
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bacterial pseudaminic acid binding to Siglec-10 induces a macrophage interleukin-10 response and suppresses phagocytosis†
I-Ming
Lee
a,
Hsing-Yu
Wu
b,
Takashi
Angata
*b and
Shih-Hsiung
Wu
 *b
*b
aDepartment of Marine Biotechnology and Resources, National Sun Yat-sen University, No. 70 Lien-hai Road, Kaohsiung 804201, Taiwan
bInstitute of Biological Chemistry, Academia Sinica, 128 Academia Road, Section 2, Nankang, 11529, Taipei, Taiwan. E-mail: shwu@gate.sinica.edu.tw
First published on 12th February 2024
Abstract
Pseudaminic acid (Pse) on pathogenic bacteria exopolysaccharide engages with the sialic acid-binding immunoglobulin-type lectin (Siglec)-10 receptor on macrophages via the critical 7-N-acetyl group. This binding stimulates macrophages to secrete interleukin 10 that suppresses phagocytosis against bacteria, but can be reverted by blocking Pse-Siglec-10 interaction with Pse-binding protein as a promising therapy.
Nonulsonic acids, a family of nine-carbon α–keto acid monosaccharides, are components of cell surface-associated glycans and are involved in various cellular interactions responsible for cell adhesion, signaling and receptor regulation.1–4 Pseudaminic acid (Pse), a member of the non-mammalian nonulsonic acids, has been regarded as a critical virulence factor of pathogenic bacteria. In Campylobacter jejuni and Helicobacter pylori, post-translational Pse decoration is necessary for functional flagellin assembly.5,6 The exopolysaccharides (EPSs) of multi-drug resistant (MDR) pathogens, such as Acinetobacter baumannii and Enterobacter species, also contain Pse that assists bacteria to evade the host immune response, causing the therapeutic affliction.7–11 Therefore, the mechanism by which Pse modulates the host immune response has drawn tremendous attention in recent years.
Pse is structurally similar to sialic acid (Sia), which modulates the immune responses of vertebrates by binding with the sialic acid-binding immunoglobulin-type lectin (Siglec) receptors on immune cells.12 The N-terminal variable (V)-set domain of Siglecs recognizes ligands to trigger signaling through regulatory motifs in the cytoplasmic tail.13 Most Siglecs have immunoreceptor tyrosine-based inhibitory motifs (ITIMs) that recruit tyrosine phosphatase SHP-1 and SHP-2 to attenuate immune system activation.14,15 With diverse Sia-related glycans, Siglecs can regulate the innate/adaptive immune system and discriminate between “self” and “non-self” cells.16–18 Hence, the Pse on the bacterial pathogens probably also modulates the host immune system by binding with the specific Siglecs.
When a ligand binds its cognate receptor on immune cells, immune signaling orchestrates the response by cytokine release. Cytokines are the small secreted proteins that regulate host response to infections and inflammation, including interleukins, interferons, colony-stimulating factors, and many growth factors.19,20 Cytokines can be mainly classified into pro-inflammatory and anti-inflammatory types. Pro-inflammatory cytokines are predominantly secreted from activated immune cells to up-regulate inflammatory reactions. Conversely, anti-inflammatory cytokines may either inhibit the pro-inflammatory cytokine synthesis or control pro-inflammatory cytokine-mediated cellular activities.21,22 Most pathogens result in an imbalance of pro and anti-inflammatory cytokines upon invading the host, which is one of the critical mechanisms to cause diseases.23
Pse on the flagella of Campylobacter jejuni was reported to stimulate the secretion of anti-inflammatory cytokine interleukin-10 (IL-10) from macrophages via Siglec-10,24 but we investigated whether Pse-containing polysaccharides exhibit a similar effect in this study. Our previous studies revealed that Pse was involved in the EPS of A. baumannii strain 54149 (Ab-54149 EPS), which can be digested by phage ΦAB6 tail-spike protein (ΦAB6TSP).25,26 To test the involvement of Siglec-10 in the recognition and response to Pse-containing Ab-54149 EPS, we prepared human macrophages THP-1 in which Siglec-10 was knocked-down by shRNA (Fig. 1A and Fig. S1, ESI†). Ab-54149 EPS can stimulate THP-1 to release IL-10 but not the Siglec-10 knock-down THP-1 (Fig. 1B and Fig. S2, ESI†). Moreover, IL-10 production by THP-1 macrophages was attenuated when the cells were treated with Pse-depleted Ab-54149 EPS (de-Pse Ab-54149 EPS)27 (Fig. 1C). All observations were reproduced when ΦAB6TSP digested oligosaccharide was used in place of Ab-54149 EPS. As two repeat units of Ab-54149 EPS, the ΦAB6TSP digested oligosaccharide induced a similar IL-10 level to Ab-54149 EPS, but it decreased when Pse on the oligosaccharide was diminished or Siglec-10 was knocked-down in the THP-1 cells (Fig. S3, ESI†). With these results, the compelling evidence confirmed that Pse served as a ligand of Siglec-10 to modulate macrophage response.
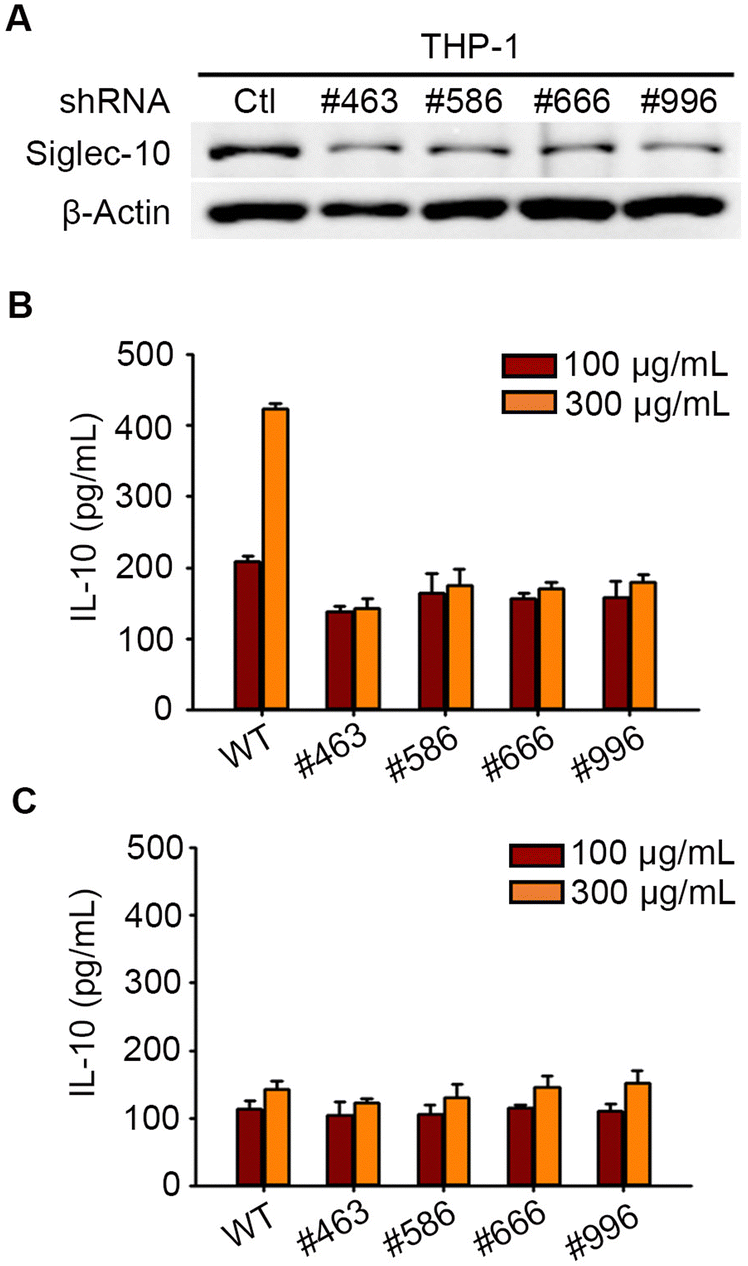 | ||
| Fig. 1 (A) Western blot revealed that shRNA #463, #586, #666, and #996 can knock-down the expression of Siglec-10 on THP-1 but not β-Actin. The wild type (WT) THP-1 and the Siglec-10 knock-down cells were treated with Ab-54149 EPS (B) and de-Pse Ab-54149 EPS (C). | ||
Like C. jejuni, Pse-coated pathogens Ab-54149 and Enterobacter cloacae 13047 (Ec-13047) can activate IL-10 release from THP-1 cells, but not the non Pse-coated strain Ab-SK44 (Fig. S4, ESI†). For comparing the bio-activity of Pse to Sia, we also challenged the THP-1 with five Sia-derived glycans generated by chemical synthesis, including Sia-(2,3)-α-Gal-(1,4)-β-Glc (L1), Sia-(2,3)-α-Gal-(1,4)-β-GlcNAc (L2), Sia-(2,3)-α-Gal-(1,3)-β-GalNAc (L3), Sia-(2,6)-α-Gal-(1,4)-β-GlcNAc (L4) and Sia-(2,8)-α-Sia (L5) (Gal: galactose, Glc: glucose, GalNAc: N-acetyl galactosamine, GlcNAc: N-acetyl glucosamine) (Fig. S5, ESI†). Sia-(2,6)-α-Gal-(1,4)-β-GlcNAc was reported to be a preferred ligand of Siglec-10 and could induce a similar degree of IL-10 from THP-1 macrophages as the ΦAB6TSP digested product and other Sia-Gal derived glycans (Fig. S6, ESI†). However, the IL-10 release caused by Sia-(2,8)-α-Sia was apparently minor compared to the ΦAB6TSP digested oligosaccharide (Fig. 2A). Several studies have indicated that the binding affinity of the ligand toward the receptor contributes to the difference of immune response.28,29 Indeed, the binding affinity of ΦAB6TSP digested oligosaccharide toward Siglec-10 was nearly 6-fold higher than that of Sia-(2,8)-α-Sia (Fig. 2B and C).
 | ||
| Fig. 2 (A) The IL-10 secretion from THP-1 by treating with ΦAB6TSP digested oligosaccharide and Sia-(2,8)-α-Sia. The binding affinities of the ΦAB6TSP digested product (B) and Sia-(2,8)-α-Sia (C) toward Siglec-10 were 0.488 ± 0.186 μM and 3.13 ± 1.23 μM, respectively. The binding affinity was detected by fluorescence titration and the different concentrations of the ligand were recorded in a curve with different colors. | ||
To understand the binding difference at the atomic level, we aimed to analyze the structure of Siglec-10 in complex with Pse/Sia glycans. Since the real structure of Siglec-10 has not been solved yet, we predicted the structure of the N-terminal V-set domain of Siglec-10 by homology modeling, and simulated its interaction with oligosaccharide by computational simulation. The sequence alignment of Siglec-10 with respect to other known structure CD-33 family members, including Siglec-5, Siglec-7 and Siglec-8, revealed high similarity (Fig. S7A, ESI†).30 By using the structures of these Siglecs as homology modeling templates, the 3D structural model of Siglec-10 was constructed (Fig. S7B, ESI†). The V-set domain of Siglec-10, like the canonical Siglec lectin domain fold, displayed the Ig-like β-sandwich of two antiparallel β-sheets formed by strands ABED and C′CFG (Fig. S7C, ESI†). Notably, the C and G strands were split into two shorter strands, which are connected by a CC′ loop and GG′ loop, respectively (Fig. S7D, ESI†).
Most carbohydrate binding sites of Siglecs are located in the groove between the CC′ and GG′ loops. The CC′ loop protrudes away from the main body of these Siglec structures and its conformation allows the entrance of sialo-glycans into the binding groove. Part of the CC′ loop contributes the interactions to the carbohydrate moiety of the ligand and thus the residue diversity of the CC′ loop determines the substrate specificity of individual Siglecs. Besides, the known structure of Siglecs in complex with oligosaccharides demonstrates that the conserved arginine plays a critical role in interaction with the carboxyl group on the terminal Sia. Here, the one repeat unit of Ab-54149 EPS (Ab-OU) and five Sia-derived glycans were docked into the binding groove between the CC′ and GG′ loops on Siglec-10. With the optimized docking parameters, we found that the carboxyl groups on both Pse and Sia have a polar interaction with R119, which is an equivalent of Sia-engaged R124, R124 and R119 on Siglec-7, 5 and 8, respectively (Fig. 3A and Fig. S7E and S8, ESI†). R119 may slightly change its orientation while Siglec-10 has substantial binding with Pse/Sia containing glycans but can be not shown in the docking structure.31,32 Near the CC′ loop, the locations of R127, Y128 and N129 of Siglec-10 are highly similar to Sia-engaged residues in Siglec-5, 7 and 8 (Fig. S7E, ESI†). R127 formed hydrogen bonds with the 7-N-acetyl group on Pse of Ab-OU that is different from the 5-N-acetyl group on Sia of L2, L3 and L4, binding with K131 in Siglec-7, K132 in Siglec-5 and K116 in Siglec-8, respectively (Fig. 3A). Y128 forms a hydrophobic interaction with the methyl group on the 7-N-acetyl of Pse in contrast to the C9 methylene on Sia of L1–L4, like W132 in Siglec-7, Y133 in Siglec-5 and W117 in Siglec-8. N129 also formed hydrogen bonds with the Pse C4 hydroxyl group but majorly interacted with the Sia C9 hydroxyl group of L1–L4, like N133 in Siglec-7, S134 in Siglec-5 and N118 in Siglec-8. Other unique interactions between Siglec-10 and ligands occurred in the 3-hydroxyl group on Gal of Ab-OU with T68, the 1-hydroxyl group on Glc of L1 with T68, the 2-N-acetyl group on GalNAc of L3 with Y125 and the 6-hydroxyl group on L4 with T67, respectively (Fig. S8, ESI†). On L5, only the C9 on the second Sia engages with R127 and Y128 besides the R119 interaction (Fig. 3B). Ligand binding of Siglec-10 can probably be affected by the linkage between Pse/Sia and the penultimate sugar. The ligand with Sia in the (2,3) and (2,6) linkages seems to fit well in the groove between the CC′/GG′ loop and have more contacts with the non-conventional binding residues on Siglec, like T67, T68 and Y125, but the ligand with Sia in the (2,8) linkage likely projects out from the surface of Siglec-10. Accordingly, more interactions of Ab-OU and L1–L4 toward Siglec-10 than L5 may explain the stronger binding and more release of IL-10.
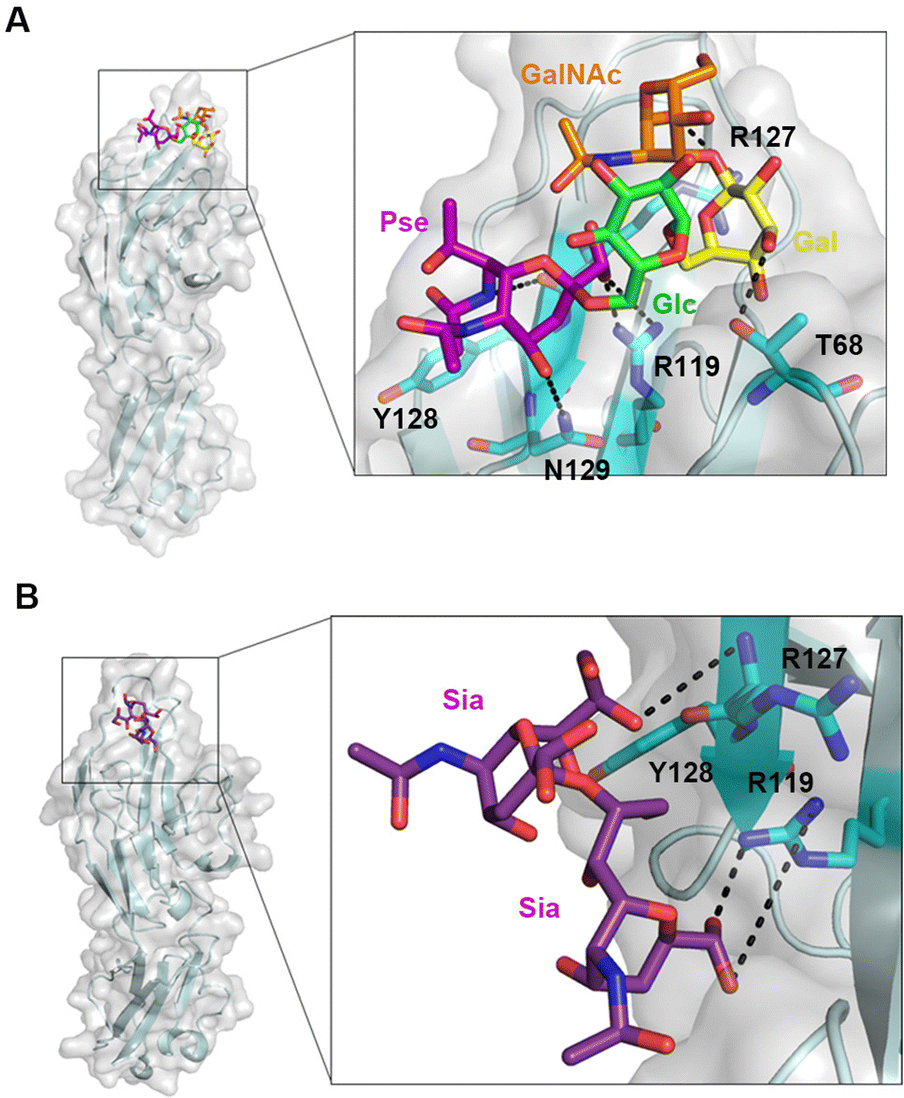 | ||
| Fig. 3 (A) The interactions between Ab-OU and Siglec-10. (B) The interactions between L5 and Siglec-10. The engaged residues on Siglec-10 were colored in cyan and labeled in black. The interactions were represented by a black dashed line. | ||
Moreover, IL-10 is known to modulate bacterial phagocytosis by macrophages. Here, we labeled Ab-54149 with fluorescein isothiocyanate (FITC) and then incubated it with THP-1 macrophages. The ability of THP-1 macrophages to phagocytose Ab-54149 was tested by analyzing the fluorescence intensity of the THP-1 cell lysate after the removal of bacteria in the medium. Upon treatment with FITC-Ab-54149, WT THP-1 has apparently lower fluorescence than Siglec-10 knock-down THP-1, indicating that IL-10 induced by Pse and Siglec-10 binding reduced the phagocytosis (Fig. 4A). However, the phagocytosis of THP-1 can be restored when Ab-54149 was first incubated with catalytic residues mutated in ΦAB6TSP that can particularly recognize the Pse but are deficient in glycosidase activity to block its interaction with Siglec-10.27 A similar result was observed in the case of Enterobacter cloacae ATCC 13047 that contained the Pse on the surface EPS as well (Fig. 4B).9 IL-10 release caused by Pse-derived glycan can also attenuate macrophage phagocytosis toward other non-Pse coated bacteria. THP-1 macrophages could phagocytose FITC-labeled Escherichia coli BL-21 and Pseudomonas aeruginosa PA14 in a concentration dependent manner (Fig. S9, ESI†). However, the phagocytosis of these bacteria dramatically reduced when THP-1 macrophages were treated with Ab-54149 EPS but the Siglec-10 knockdown THP-1 macrophages were insensitive to EPS (Fig. S10, ESI†).
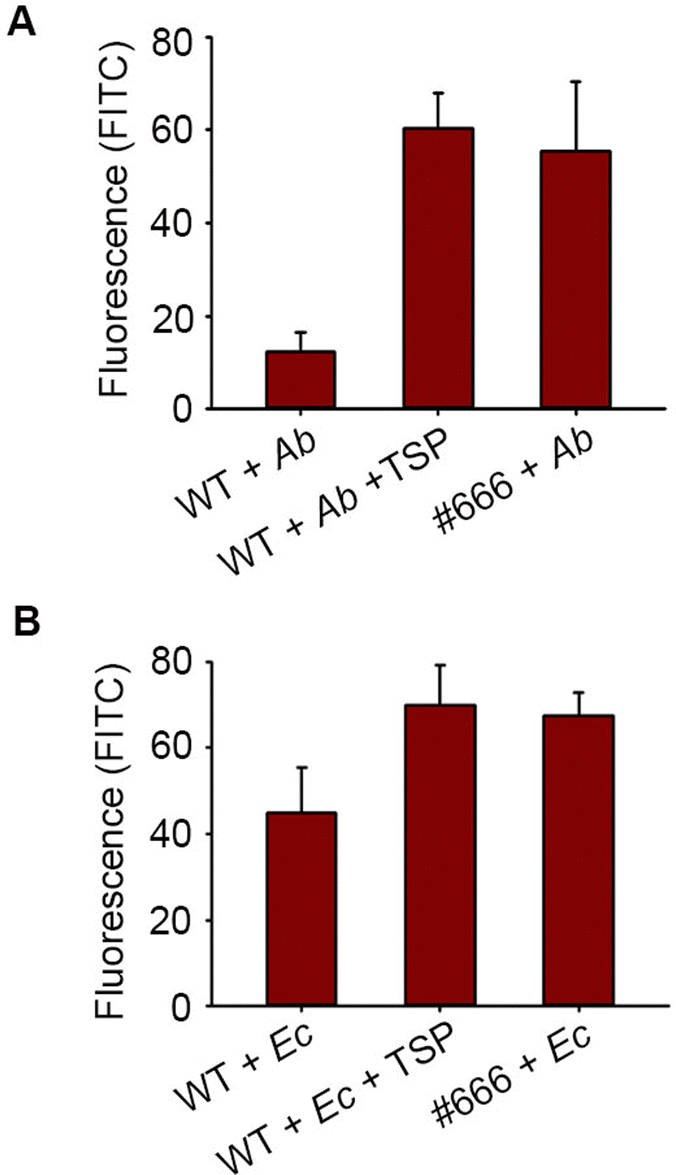 | ||
| Fig. 4 (A) Phagocytosis of Ab-54149 by THP-1. (B) Phagocytosis of Ec-13047 by THP-1 (Ab: Ab-54149, Ec: Ec-13047, WT: THP-1, #666: Siglec-10 knockdown THP-1 with shRNA 666, TSP: enzyme-inactive ΦAB6TSP). | ||
Taken together, our results revealed a unique survival mechanism by pathogenic bacteria in the host involving Pse and Siglec-10 that diminish the host immune response, like macrophage phagocytosis. Other bacterial surface glycans containing Pse/Sia may also engage cognate Siglec member(s), with varying outcomes. Notably, blocking the interaction between Pse and Siglec-10 may restore the immune defense (i.e., bacterial phagocytosis), and might represent a unique therapeutic modality to combat the infection by Pse-coated pathogenic bacteria, especially those with antibiotic resistance.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. B. Tomek, B. Janesch, D. Maresch, M. Windwarder, F. Altmann, P. Messner and C. Schaffer, Glycobiology, 2017, 27, 555–567 CrossRef CAS PubMed.
- S. Bloch, M. B. Tomek, V. Friedrich, P. Messner and C. Schäffer, Interface Focus, 2019, 9, 20180064–20180075 CrossRef PubMed.
- L. Kandiba and J. Eichler, FEMS Microbiol. Lett., 2013, 345, 110–120 CrossRef CAS PubMed.
- N. D. McDonald and E. F. Boyd, Trends Microbiol., 2021, 29, 142–157 CrossRef CAS PubMed.
- A. I. M. Salah Ud-Din and A. Roujeinikova, Cell. Mol. Life Sci., 2018, 75, 1163–1178 CrossRef CAS PubMed.
- P. Hitchen, J. Brzostek, M. Panico, J. A. Butler, H. R. Morris, A. Dell and D. Linton, Microbiology, 2010, 156, 1953–1962 CrossRef CAS PubMed.
- J. J. Kenyon, A. M. Marzaioli, R. M. Hall and C. De Castro, Glycobiology, 2014, 24, 554–563 CrossRef CAS PubMed.
- A. Davin-Regli and J. M. Pages, Front. Microbiol., 2015, 6, 392 Search PubMed.
- A. V. Perepelov, M. Wang, A. V. Filatov, X. Guo, A. S. Shashkov, L. Wang and Y. A. Knirel, Carbohydr. Res., 2015, 407, 59–62 CrossRef CAS PubMed.
- A. M. Gonzalez-Villoria and V. Valverde-Garduno, J. Pathog., 2016, 2016, 7318075 Search PubMed.
- Z. A. Qureshi, L. E. Hittle, J. A. O'Hara, J. I. Rivera, A. Syed, R. K. Shields, A. W. Pasculle, R. K. Ernst and Y. Doi, Clin. Infect. Dis., 2015, 60, 1295–1303 CrossRef PubMed.
- K. F. Bornhofft, T. Goldammer, A. Rebl and S. P. Galuska, Dev. Comp. Immunol., 2018, 86, 219–231 CrossRef PubMed.
- P. R. Crocker, J. C. Paulson and A. Varki, Nat. Rev. Immunol., 2007, 7, 255–266 CrossRef CAS PubMed.
- Y. C. Chang and V. Nizet, Glycobiology, 2014, 24, 818–825 CrossRef CAS PubMed.
- Y. C. Liu, M. M. Yu, Y. F. Chai and S. T. Shou, Front. Immunol., 2017, 8, 1601 CrossRef PubMed.
- A. Varki and P. Gagneux, Ann. N. Y. Acad. Sci., 2012, 1253, 16–36 CrossRef CAS PubMed.
- T. Angata, C. M. Nycholat and M. S. Macauley, Trends Pharmacol. Sci., 2015, 36, 645–660 CrossRef CAS PubMed.
- T. Angata, Mol. Diversity, 2006, 10, 555–566 CrossRef CAS PubMed.
- J. M. Zhang and J. An, Int. Anesthesiol. Clin., 2007, 45, 27–37 CrossRef PubMed.
- A. Ray, K. Gulati, J. Joshi, S. Guhathakurta and N. Rai, MOJ Immunol., 2016, 4, 00121–00129 Search PubMed.
- P. Wojdasiewicz, L. A. Poniatowski and D. Szukiewicz, Mediators Inflammation, 2014, 2014, 561459 Search PubMed.
- M. Liu, J. Saredy, R. Zhang, Y. Shao, Y. Sun, W. Y. Yang, J. Wang, L. Liu, C. T. Drummer, C. Johnson, F. Saaoud, Y. Lu, K. Xu, L. Li, X. Wang, X. Jiang, H. Wang and X. Yang, Front. Immunol., 2020, 11, 554301 CrossRef CAS PubMed.
- F. Cristofori, V. N. Dargenio, C. Dargenio, V. L. Miniello, M. Barone and R. Francavilla, Front. Immunol., 2021, 12, 578386 CrossRef CAS PubMed.
- H. N. Stephenson, D. C. Mills, H. Jones, E. Milioris, A. Copland, N. Dorrell, B. W. Wren, P. R. Crocker, D. Escors and M. Bajaj-Elliott, J. Infect. Dis., 2014, 210, 1487–1498 CrossRef CAS PubMed.
- I. M. Lee, I. F. Tu, F. L. Yang, T. P. Ko, J. H. Liao, N. T. Lin, C. Y. Wu, C. T. Ren, A. H. Wang, C. M. Chang, K. F. Huang and S. H. Wu, Sci. Rep., 2017, 7, 42711 CrossRef CAS PubMed.
- I. M. Lee, F. L. Yang, T. L. Chen, K. S. Liao, C. T. Ren, N. T. Lin, Y. P. Chang, C. Y. Wu and S. H. Wu, J. Am. Chem. Soc., 2018, 140, 8639–8643 CrossRef CAS PubMed.
- I. M. Lee, I. F. Tu, F. L. Yang and S. H. Wu, J. Am. Chem. Soc., 2020, 142, 19446–19450 CrossRef CAS PubMed.
- S. V. Kundapura and U. A. Ramagopal, Sci. Rep., 2019, 9, 19191 CrossRef CAS PubMed.
- N. Jiang, W. Chen, P. Jothikumar, J. M. Patel, R. Shashidharamurthy, P. Selvaraj and C. Zhu, Mol. Biol. Cell, 2016, 27, 3449–3458 CrossRef CAS PubMed.
- R. E. Forgione, C. Di Carluccio, J. Guzman-Caldentey, R. Gaglione, F. Battista, F. Chiodo, Y. Manabe, A. Arciello, P. Del Vecchio, K. Fukase, A. Molinaro, S. Martin-Santamaria, P. R. Crocker, R. Marchetti and A. Silipo, iScience, 2020, 23, 101231 CrossRef CAS PubMed.
- J. M. Propster, F. Yang, S. Rabbani, B. Ernst, F. H. Allain and M. Schubert, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E4170–4179 CrossRef CAS PubMed.
- M. S. Alphey, H. Attrill, P. R. Crocker and D. M. van Aalten, J. Biol. Chem., 2003, 278, 3372–3377 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc00077c |
| This journal is © The Royal Society of Chemistry 2024 |