
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fusion then fission: splitting and reassembly of an artificial fusion-protein nanocage†
Naoya
Ohara
 a,
Norifumi
Kawakami
*a,
Ryoichi
Arai
bc,
Naruhiko
Adachi
de,
Akihito
Ikeda
d,
Toshiya
Senda
d and
Kenji
Miyamoto
*a
a,
Norifumi
Kawakami
*a,
Ryoichi
Arai
bc,
Naruhiko
Adachi
de,
Akihito
Ikeda
d,
Toshiya
Senda
d and
Kenji
Miyamoto
*a
aDepartment of Bioscience and Informatics, Faculty of Science and Technology, Keio University, Yokohama, Kanagawa 223-8522, Japan. E-mail: norikawakami@bio.keio.ac.jp; kmiyamoto@bio.keio.ac.jp
bDepartment of Biomolecular Innovation, Institute for Biomedical Sciences, Interdisciplinary Cluster for Cutting Edge Research, Shinshu University, Ueda, Nagano 386-8567, Japan
cDepartment of Applied Biology, Faculty of Textile Science and Technology, Shinshu University, Ueda, Nagano 386-8567, Japan
dStructural Biology Research Center, Institute of Materials Structure Science, High Energy Accelerator Research Organization (KEK), Oho, Tsukuba 305-0801, Japan
eLife Science Center for Survival Dynamics, Tsukuba Advanced Research Alliance (TARA), University of Tsukuba, 1-1-1 Tennodai, Tsukuba, Ibaraki 305-8577, Japan
First published on 8th April 2024
Abstract
A split-protein system is a simple approach to introduce new termini which are useful as modification sites in protein engineering, but has been adapted mainly for monomeric proteins. Here we demonstrate the design of split subunits of the 60-mer artificial fusion-protein nanocage TIP60. The subunit fragments successfully reformed the cage structure in the same manner as prior to splitting. One of the newly introduced terminals at the interior surface can be modified using a tag peptide and green fluorescent protein. Therefore, the termini could serve as a versatile modification site for incorporating a wide variety of functional peptides and proteins.
Proteins generally have N- and C-termini, which are often targeted in protein engineering as sites for chemical and genetic modification.1–5 These modifications may serve a wide variety of purposes, such as making a protein more easily detectable,5–7 creating simple purification systems,8,9 improving the native function of a protein,10 transporting the modified molecules,11,12 or designing new structures.13–15 However, the native N- and C-termini are not always located at the desired positions, thus motivating attempts to develop methods for their relocation. Circular permutation is the most frequently used approach to relocate protein termini.16–20 In this method, the native N- and C-termini are connected by a linker peptide followed by cleavage at a desired position of the sequence. Although most proteins subjected to circular permutation retain their original three-dimensional (3D) structure,21,22 this structure may not be restored if the native N- and C-termini are far apart or if the template protein possesses a flexible structure. Furthermore, circular permutation may induce domain swapping oligomerization by destabilizing the monomeric structure.23,24 This risk may be further increased if a protein that forms homo-oligomers in nature is adopted as the template.
The split-protein system provides a simpler approach to introduce new N- and C-termini compared with circular permutation.25–29 In this strategy, a protein is split into two or more fragments, which can then be assembled in vivo or in vitro into the same structure as that prior to splitting. For example, green fluorescent protein (GFP) was split into multiple β-strand fragments by cleaving the loop regions.30,31 These split fragments are capable of forming a structure similar to the native one that retains the ability to fluoresce. This approach does not require connection of the native N- and C-termini, thus making it applicable to molecules for which circular permutation cannot be used.
We previously designed a soccer-ball-shaped 60-mer protein nanocage referred to as TIP60.32,33 Each single subunit of TIP60 is a fusion protein of pentameric and dimeric proteins (LSm![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 34 and MyoX-coil,35 respectively). By linking these proteins, two interaction sites are created on the subunit, enabling the formation of the 60-mer. The fusion protein is expressed in Escherichia coli cells and is presumed to form the 60-mer structure inside the cells. Furthermore, we recently reported the functionalization of TIP60 based on the chemical modification of both the interior and exterior surfaces.36,37 In these cases, Cys residues were used for the modification reactions, thus leaving both the N- and C-termini still available for further functionalization. However, the N- and C-termini of TIP60 are located on the outer surface of the cage,33 limiting their suitability for modification of the inner cavity (Fig. 1a). If new termini can be introduced within the inner space, it would be possible to encapsulate macromolecules such as enzymes simply by designing fusion proteins, thereby holding promise for catalytic applications. Although similar issues have been addressed for several protein cages by changing the terminus positions through circular permutation,20,38,39 this strategy is not expected to be applicable to TIP60 owing to its distant locations of the N- and C-termini. We thus attempted to apply the split-protein system to TIP60 to introduce additional N- and C-termini inside the cage.
34 and MyoX-coil,35 respectively). By linking these proteins, two interaction sites are created on the subunit, enabling the formation of the 60-mer. The fusion protein is expressed in Escherichia coli cells and is presumed to form the 60-mer structure inside the cells. Furthermore, we recently reported the functionalization of TIP60 based on the chemical modification of both the interior and exterior surfaces.36,37 In these cases, Cys residues were used for the modification reactions, thus leaving both the N- and C-termini still available for further functionalization. However, the N- and C-termini of TIP60 are located on the outer surface of the cage,33 limiting their suitability for modification of the inner cavity (Fig. 1a). If new termini can be introduced within the inner space, it would be possible to encapsulate macromolecules such as enzymes simply by designing fusion proteins, thereby holding promise for catalytic applications. Although similar issues have been addressed for several protein cages by changing the terminus positions through circular permutation,20,38,39 this strategy is not expected to be applicable to TIP60 owing to its distant locations of the N- and C-termini. We thus attempted to apply the split-protein system to TIP60 to introduce additional N- and C-termini inside the cage.
 | ||
| Fig. 1 Design for splitting the subunit of the TIP60 cage. (a) 3D structure of the original TIP60 (PDB ID: 7EQ9). An individual subunit and its N- and C-termini are highlighted in green and blue, respectively. A close-up view is depicted on the right of the panel. (b) Side view of the subunit structure of TIP60. (c) Schematic illustration of the design of the split TIP60 (TIP120). The structural models of CoreN and CoreC were extracted from the corresponding regions of TIP60. | ||
In this study, we demonstrate that the soccer-ball-shaped cage is also produced from the two fragment proteins obtained by splitting the subunit protein of TIP60. The cage structure was found to be successfully generated both in a co-expression system of the two fragments in E. coli cells and upon mixing the separately purified fragment proteins in vitro.
We initially identified the optimal splitting position based on the 3D structure of TIP60 (PDB ID: 7EQ9).33 The criteria considered were as follows: the position must be inside the cage and located at a loop to minimize the negative impact on the fragment structure, and the split fragments should interact with each other by β-strand–β-strand interactions, which are frequently used in this system. We found that the disordered β3–4 loop (M37 to Q46) fulfilled these criteria (Fig. 1a and b). Based on these structural features, we split the subunit protein of TIP60 between P41 and G42. The G42-containing protein contains the amino acids that comprise the interaction site necessary for forming the LSm and MyoX-coil. To obtain the expression system for the fragments, the DNA sequences encoding residues 1–41 (CoreN) and 42–140 (CoreC) of the single subunit of TIP60 were placed bicistronic on the same plasmid (Fig. 1c and Table S2, ESI†).
The fragments were co-expressed in E. coli cells and purified by Ni-NTA column chromatography, in the same manner as the original TIP60. The purified split proteins were separated by native polyacrylamide gel electrophoresis (PAGE), and a single band appeared at the position corresponding to that of the original TIP60 (Fig. 2a). Tricine sodium dodecyl sulfate (SDS)-PAGE analysis of the Ni-NTA purified protein revealed two bands at ∼6.5 and 10 kDa corresponding to the theoretical molecular weights of CoreN (6.0 kDa) and CoreC (11.8 kDa), respectively, although only CoreN contained the His-tag (Fig. 2b and Fig. S1, ESI†). This suggests that the split proteins formed an assembled structure similar to the original TIP60. The purified split proteins were further analyzed by transmission electron microscopy (TEM). The images revealed spherical nanoparticles as in the case of the original TIP60 (Fig. S2, ESI†). Since TIP60 was named based on its number of subunits, we similarly refer to the split TIP60 as TIP120. Small-angle X-ray scattering (SAXS) of TIP120 showed oscillating scattering typical of monodisperse large spherical particles40 as with TIP60 (Fig. 2c). The pair distance distribution (P(r)) showed slightly right-shifted Gaussian distribution, indicating a hollow spherical structure, which was also supported by ab initio modelling (Fig. 2d and Fig. S3a, ESI†). The radius of gyration (Rg) and maximum dimension (Dmax) were estimated as 9.2 and 22.5 nm, respectively, in good agreement with those obtained for TIP6032 (Table S3 and Fig. S4, ESI†). All of these data indicated that the two split fragments of 60 subunits each interacted to form a structure similar to the original TIP60.
 | ||
| Fig. 2 Analysis of the co-expressed split TIP60 fragments. (a) Native PAGE and (b) tricine SDS-PAGE analysis of the purified split protein (TIP120) and TIP60. (c) Scattering curve and (d) pair distance distribution obtained from the SAXS analysis of TIP120. | ||
Interestingly, the thermostability of TIP120 was slightly improved compared with TIP60, according to the results of the native PAGE analysis after heat treatment at various temperatures (Fig. S5, ESI†). The stability against protease was also improved in TIP120 (Fig. S6, ESI†). In considering that the proteolytic degradation is known to proceed more quickly in flexible regions or loops,41 TIP120 would have fewer unstable regions than TIP60. This could be due to the higher uniformity shown in Fig. 2a. We speculate that the uniformity would limit the access of the protease and improve the thermostability.
We next examined the in vitro assembly of TIP120 by simple mixing of the independently expressed and purified CoreN and CoreC (Fig. S7, ESI†). An additional His-tag was inserted in the N-terminus of CoreC for purification. Although CoreC contains both of the regions required for pentamer and dimer formation (Fig. 1), CoreC alone showed no high-molecular-weight band in native PAGE (Fig. S8a, ESI†). This suggests that CoreN is necessary for the assembly. The in vitro assembly of TIP120 was then evaluated by mixing the purified CoreN and CoreC in a molar ratio of 2:1, which was calculated from the SDS-PAGE band intensity of TIP120 observed for the in vivo co-expression system (Fig. 2b). In principle, the stoichiometry of the interaction between the split proteins should be 1:1. This discrepancy could be explained by the fact that the band intensity after staining with Coomassie Brilliant Blue depends on the cationic amino acid residues of the proteins, and thus the concentration of CoreN (pI = 8.30) would be overestimated in this analysis.42 The mixture was incubated for 16 h then analysed by native PAGE. As expected, a band was observed at the position corresponding to TIP120 (Fig. S8a, ESI†). Furthermore, TEM analysis revealed the spherical shape of TIP120 (Fig. S8b, ESI†).
We previously reported that TIP60 underwent dissociation to a dimeric structure upon introducing a mutation at the interface of the pentameric domain LSm (K67E).43 This mutation prevented pentamer formation owing to the electronic repulsion caused by the negatively charged carboxyl group. We demonstrated that the addition of alkaline-earth metal ions such as Ca2+, Sr2+, and Ba2+ induced 60-mer formation (mTIP60). Cryo-electron microscopy (cryo-EM) analysis indicated that the overall structure was almost identical to that of the original TIP60, where the metal ions were bound at the interface of the pentamer corresponding to the mutated region (PDB ID: 7XM1). Based on these findings, we speculated that the metal-ion-dependent reassociation system may also be applicable to TIP120, which allow reversible assembly and disassembly useful for future applications (Fig. 3a). Thus, the corresponding K26E mutation was introduced into CoreC (Fig. S7, ESI†). The mixture of CoreN and CoreC(K26E) did not afford any large structure according to the results of native PAGE analysis (Fig. 3b). By contrast, the addition of Ca2+, Sr2+, or Ba2+ ions to the mixture of the fragments resulted in a band located at the position corresponding to TIP120 (Fig. 3c). Other metal ions, including Mg2+, Na+, K+, and various transition metal ions (Mn2+, Fe2+, Fe3+, Co2+, Ni2+, Cu2+, and Zn2+), did not induce the association (Fig. 3c and Fig. S9, ESI†). The estimated binding affinity based on Trp fluorescence measurements was in the order Ba2+ > Sr2+ > Ca2+ (Fig. S10, ESI†). TEM images of the assembled protein revealed a hollow spherical shape (Fig. S11, ESI†). The assembly yield was quantified by size-exclusion chromatography as 91% with 4 mM BaCl2 (Fig. S12, ESI†). Furthermore, the addition of ethylenediaminetetraacetate (EDTA) induced the dissociation of this large structure as shown in Fig. S13 (ESI†), strongly indicating that the metal ions played a similar role as in the case of mTIP60. The metal-induced structure of the split mTIP60 is thus referred to as mTIP120. We next performed SAXS analysis of mTIP120 obtained in the presence of Ba2+ ions (mTIP120-Ba). The resulting scattering pattern was similar to that observed for TIP120 (Fig. 4a). In addition, the Rg and Dmax values were estimated as 9.4 and 22.4 nm, respectively, which were very similar to those of TIP6032 and TIP120 (Table S3 and Fig. S4, ESI†). The P(r) was more shifted to the right than in the case of TIP120, it was closer to the ideal P(r) for a hollow spherical structure (Fig. 4b). Ab initio modelling also indicated an almost empty interior (Fig. S3, ESI†). This difference was due to the encapsulation of foreign molecules from E. coli cells, as also observed in mTIP60.43 The oscillating scattering pattern disappeared upon the addition of EDTA and the Rg value decreased to 4.7 nm, suggesting the disassembly of mTIP120 (Fig. 4a and Fig. S4, Table S3, ESI†).
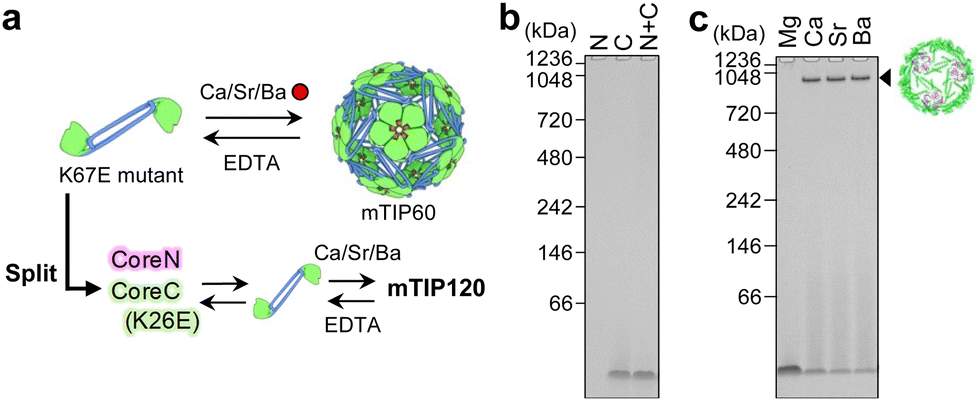 | ||
| Fig. 3 Metal-induced assembly of the split mTIP60 fragments. (a) Schematic illustration of the design of mTIP120. (b) Native PAGE analysis of the purified fragments (CoreN and CoreC(K26E)) and a mixture of them. (c) Native PAGE analysis of mixtures of the fragments with a series of alkaline-earth metal ions. | ||
 | ||
| Fig. 4 Structural analysis of mTIP120-Ba. (a) Scattering curves and (b) pair distance distributions of mTIP120 with Ba2+ (red) and excess EDTA (blue) from SAXS analysis. (c) Cryo-EM map (C1) of mTIP120-Ba. The model structure of mTIP60-Ba is superimposed. One CoreN and CoreC(K26E) unit are highlighted in green and magenta, respectively. | ||
The structure of mTIP120-Ba was further examined by cryo-EM single-particle analysis. The final map was obtained from 81,967 particles at a resolution of 7.44 Å (gold-standard Fourier shell correlation (FSC) criterion of 0.143; Fig. S14 and Fig. S15, ESI†). Although this resolution is not sufficient to discuss the atomic structure in detail, the map was well matched to the structures of the original TIP60 and mTIP60-Ba (Fig. 4c). The splitting appeared to have almost no impact on the overall structure. These data indicate that the newly incorporated N- and C-termini were introduced on the inner surface. Indeed, Hrv 3C Protease recognition sequence introduced at the new N-terminus of CoreC(K26E) was protected from digestion by the protease only when the cage structure was formed (Fig. S16, ESI†). Since the obtained map of mTIP120-Ba exhibited icosahedral symmetry, we further performed 3D refinement with I symmetry constraints. The resolution was improved to 4.58 Å from 54,925 particles, while the sphericity was 0.73 and a noisy FSC curve was observed around 5 Å (Fig. S17, ESI†). Although this suggests that the data quality was not sufficient to discuss the structure in detail, the map almost matched with the cryo-EM structure of mTIP60-Ba (Fig. S18a and b, ESI†) and a potential map was observed at a position similar to the Ba2+ binding site of mTIP60-Ba (Fig. S18c, ESI†).
Finally, we attempted to incorporate GFP in the inner cavity of mTIP120-Ba by designing a fusion protein GFP-CoreC(K26E). However, no cage formation was observed by simply mixing with CoreN, and Ba2+, suggesting that 60 GFP molecules were too many to be incorporated (50 GFPs per cage is the maximum based on volume ratio). To reduce the number of GFP molecules, we optimized the mixing ratio of CoreC(K26E):GFP-CoreC(K26E) and found that only a 58:2 ratio produced the cage with GFP fluorescence (Fig. S19, ESI†). The cage was not observed with 10 GFPs per cage which is well below the 50 GFPs per cage. The fused GFP likely inhibits assembly, possibly by causing structural distortion of the LSm pentamer or reducing the affinity for metal binding.
In conclusion, we redesigned the artificial protein cage TIP60 by splitting the subunit into two fragments which spontaneously reassociate into the same cage structure (TIP120). Furthermore, the split-protein system was applicable to mTIP60 (the K67E mutant), which associates and dissociates in response to alkaline-earth metal ions. We showed that the newly introduced termini after splitting were oriented toward the interior of the cage. One of the termini can be used as a genetic modification site for the encapsulation of GFP in the mTIP120 cage. We thus believe that the split-protein system not only allows the incorporation of various enzymes but also enables the simultaneous encapsulation of multi-species enzymes, holding promise for future catalytic cascade applications.
We thank the staff at the Structural Biology Research Center (SBRC) in KEK for cryo-EM grid preparation and data collection, and Prof. Nobutaka Shimizu and the staff of the Photon Factory (PF) for their help in synchrotron SAXS experiments, which were performed at PF, KEK, under the approval of the PF program advisory committee (proposal number 2022G667). This research was supported by the Research Support Project for Life Science and Drug Discovery (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under Grant Number JP21am0101071/JP23ama121001 (support number 1582), JSPS KAKENHI (Grant Numbers JP18K05324 to N. K., JP17KK0104 and JP19H02522 to R. A., and JP22J13988 to N. O.), the Japan Association for Chemical Innovation (JACI) 8th Research Encouragement Award to N. K., and the Keio University Doctorate Student Grant-in-Aid Program from Ushioda Memorial Fund to N. O.
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. B. Rosen and M. B. Francis, Nat. Chem. Biol., 2017, 13, 697–705 CrossRef CAS PubMed.
- S. Bloom, C. Liu, D. K. Kölmel, J. X. Qiao, Y. Zhang, M. A. Poss, W. R. Ewing and D. W. C. Macmillan, Nat. Chem., 2018, 10, 205–211 CrossRef CAS PubMed.
- E. A. Hoyt, P. M. S. D. Cal, B. L. Oliveira and G. J. L. Bernardes, Nat. Rev. Chem., 2019, 3, 147–171 CrossRef CAS.
- H. Mao, S. A. Hart, A. Schink and B. A. Pollok, J. Am. Chem. Soc., 2004, 126, 2670–2671 CrossRef CAS PubMed.
- D. M. Chudakov, S. Lukyanov and K. A. Lukyanov, Trends Biotechnol., 2005, 23, 605–613 CrossRef CAS PubMed.
- S. R. Adams, R. E. Campbell, L. A. Gross, B. R. Martin, G. K. Walkup, Y. Yao, J. Llopis and R. Y. Tsien, J. Am. Chem. Soc., 2002, 124, 6063–6076 CrossRef CAS PubMed.
- C. Jing and V. W. Cornish, Acc. Chem. Res., 2011, 44, 784–792 CrossRef CAS PubMed.
- E. Hochuli, W. Bannwarth, H. Dobeli, R. Gentzi and D. Stuber, Nat. Biotechnol., 1988, 6, 1321–1325 CrossRef CAS.
- C. L. Young, Z. T. Britton and A. S. Robinson, Biotechnol. J., 2012, 7, 620–634 CrossRef CAS PubMed.
- M. Si, Q. Xu, L. Jiang and H. Huang, PLoS One, 2016, 11, e0162318 CrossRef PubMed.
- J. Wang, T. Fang, M. Li, W. Zhang, Z. P. Zhang, X. E. Zhang and F. Li, J. Mater. Chem. B, 2018, 6, 3716–3726 RSC.
- T. Inoue, M. Kawano, R. Takahashi, H. Tsukamoto, T. Enomoto, T. Imai, K. Kataoka and H. Handa, J. Biotechnol., 2008, 134, 181–192 CrossRef CAS PubMed.
- J. E. Padilla, C. Colovos and T. O. Yeates, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 2217–2221 CrossRef CAS PubMed.
- N. Kobayashi, K. Yanase, T. Sato, S. Unzai, M. H. Hecht and R. Arai, J. Am. Chem. Soc., 2015, 137, 11285–11293 CrossRef CAS PubMed.
- J. Marcandalli, B. Fiala, S. Ols, M. Perotti, W. de van der Schueren, J. Snijder, E. Hodge, M. Benhaim, R. Ravichandran, L. Carter, W. Sheffler, L. Brunner, M. Lawrenz, P. Dubois, A. Lanzavecchia, F. Sallusto, K. K. Lee, D. Veesler, C. E. Correnti, L. J. Stewart, D. Baker, K. Loré, L. Perez and N. P. King, Cell, 2019, 176, 1420–1431.e17 CrossRef CAS PubMed.
- D. P. Goldenberg and T. E. Creighton, J. Mol. Biol., 1983, 165, 407–413 CrossRef CAS PubMed.
- Y. Yu and S. Lutz, Trends Biotechnol., 2011, 29, 18–25 CrossRef CAS PubMed.
- T. Zhang, E. Bertelsen, D. Benvegnu and T. Alber, Biochemistry, 1993, 32, 12311–12318 CrossRef CAS PubMed.
- G. S. Baird, D. A. Zacharias and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11241–11246 CrossRef CAS PubMed.
- Y. Lee, M. Kim, J. Y. Kang and Y. Jung, ACS Appl. Mater. Interfaces, 2022, 14, 35357–35365 CrossRef CAS.
- A. R. Viguera, F. J. Blanco and L. Serrano, J. Mol. Biol., 1995, 247, 670–681 CAS.
- J. Ay, M. Hahn, K. Decanniere, K. Piotukh, R. Borriss and U. Heinemann, Proteins: Struct., Funct., Genet., 1998, 30, 155–167 CrossRef CAS.
- Z. Qian, J. R. Horton, X. Cheng and S. Lutz, J. Mol. Biol., 2009, 393, 191–201 CrossRef CAS PubMed.
- K. Ko, I. Hu, K. Huang, P. Lyu and S. D. Hsu, Structure, 2019, 27, 1224–1233 CrossRef CAS PubMed.
- S. S. Shekhawat and I. Ghosh, Curr. Opin. Chem. Biol., 2011, 15, 789–797 CrossRef CAS.
- J. N. Pelletier, F. X. Campbell-Valois and S. W. Michnick, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 12141–12146 CrossRef CAS PubMed.
- N. Johnsson and A. Varshavsky, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 10340–10344 CrossRef CAS PubMed.
- K. Camacho-Soto, J. Castillo-Montoya, B. Tye, L. O. Ogunleye and I. Ghosh, J. Am. Chem. Soc., 2014, 136, 17078–17086 CrossRef CAS.
- A. Walker, C. Skamel and M. Nassal, Sci. Rep., 2011, 1, 1–8 CrossRef.
- I. Ghosh, A. D. Hamilton and L. Regan, J. Am. Chem. Soc., 2000, 122, 5658–5659 CrossRef CAS.
- T. J. Magliery, C. G. M. Wilson, W. Pan, D. Mishler, I. Ghosh, A. D. Hamilton and L. Regan, J. Am. Chem. Soc., 2005, 127, 146–157 CrossRef CAS PubMed.
- N. Kawakami, H. Kondo, Y. Matsuzawa, K. Hayasaka, E. Nasu, K. Sasahara, R. Arai and K. Miyamoto, Angew. Chem., Int. Ed., 2018, 57, 12400–12404 CrossRef CAS PubMed.
- J. Obata, N. Kawakami, A. Tsutsumi, E. Nasu, K. Miyamoto, M. Kikkawa and R. Arai, Chem. Commun., 2021, 57, 10226–10229 RSC.
- D. Das, P. Kozbial, H. L. Axelrod, M. D. Miller, D. McMullan, S. S. Krishna, P. Abdubek, C. Acosta, T. Astakhova, P. Burra, D. Carlton, C. Chen, H. J. Chiu, T. Clayton, M. C. Deller, L. Duan, Y. Elias, M. A. Elsliger, D. Ernst, C. Farr, J. Feuerhelm, A. Grzechnik, S. K. Grzechnik, J. Hale, G. W. Han, L. Jaroszewski, K. K. Jin, H. A. Johnson, H. E. Klock, M. W. Knuth, A. Kumar, D. Marciano, A. T. Morse, K. D. Murphy, E. Nigoghossian, A. Nopakun, L. Okach, S. Oommachen, J. Paulsen, C. Puckett, R. Reyes, C. L. Rife, N. Sefcovic, S. Sudek, H. Tien, C. Trame, C. V. Trout, H. Van Den Bedem, D. Weekes, A. White, Q. Xu, K. O. Hodgson, J. Wooley, A. M. Deacon, A. Godzik, S. A. Lesley and I. A. Wilson, Proteins: Struct., Funct., Bioinf., 2009, 75, 296–307 CrossRef CAS PubMed.
- Q. Lu, F. Ye, Z. Wei, Z. Wen and M. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17388–17393 CrossRef CAS PubMed.
- E. Nasu, N. Kawakami and K. Miyamoto, ACS Appl. Nano Mater., 2021, 4, 2434–2439 CrossRef CAS.
- M. Yamashita, N. Kawakami and K. Miyamoto, ChemPlusChem, 2023, 88, e202200392 CrossRef CAS.
- Y. Azuma, M. Herger and D. Hilvert, J. Am. Chem. Soc., 2018, 140, 558–561 CrossRef CAS.
- Y. Azuma, T. G. W. Edwardson, N. Terasaka and D. Hilvert, J. Am. Chem. Soc., 2018, 140, 566–569 CrossRef CAS PubMed.
- O. Glatter and O. Kratky, Small angle X-ray scattering, Academic Press, 1982 Search PubMed.
- K. Tsuboyama, J. Dauparas, J. Chen, E. Laine, Y. Mohseni Behbahani, J. J. Weinstein, N. M. Mangan, S. Ovchinnikov and G. J. Rocklin, Nature, 2023, 620, 434–444 CrossRef CAS PubMed.
- H. K. Ku, H. M. Lim, K. H. Oh, H. J. Yang, J. S. Jeong and S. K. Kim, Anal. Biochem., 2013, 434, 178–180 CrossRef CAS PubMed.
- N. Ohara, N. Kawakami, R. Arai, N. Adachi, T. Moriya, M. Kawasaki and K. Miyamoto, J. Am. Chem. Soc., 2023, 145, 216–223 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods and supplementary figures. See DOI: https://doi.org/10.1039/d4cc00115j |
| This journal is © The Royal Society of Chemistry 2024 |