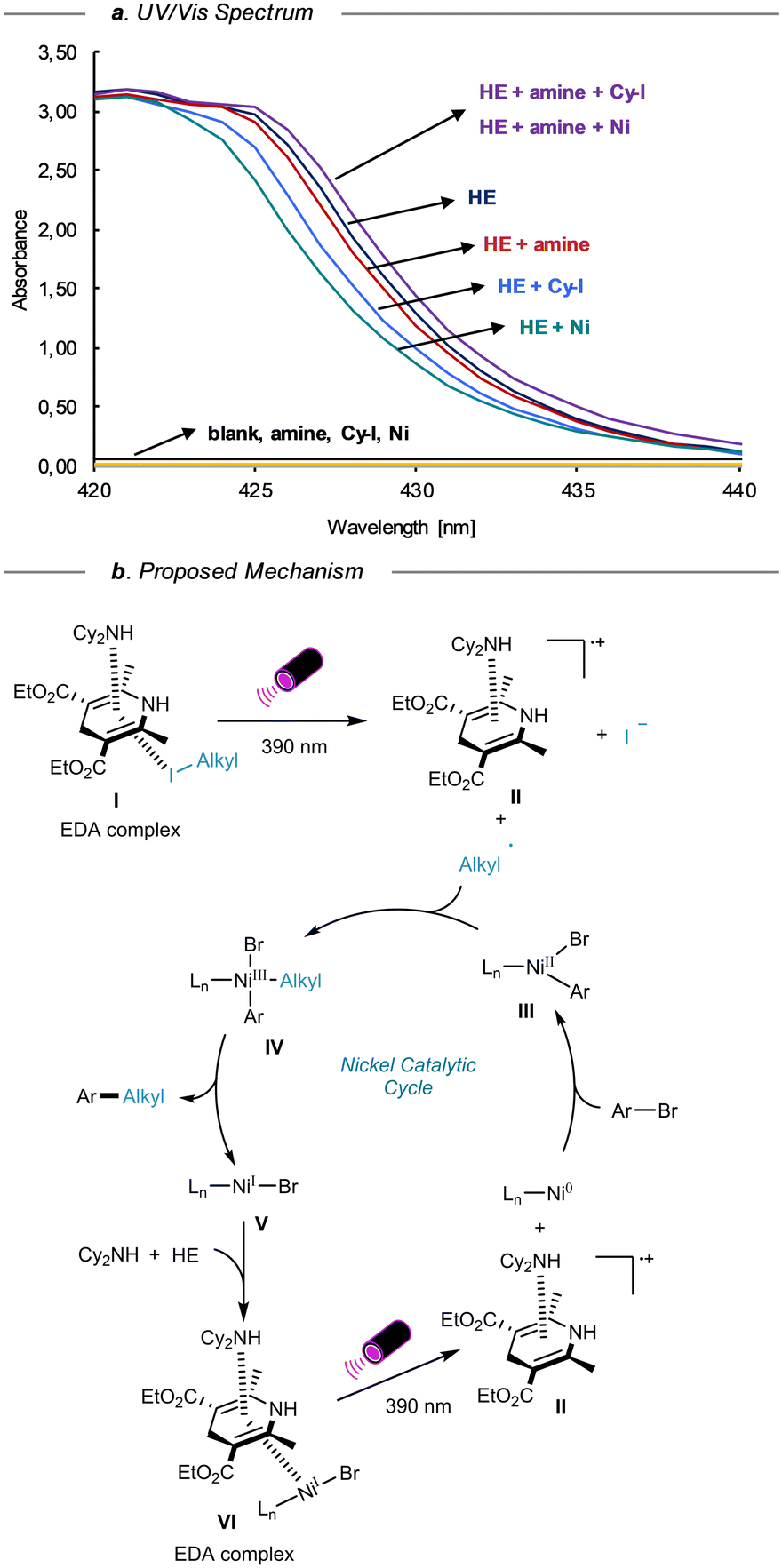 | ||
| Scheme 3 UV-vis absorption study and proposed mechanism. | ||
In conclusion, an efficient and low-cost photo-induced methodology for the formation of C(sp2)–C(sp3) bonds has been reported. The newly developed protocol proceeds via photoactive intermolecular change transfer between the EDA complex of alkyl iodide and HE, followed by nickel catalytic cycle. Notably, the method bypasses the need for precious metal photocatalysts, operates under mild reaction conditions, and possesses a broad substrate scope. The successful application of this approach to complex molecules underscores its practical potential. Furthermore, UV-vis absorption measurements support the involvement of the EDA complexes as reactive intermediates during the reaction.
This work was supported by the King Abdullah University of Science and Technology, Saudi Arabia, Office of Sponsored Research (URF/1/4384-01-01).
Conflicts of interest
There are no conflicts to declare.Notes and references
- T. J. Colacot, New trends in cross-coupling: theory and applications, Royal Society of Chemistry, 2014 Search PubMed.
- S. B. Armin de Meijere and Martin Oestreich, Metal-Catalyzed Cross-Coupling Reactions and More, Wiley-VCH, Weinheim, 2014 Search PubMed.
- C. E. Knappke, S. Grupe, D. Gärtner, M. Corpet, C. Gosmini and A. J. V. Wangelin, Chem. – Eur. J., 2014, 20, 6828–6842 CrossRef CAS PubMed.
- J. Gu, X. Wang, W. Xue and H. Gong, Org. Chem. Front., 2015, 2, 1411–1421 RSC.
- D. J. J. A. o C. R. Weix, Acc. Chem. Res., 2015, 48, 1767–1775 CrossRef CAS PubMed.
- D. Weix, Acc. Chem. Res., 2015, 48, 1767–1775 CrossRef CAS PubMed.
- Y. Ye, H. Chen, J. L. Sessler and H. Gong, J. Am. Chem. Soc., 2019, 141, 820–824 CrossRef CAS PubMed.
- M. Amatore and C. Gosmini, Angew. Chem., Int. Ed., 2008, 47, 2089–2092 CrossRef CAS PubMed.
- G. A. Molander, S. R. Wisniewski and K. M. Traister, Org. Lett., 2014, 16, 3692–3695 CrossRef CAS PubMed.
- (a) Z. Duan, W. Li and A. Lei, Org. Lett., 2016, 18, 4012–4015 CrossRef CAS PubMed; (b) X. Tian, J. Kaur, S. Yakubov and J. P. Barham, ChemSusChem, 2022, 15, e202200906 CrossRef CAS PubMed.
- B. L. Truesdell, T. B. Hamby and C. S. Sevov, J. Am. Chem. Soc., 2020, 142, 5884–5893 CrossRef CAS PubMed.
- M. C. Franke, V. R. Longley, M. Rafiee, S. S. Stahl, E. C. Hansen and D. J. Weix, ACS Catal., 2022, 12, 12617–12626 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS PubMed.
- C. G. S. Lima, T. M. Lima, M. Duarte, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389–1407 CrossRef CAS.
- S. V. Rosokha and J. K. Kochi, Acc. Chem. Res., 2008, 41, 641–653 CrossRef CAS PubMed.
- E. Arceo, I. D. Jurberg, A. Álvarez-Fernández and P. Melchiorre, Nat. Chem., 2013, 5, 750–756 CrossRef CAS PubMed.
- Z.-Y. Cao, T. Ghosh and P. Melchiorre, Nat. Commun., 2018, 9, 3274 CrossRef PubMed.
- J. Davies, S. G. Booth, S. Essafi, R. A. W. Dryfe and D. Leonori, Angew. Chem., Int. Ed., 2015, 54, 14017–14021 CrossRef CAS PubMed.
- J. Zhang, Y. Li, R. Xu and Y. Chen, Angew. Chem., Int. Ed., 2017, 56, 12619–12623 CrossRef CAS PubMed.
- B. Liu, C.-H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2017, 139, 13616–13619 CrossRef CAS PubMed.
- L. Marzo, S. Wang and B. König, Org. Lett., 2017, 19, 5976–5979 CrossRef CAS PubMed.
- J. Wu, R. M. Bär, L. Guo, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2019, 58, 18830–18834 CrossRef CAS PubMed.
- D. Chen, L. Xu, T. Long, S. Zhu, J. Yang and L. Chu, Chem. Sci., 2018, 9, 9012–9017 RSC.
- M.-C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Science, 2019, 363, 1429–1434 CrossRef CAS PubMed.
- L. M. Kammer, S. O. Badir, R.-M. Hu and G. A. Molander, Chem. Sci., 2021, 12, 5450–5457 RSC.
- V. C. Polites, S. O. Badir, S. Keess, A. Jolit and G. A. Molander, Org. Lett., 2021, 23, 4828–4833 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc00217b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |