
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStrain-induced carbon–carbon bond cleavage of bowl-shaped sumanenone†‡
Mikey
Nishimoto
a,
Yuta
Uetake
 ab,
Yumi
Yakiyama
ab and
Hidehiro
Sakurai
*ab
ab,
Yumi
Yakiyama
ab and
Hidehiro
Sakurai
*ab
aDivision of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: hsakurai@chem.eng.osaka-u.ac.jp
bInnovative Catalysis Science Division, Institute for Open and Transdisciplinary Research Initiatives (ICS-OTRI), Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan
First published on 13th March 2024
Abstract
This study details a highly effective ring-opening reaction that involves acid-mediated carbon–carbon bond cleavage of the buckybowl, sumanenone. The reaction of the bowl-shaped sumanenone with AcOH and TfOH results in the formation of a planar carboxylic acid. The examination of reactivity in comparison to planar analogues, along with theoretical calculations, suggests that the release of curved strain is a crucial factor for the success of this reaction.
The inherent strain in a molecule has a significant influence on its reactivity. As a representative example, Bertozzi et al. reported that the azide–alkyne cycloaddition reaction proceeds without a copper catalyst using strained alkynes.1 Curved π-conjugated molecules such as fullerenes2,3 and buckybowls4–6 are also regarded as destabilized substrates in comparison with planar π-conjugated molecules; therefore, they exhibit unique reactivities (Fig. 1a). Previous reports demonstrated that addition reactions involving dearomatization, which are challenging to initiate in planar molecules, successfully occur in the context of curved π-conjugated molecules.7–13 For instance, the ring-opening of fullerenes arising from cycloaddition reactions has been employed to synthesize azafullerene dimer (C59N)2,14 and diverse endohedral fullerenes by “molecular surgery” methods.15–17 Although reports are more limited than for addition reactions, C–C bond cleavage reactions are also initiated by curved strain in several buckybowls (Scheme S1, ESI‡). Shionoya et al. demonstrated an iridium-catalyzed C–C bond cleavage of corannulene bearing a 2-pyridyl group as a directing group.18 Shao et al. observed cleavage at the peripheral C–C double bond of trithiasumanene with electron-donating substituents, achieved through Oxone® oxidation, leading to the synthesis of planar π-conjugated molecules. These molecules were then employed to construct [5-6-7] fused poly-heterocycles, exhibiting unique photophysical properties derived from a donor–acceptor system.19 Consequently, the progression of reactions harnessing curved strain holds the potential to offer a pathway to access unique molecular skeletons that pose challenges in synthesis from planar molecules.
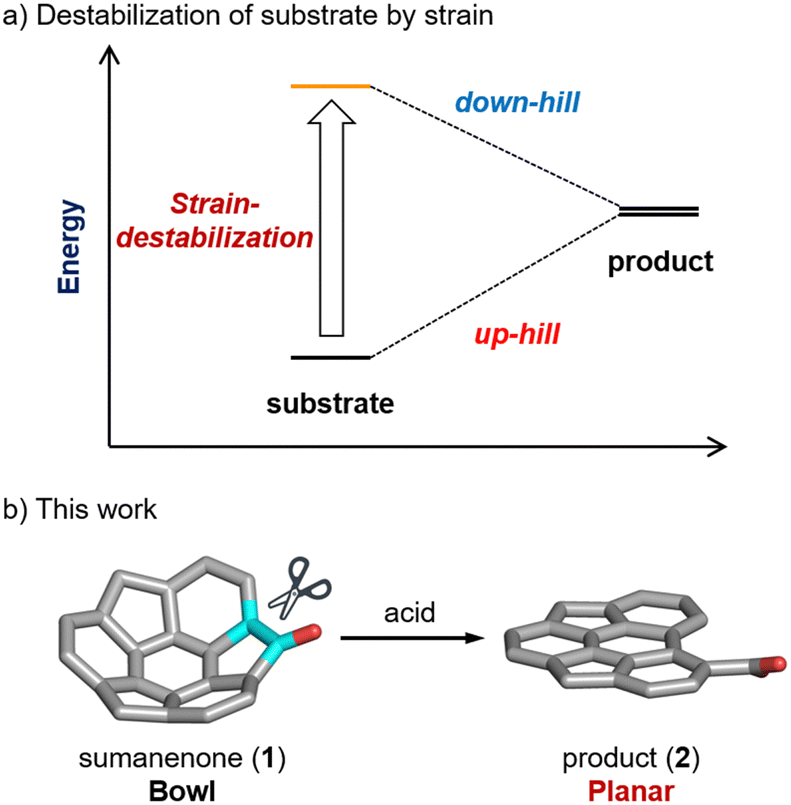 | ||
| Fig. 1 (a) Energy diagram about the destabilization of the substrate by strain. (b) Strain-induced C–C bond cleavage of sumanenone (1). | ||
Sumanene,20,21 a C60 fragment buckybowl as well as corannulene, possesses aromatic rings and reactive benzyl positions on its periphery. Investigations into both peripheral and internal functionalisation have been conducted. The sumanene bowl structure is more distorted than that of corannulene. Our group has reported osmylation to aromatic ring in sumanene13 and the mild C–C bond cleavage of 2-hydroxyphenyl sumanenetrione.22 In the latter case, the presence of the 2-hydroxyphenyl group is essential for the reaction to proceed. Herein, we report the Brønsted acid-mediated five-membered ring-opening of sumanenone (1) via C–C bond cleavage (Fig. 1b). Experimental and theoretical approaches were employed to investigate the origin of the difference in reactivity between a bowl-shaped molecule and a planar compound having a similar π-conjugation. In contrast to prior examples, this reaction advances without the need for directing groups or electron-donating substituents, clearly demonstrating the influence of inherent strain in the molecule on reactivity.
We have previously reported that Baeyer–Villiger oxidation of sumanenone (1) was accelerated by the addition of Brønsted acids.23 In the current study, we observed that the five-membered ring-opening reaction of 1 led to the formation of carboxylic acid (2) in the presence of trifluoromethanesulfonic acid (TfOH) (500 mol%) in acetic acid under heating (Scheme 1). The 1H NMR spectrum of the crude mixture indicated the complete consumption of 1, with only product peaks observed, suggesting the clean formation of 2. After extraction and washing with n-hexane, the pure product was isolated in 91% yield. Notably, this reaction did not proceed without TfOH, and 1 was fully recovered, suggesting the essential role of a strong acid.
 | ||
| Scheme 1 Strain-induced ring-opening of 1. | ||
The thus-obtained ring-opened product 2 was characterized by high-resolution mass spectrometry (HRMS) and NMR spectroscopy. The HRMS displayed m/z = 296.0837 ([M+]), indicating the formation of 2. The DFT-optimised structure of 2 exhibited a planar structure due to the ring-opening process. The 1H NMR spectra and their assignments for 1 and 2 are shown in Fig. 2, corroborated by DFT calculation (Table S1, ESI‡). A comparison of the 1H NMR spectra between 1 and 2 revealed an increase in signals in the aromatic region of 2, accounting for seven protons (Fig. 2). The signal for Hi in Fig. 2b appeared at a lower magnetic field than other aromatic protons, suggesting a deshielding effect from the adjacent carboxyl group. In the case of 1, the proton signals at the benzyl position are observed as a doublet with the J-coupling constant of approx. 20 Hz, reflecting the geminal coupling between endo- and exo-protons due to the deep bowl structure.24 In contrast, those of 2 appeared as a singlet peak (Hc: 4.48 ppm, Hf: 4.51 ppm) owing to the planarisation of the molecular framework. This planarised ring-opened framework was also supported by the single-crystal X-ray structure analysis by converting from 2 to the corresponding 4-bromophenyl ester (3) (Scheme 1 and Fig. S1, ESI‡).
 | ||
| Fig. 2 1H NMR spectra of (a) 1 in CDCl3 and (b) 2 in DMSO-d6. | ||
The ring-opening hydrolysis of fluorenone (4), a substructure of 1, has been reported to occur under high-temperature conditions and with the addition of an excess amount of base.25 Although ring-opening reactions of a particular fluorenone derivative have been reported to proceed with an excess amount of Lewis acid,26 ring-opening reactions with simple Brønsted acids have not been reported. However, given our prior demonstration that the ring-opening reaction occurred at room temperature when employing a sumanenetrione derivative with significant strain, it is plausible that the acid-promoted ring-opening reaction also takes place influenced by steric or electronic effect. To test this hypothesis, we conducted the reaction under the same conditions using 4, and triphenylenone (5), another planar molecule with the same π-conjugated structure as 1. However, in both cases, the ring-opening reaction did not proceed, and the starting materials were recovered (Scheme 2). Therefore, the curved distortion of the bowl skeleton in 1 should play a crucial role in facilitating this reaction.
 | ||
| Scheme 2 Control experiments on the strain-induced ring-opening reaction. | ||
Based on these results, we proposed a possible reaction mechanism for the acid-mediated ring opening reaction (Fig. 3). The carbonyl group of 1 undergoes protonation and reacts with acetic acid to form a corresponding hemiketal intermediate. The α-carbon (sp2) at the quaternary carbon centre is protonated by the superacid TfOH, generating a cationic intermediate. A similar electrophilic addition of bromonium ions to the quaternary carbon on sumanene skeleton has been previously reported.27 The cationic intermediate undergoes ring-opening involving the release of curved strain, followed by hydrolysis to give the target compound. In this proposed mechanism, the reaction is considered to proceed efficiently due to the irreversibility of the ring-opening step.
 | ||
| Fig. 3 Possible reaction mechanism. | ||
To understand the reaction mechanism and the impact of bowl structure on reactivity, the density functional theory (DFT) calculations were performed for 1, 4, and 5 (Fig. 4) at the ωB97X-D/aug-cc-PVTZ/SMD(AcOH)//ωB97X-D/aug-cc-PVDZ/SMD(AcOH) level of theory. There is no substantial difference in ΔG‡ in the formation of hemiketals B by nucleophilic addition of acetic acid at 1, 4, and 5 (1: ΔG‡ = 21.7 kcal mol−1; 4: ΔG‡ = 22.5 kcal mol−1; 5: ΔG‡ = 22.0 kcal mol−1). In contrast, comparing the ΔΔG values for the protonation step, all the compounds were significantly destabilized due to dearomatization. Nevertheless, the calculated ΔΔG value for 1 was 16.7 kcal mol−1, considerably lower than that of 4 and 5 (29.8 kcal mol−1 for 4, 28.8 kcal mol−1 for 5). This can be attributed to the resonance effect and the hyperconjugation that exclusively benefits the carbocation intermediate 1-C due to the presence of methylene bridges (Fig. S2, ESI‡). The maximum ΔG‡ for the reaction mechanism was determined to be +34.7 kcal mol−1 for the ring-opening step (TS2), a value conducive to the reaction occurring at 100 °C. In contrast, the corresponding values for 4 and 5 were evaluated to be +46.0 and +46.6 kcal mol−1, respectively. This result confirms that the ring-opening reaction is unlikely to take place even at high temperatures in the case of 4 and 5. For compound 1, the ΔG of the final product 1-D was found to be a negative value (–9.4 kcal mol−1), attributed to substantial stabilization by strain-release, rendering the overall reaction process exothermic. Meanwhile, those of 4 and 5 were +24.6 and +24.1 kcal mol−1, respectively, indicating an endothermic nature and supporting the notion that the reaction does not proceed. In summary, these DFT calculations provide a good explanation of the validity of the reaction mechanism and are in good agreement with the experimental results. The key to this reaction is the capacity of compound 1 to stabilize the cationic intermediate using its molecular framework, mitigating the destabilizing impact of dearomatization caused by protonation. Furthermore, the substantial contribution to stabilization arising from the release of curved strain is considered crucial. Both factors are posited to play a pivotal role in facilitating the cleavage of a stable C–C bond.
 | ||
| Fig. 4 Energy profile of the acid-mediated ring-opening reaction of 1, 4, and 5. The calculated Gibbs free energies (ΔG and ΔG‡, 298.15 K, 1 atm) are shown in kcal mol−1. All calculations were performed at the ωB97X-D/aug-cc-PVTZ/SMD(AcOH)//ωB97X-D/aug-cc-PVDZ/SMD(AcOH) level of theory. To compare energies in all steps of the reaction, the energy of A, B and TS1 is denoted by the sum of ΔG of TfOH and 1-A, 1-B, and TS1, respectively. | ||
In conclusion, we have demonstrated an effective ring-opening reaction involving the C–C bond cleavage of the sumanenone backbone. A comparative analysis of 1 and planar analogs via experimental and computational methods highlights the critical role of the release of curved strain. The findings illuminate the distinctive reactivity conferred by the strained structure of the curved π-conjugated system. Moreover, the resulting carboxylic acid holds promise as a precursor for constructing unique molecular skeletons with a bowl/planar π-conjugated configuration, such as heterobuckybowls.
This study was supported by a Grant-in-Aid for Transformative Research Areas “Science of 2.5 Dimensional Materials” (No. JP21H05233), “Hyper-Ordered Structures Sciences” (No. JP21H05563 and JP23H04112), and JSPS KAKENHI (JP19H00912, JP20H00400 and JP20K15279), and JST SPRING (JPMJSP2138). The theoretical calculations were performed at the Research Centre for Computational Science, Okazaki, Japan (Project: 22-IMSC068).
Conflicts of interest
There are no conflicts to declare.Notes and references
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- H. W. Kroto, J. R. Heath, S. C. O’Brien, R. F. Curl and R. D. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS.
- R. C. Haddon, Science, 1993, 261, 1545–1550 CrossRef CAS PubMed.
- Y.-T. Wu and J. S. Siegel, Chem. Rev., 2006, 106, 4843–4867 CrossRef CAS PubMed.
- V. M. Tsefrikas and L. T. Scott, Chem. Rev., 2006, 106, 4868–4884 CrossRef CAS PubMed.
- M. Saito, H. Shinokubo and H. Sakurai, Mater. Chem. Front., 2018, 2, 635–661 RSC.
- D. V. Preda and L. T. Scott, Tetrahedron Lett., 2000, 41, 9633–9637 CrossRef CAS.
- A. V. Zabula, S. N. Spisak, A. S. Filatov, A. Y. Rogachev and M. A. Petrukhina, Angew. Chem., Int. Ed., 2011, 50, 2971–2974 CrossRef CAS PubMed.
- M. Yanney, F. R. Fronczek and A. Sygula, Org. Lett., 2012, 14, 4942–4945 CrossRef CAS PubMed.
- H. E. Bronstein and L. T. Scott, J. Org. Chem., 2008, 73, 88–93 CrossRef CAS PubMed.
- J. A. Steckel and K. D. Jordan, J. Phys. Chem. A, 2002, 106, 2572–2579 CrossRef CAS.
- H. Y. Cho, R. B. M. Ansems and L. T. Scott, Beilstein J. Org. Chem., 2014, 10, 956–968 CrossRef PubMed.
- N. Ikuma, Y. Yoshida, Y. Yakiyama, N. Ngamsomprasert and H. Sakurai, Chem. Lett., 2018, 47, 736–739 CrossRef.
- J. C. Hummelen, B. Knight, J. Pavlovich, R. González and F. Wudl, Science, 1995, 269, 1554–1556 CrossRef CAS PubMed.
- K. Komatsu, M. Murata and Y. Murata, Science, 2005, 307, 238–240 CrossRef CAS PubMed.
- M. Murata, Y. Murata and K. Kumatsu, Chem. Commun., 2008, 6083–6094 RSC.
- K. Kurotobi and Y. Murata, Science, 2011, 333, 613–616 CrossRef CAS PubMed.
- S. Tashiro, M. Yamada and M. Shionoya, Angew. Chem., Int. Ed., 2015, 54, 5351–5354 CrossRef CAS PubMed.
- X. Li, Y. Zhu, J. Shao, L. Chen, S. Zhao, B. Wang, S. Zhang, Y. Shao, H.-L. Zhang and X. Shao, Angew. Chem., Int. Ed., 2015, 54, 267–271 CrossRef CAS PubMed.
- H. Sakurai, T. Daiko and T. Hirao, Science, 2003, 301, 1878 CrossRef CAS PubMed.
- H. Sakurai, Bull. Chem. Soc. Jpn., 2021, 94, 1579–1587 CrossRef.
- J. Han, Y. Uetake, Y. Yakiyama and H. Sakurai, Chem. Commun., 2023, 59, 4632–4635 RSC.
- M. Nishimoto, Y. Uetake, Y. Yakiyama, F. Ishiwari, A. Saeki and H. Sakurai, J. Org. Chem., 2022, 87, 2508–2519 CrossRef CAS PubMed.
- T. Amaya, M. Hifumi, M. Okada, Y. Shimizu, T. Moriuchi, K. Segawa, Y. Ando and T. Hirao, J. Org. Chem., 2011, 76, 8049–8052 CrossRef CAS PubMed.
- R. Fittig and E. Ostermayer, Justus Liebigs Ann. Chem., 1873, 166, 361–382 CrossRef.
- R. A. Haggam, Synth. Commun., 2022, 52, 1490–1499 CrossRef CAS.
- N. Ngamsomprasert, J.-S. Dang, S. Higashibayashi, Y. Yakiyama and H. Sakurai, Chem. Commun., 2017, 53, 697–700 RSC.
Footnotes |
| † Dedicated to Professor Koichi Narasaka in celebration of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2323143. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc00008k |
| This journal is © The Royal Society of Chemistry 2024 |