
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of easily-modified and useful dibenzo-[b,d]azepines by palladium(II)-catalyzed cyclization/addition with a green solvent†
Hua
Cheng‡
 abd,
Rongqi
Liu‡
a,
Shengyang
Fang‡
a,
Zixiang
Li
a,
Denggao
Zhang
a,
Xi
Zhang
d,
Wenfei
Chen
c,
Huixin
Chen
c,
Leyi
Kang
d,
Juan
Wang
*c,
Yulong
Xu
*d,
Shaoli
Song
*b and
Liming
Shao
*a
abd,
Rongqi
Liu‡
a,
Shengyang
Fang‡
a,
Zixiang
Li
a,
Denggao
Zhang
a,
Xi
Zhang
d,
Wenfei
Chen
c,
Huixin
Chen
c,
Leyi
Kang
d,
Juan
Wang
*c,
Yulong
Xu
*d,
Shaoli
Song
*b and
Liming
Shao
*a
aSchool of Pharmacy, Fudan University, 826 Zhangheng Road, Zhangjiang Hi-tech Park, Pudong, Shanghai, 201203, China. E-mail: limingshao@fudan.edu.cn
bChina Department of Nuclear Medicine, Fudan University Shanghai Cancer Center, No. 270, Dong'an Road, Xuhui District, Shanghai 200032, China. E-mail: shaoli-song@163.com
cSchool of Medicine, Shanghai University, Shanghai 200444, China. E-mail: juanw@shu.edu.cn
dMassachusetts General Hospital, Harvard Medical School, Boston, MA 02129, USA. E-mail: yulong.xu@mgh.harvard.edu
First published on 5th March 2024
Abstract
A novel strategy in which palladium(II)-catalyzed tandem cyclization is used to obtain N-heterocyclic architectures containing a seven-membered ring has been developed and used to synthesize a series of derivatives. The reaction uses an eco-friendly mixed solvent (water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOH = 2:1) instead of DMSO and maintains a high yield (91%). Its potential application value and reaction mechanism have also been explored.
:EtOH = 2:1) instead of DMSO and maintains a high yield (91%). Its potential application value and reaction mechanism have also been explored.
Transition-metal-catalyzed transformation has emerged as a highly efficient approach for accessing medicinal chemistry compounds and various functional materials.1 As a versatile method, the classical synthetic method plays an important role in activating organonitriles.2 Larock's group pioneered a novel approach involving the catalytic carbopalladation of nitriles, which has paved the way for numerous innovative advances.3,4 Linjun Qi et al. discovered a series of crucial structural motifs attainable through this method,5 such as indoles, isoquinolines, and isoquinolones.6 Ketimine created by the coordination of aryl-Pd with nitrile is a useful intermediate for many key reactions.7–9
Dibenzoazepines are an essential class of N-containing polycyclic compounds and are crucial structural motifs in many medicines, natural products, and other vital bioactive substances.10 For instance (Fig. 1), molecule 1, a dibenzoazepine derivative, is a potent and selective allosteric PAK1 inhibitor that possesses excellent inhibitory activity (5 nm).11 Molecule 2, natural products 3 and 4 containing dibenzoazepines were identified as effective potassium channels12 and serotonin (5-HT) receptors,13 and dimeric erythrivarine B,14 respectively. However, owing to the limited synthetic approach to obtaining dibenzoazepines, this useful motif cannot be explored further. Therefore, a novel way to obtain dibenzoazepines is needed.
 | ||
| Fig. 1 Some examples containing dibenzo[b,d]azepine. | ||
Recently, some impressive approaches to synthesizing dibenzoazepines have been reported.16–18 Among them, Zhijun Zuo et al. reported the synthesis of imine-containing dibenzo[b,d]azepines by palladium(II)-catalyzed [5+2] oxidative annulation of o-arylanilines with alkynes,15 achieving a high yield (91%) (Scheme 1). However, some problems limit the usage of this reaction. The minimum exact mass without any modification is 345.15, which means the molecule is too large to be further modified according to the classical Lipinski ‘rule of five’.19 Moreover, the high temperature of 120 °C is impractical for a standard reaction. Meanwhile, DMSO is not a friendly solvent and is difficult to remove.20 If DMSO could be replaced by H2O or another eco-friendly solvent, the reaction would have more potential for future use.21 Besides that, two different catalysts make the reaction more difficult and costly. Therefore, a method for the synthesis of dibenzoazepines from aliphatic nitriles was devised (Scheme 1) and a series of aliphatic nitriles and their derivatives were obtained through a transition-metal-catalyzed tandem addition and cyclization process. Compared with other relevant studies, this study exhibits several advantages, notably operating at reduced temperature, employing a single catalyst, and utilizing environmentally friendly solvents.
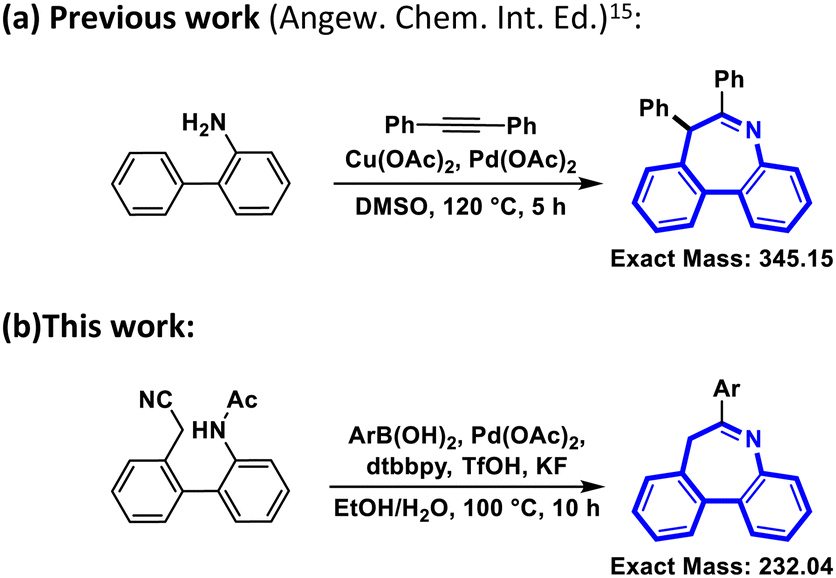 | ||
| Scheme 1 Synthesis of dibenzo[b,d]azepine core structures. (a) Previous work (Angew. Chem. Int. Ed.).15 (b) This work. | ||
Initially, a novel molecule N-(2′-(cyanomethyl)-[1,1′-biphenyl]-2-yl) acetamide (1a) and phenylboronic acid 2a were chosen as the substrate for the reaction (Table 1). As shown in entry 1, the desired product 3a was obtained with a 65% yield using Pd(acac)2, o-phenanthroline (L1), and TFA at 100 °C. When the catalyst was replaced by Pd(PPh3)4 or Pd(dppf)Cl2, the yield significantly decreased (entries 2 and 3) and the target product nearly disappeared when a 0-valent catalyst Pd(PPh3)4 was used. Fortunately, the yield increased to 74% when Pd(OAc)2 was used. Seeking a cost-effective metal catalyst, Cu and Ni with selected dominant ligands were chosen to catalyse the reaction (entries 5–7), but the yield did not improve. Next, various acids were investigated (entries 8–10), revealing higher yields with TfOH and PhSO3H (82% and 77%).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Ligand | Acid | Solvent | Yieldb (%) |
| a Some general reaction conditions: 1a (0.4 mmol), 2a (0.6 mmol), Pd-catalyst (5 mol%), ligand (10 mol%), acid (10 equiv.), KF (2 equiv.), 100 °C, 10 h, Ar. b Isolated yield. TFA = CF3COOH, TsOH = p-MeC6H4SO3H, and TfOH = CF3SO3H. The reaction is a tube sealing reaction. | |||||
| 1 | Pd(acac)2 | L1 | TFA | THF–H2O (2:1) |
65 |
| 2 | Pd(PPh3)4 | L1 | TFA | THF–H2O (2:1) |
0 |
| 3 | Pd(dppf)2Cl2 | L1 | TFA | THF–H2O (2:1) |
10 |
| 4 | Pd(OAc)2 | L1 | TFA | THF–H2O (2:1) |
74 |
| 5 | Ni(acac)2 | L1 | TFA | THF–H2O (2:1) |
Trace |
| 6 | Ni(OAc)2 | L1 | TFA | THF–H2O (2:1) |
15 |
| 7 | Cu(OAc)2 | L1 | TFA | THF–H2O (2:1) |
Trace |
| 8 | Pd(OAc)2 | L1 | TfOH | THF–H2O (2:1) |
82 |
| 9 | Pd(OAc)2 | L1 | PhSO3H | THF–H2O (2:1) |
77 |
| 10 | Pd(OAc)2 | L1 | HCl | THF–H2O (2:1) |
Trace |
| 11 | Pd(OAc)2 | L1 | TfOH | Tol–H2O (2:1) |
74 |
| 12 | Pd(OAc)2 | L1 | TfOH | Dioxane–H2O (2:1) |
82 |
| 13 | Pd(OAc)2 | L1 | TfOH | DMSO–H2O (2:1) |
Trace |
| 14 | Pd(OAc)2 | L1 | TfOH | EtOH–H2O (2:1) |
84 |
| 15 | Pd(OAc)2 | L1 | TfOH | EtOH–H2O (1:1) |
85 |
| 16 | Pd(OAc)2 | L1 | TfOH | EtOH–H2O (1:2) |
85 |
| 17 | Pd(OAc)2 | L1 | TfOH | EtOH–H2O (1:3) |
30 |
| 18 | Pd(OAc)2 | L2 | TfOH | EtOH–H2O (1:2) |
69 |
| 19 | Pd(OAc)2 | L3 | TfOH | EtOH–H2O (1:2) |
82 |
| 20 | Pd(OAc)2 | L4 | TfOH | EtOH–H2O (1:2) |
83 |
| 21 | Pd(OAc) 2 | L5 | TfOH |
EtOH–H
2
O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :2) :2)
|
91 |
| 22 | Pd(OAc)2 | L6 | TfOH | EtOH–H2O (1:2) |
87 |

The selection of an appropriate solvent, including both its type and ratio, plays a critical role in transition-metal-catalyzed reactions.22 Additionally, environmentally friendly solvents are crucial when extending the synthesis method to industrial applications.23 First, THF was replaced by dioxane or toluene, when the reaction could maintain a high conversion but one lower than THF (entries 11 and 12). However, experimental results showed that DMSO proved to be an inefficient solvent (entry 13). Compared with THF, EtOH is more beneficial to the environment, so EtOH was used and yielded an ideal 84% (entry 14). In further optimization of solvents, we explored the ratio of organic solvent to water (entries 15–17), and the conversion did not appear to decline significantly until the proportion of water increased to 75%. Finally, the best solvent and ratio for the reaction were determined (EtOH:H2O = 1:2), and the yield was 85%. Ligand selection is another critical factor for transition-metal-catalyzed organic reactions. The enhancement in yield was not significant when utilizing phenanthroline monohydrate and its derivatives (entries 18 and 19). Further, when using bipyridine and its derivatives, the yield increased to 87% with L6 and to 91% with L5. The conversion rate (91%) met expectations, and subsequent work was developed under these selected conditions.
After identifying optimal conditions for dibenzoazepines through detailed exploration, we proceeded to screen various aryl boronic acids to determine the scope of the reaction substrate. In the first series of substrate expansions, raw material 1a remained unchanged (Scheme 2A). Tolylboronic acid in different positions was evaluated first. When the methyl group was placed at the para- or meta-position, corresponding products 3b and 3c were obtained with yields of 89% and 90%, respectively, whereas 3d was obtained with a yield of 78% due to steric hindrance at the ortho-position. However, the methoxy group, an electron-donating group, led to the yields of 3e and 3f decreasing to 75% and 80%, respectively. This highlighted differences between different electron-donating groups. With regard to other electron-donating groups, compounds 3g (tert-butyl), 3o (dimethyl), and 3p (bismethoxy) were isolated with yields of 91%, 84%, and 80%, respectively. Next, various electron-withdrawing groups were examined, showing differences between different groups, similar to electron-donating groups. Compounds 3i and 3j with trifluoromethyl were isolated with yields of 45% and 40%, respectively, which were lower than expected. Compound 3h was isolated with a yield of 74%. When halogen was used as a substituent, the yield could be maintained, and 3l and 3n were obtained with yields of 85% and 89%, respectively. However, due to steric hindrance at the ortho-position, 3k and 3m were isolated in yields of only 66% and 68%, respectively.
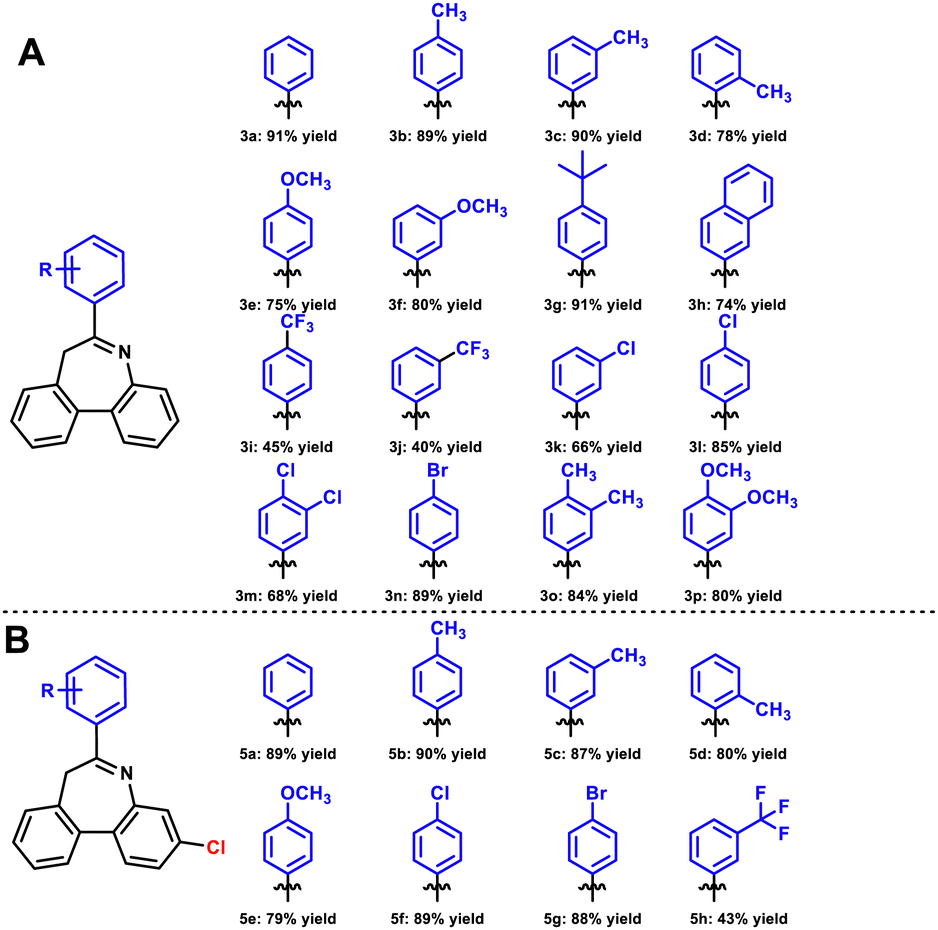 | ||
| Scheme 2 Dibenzo[b,d]azepine derivatives. (A) Derivatives without Cl, (B) chloro-derivatives. | ||
To extend the expandable site of the product, the raw material 4a (ESI†) was obtained. 4a and 2a were used as substrates to synthesize a second series of products (Scheme 2B). Several substituent groups were selected, and the yields of para-, meta-, and ortho-toluene boric acid were evaluated. Compounds 5b, 5c, and 5d were isolated in yields of 90%, 87%, and 80%, respectively. ortho-Tolylboronic acid produced corresponding product 5d with a lower yield due to steric hindrance. Meanwhile, using methoxyphenyl boronic acid did not produce an ideal yield (5e, 79%). When the electron-donating groups were replaced by electron-withdrawing groups, the yield of the products again showed differentiation. Halogen substitution products 5f and 5g were isolated in yields of 89% and 88%, respectively. We also observed that trifluoromethyl was still not a good substituent, resulting in a lower yield of 43%.
To explore the yield on a large scale, a 1-g scale reaction was studied, and the yield was found to be 85%. (Fig. S1 in ESI†). Furthermore, to demonstrate the potential for obtaining more derivatives via bromo-substituted dibenzoazepine skeletons, compounds 6a and 7a were prepared (Fig. 2A). The expandable skeleton could couple with different substituents via a palladium-catalyzed reaction and provide different kinds of molecules for subsequent skeleton applications. Compounds 5a–5h with the substitution of chlorine atoms offer possibilities for the further development of diversified substrates.
 | ||
| Fig. 2 (A) Br- and Cl-derivatives, and the design process of Cl-substituted compounds as CB2R agonists; (B) binding of WIN 55212-2 and CB2R (X-ray); (C) superposition state of each compound in the CB2R pocket; (D)–(F) superposition results of 8a, 8b and 8c with WIN 55212-2, respectively. The original ligand WIN 55212-2 is blue-violet, compound 8a is yellow, 8b is pink, and 8c is gray. | ||
Moreover, newly designed chloro-substituted derivatives have been planned for application in medical chemistry as a cannabinoid type 2 receptor (CB2R) agonist. CB2R-targeted drugs show potential in the treatment of neurodegenerative conditions, inflammation, and pain without causing CB1R-mediated psychotropic side effects.24 In a previous report, C. R. Xing developed an efficient CB2R agonist called WIN 55212-2.25 The rigid tricyclic ring group of WIN 55212-2 ensures its selectivity for CB2R. To increase its function, a similar tricyclic ring structure synthesized in the article was introduced as the core instead of the original one (Fig. 2A).
According to previous docking results for WIN 55212-2 (Fig. 2B), the compounds exhibit an L-shaped structure in the binding pocket. Therefore, leveraging the tricyclic core, lipophilic substituents were introduced on the azepine ring and one of the benzene rings to serve as an inflection point connecting the two arms of “L”. Consequently, three compounds were designed and synthesized. Molecular docking of above three compounds was performed (PDB ID: 6PT0). The binding postures of compounds 8a, 8b, and 8c closely resemble that of the original ligand, adopting an L-shaped conformation within the orthogonal binding pocket located in the transmembrane (TM) region (Fig. 2C). The docking results of each molecule with WIN 55212-2 are shown in Fig. 2D–F. Overall, the three new compounds fit well into the CB2R orthogonal binding pocket, and the multiple benzene rings help generate additional π-links with multiple phenylalanine residues in the receptor. The synthesis of compounds 8a–8c has been completed, and remaining test results will be published in the future.
A possible mechanism was proposed based on above-mentioned results and previous reports (Scheme 3a).5 First, the Pd(II) catalyst reacts with aryl-boronic acid to form Pd-aryl compounds. Next, compound 1 joins the reaction and reacts with Pd-aryl compounds to produce intermediate A. Compound B is generated owing to the intramolecular carbopalladation of the raw compound. The protonation of acid TfOH converts the imine Pd(II) complex B to ketimine intermediate C. Additionally, this process promotes the regeneration of the Pd(II) catalyst. Subsequently, intermediate D deacetylates to obtain complex C under acidic conditions. Finally, compound 3 is obtained via intramolecular cyclization. To elucidate the mechanism, a controlled experiment was designed where the reaction was quenched at 1 h, resulting in isolation of the vital intermediate C (Scheme 3b), indicating that the proposed mechanism is reasonable. Although the possible mechanism has been elucidated, details should be explored further.
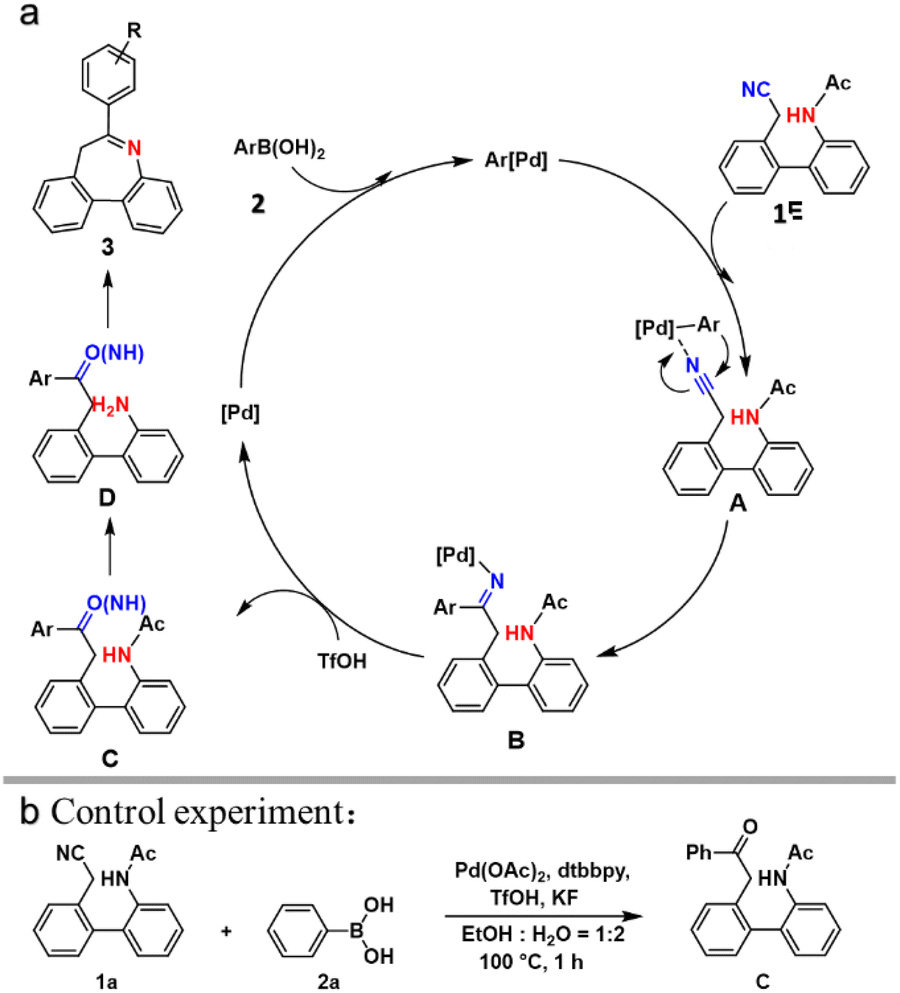 | ||
| Scheme 3 (a) Reaction mechanism. (b) Control experiment. | ||
In summary, this study has introduced a novel palladium(II)-catalyzed addition/cyclization of N-(2′-(cyanomethyl)-[1,1′-biphenyl]-2-yl)acetamide with phenylboronic acid derivatives, employing a cyano-activation method. Compared to previous methods, this approach offers a simpler and more eco-friendly route to a new series of dibenzo[b,d]azepines, achieving ideal yields using readily available phenylboronic acid derivatives as one of the raw materials. Moreover, these structures show potential application value in pharmaceutical chemistry and other fields.
L. M. Shao, S. L. Song and Y. L. Xu conceived the conceptualization. H. Cheng, R. Q. Liu, J. Wang and S. Y. Fang did the data collation. Z. X. Li, D. G. Zhang W. F. Chen and H. X. Chen did the data analysis. X. Zhang and L. Y. Kang carried out the investigation of references. All authors discussed the results of the manuscript.
The National Natural Science Foundation of China (no. 81971648), the Key Program of the National Natural Science Foundation of China (no. U23A20465), and the Project of Shanghai Science and Technology Commission Outstanding Technical Leader (21XD1431300).
Conflicts of interest
There are no conflicts to declare.References
- F. F. Fleming and Q. Wang, Chem. Rev., 2003, 103, 2035–2078 CAS.
- S. F. Rach and F. E. Kühn, Chem. Rev., 2009, 109, 2061–2080 CAS.
- R. C. Larock, Q. Tian and A. A. Pletnev, J. Am. Chem. Soc., 1999, 121, 3238–3239 CAS.
- B. Skillinghaug, C. Skold, J. Rydfjord, F. Svensson, M. Behrends, J. Savmarker, P. J. Sjoberg and M. Larhed, J. Org. Chem., 2014, 79, 12018–12032 CAS.
- S. Yu, L. Qi, K. Hu, J. Gong, T. Cheng, Q. Wang, J. Chen and H. Wu, J. Org. Chem., 2017, 82, 3631–3638 CAS.
- L. Qi, K. Hu, S. Yu, J. Zhu, T. Cheng, X. Wang, J. Chen and H. Wu, Org. Lett., 2017, 19, 218–221 CAS.
- L. Qi, K. Hu, S. Yu, J. Zhu, T. Cheng, X. Wang, J. Chen and H. Wu, Org. Lett., 2017, 19, 218–221 CAS.
- B. Zhao and X. Lu, Org. Lett., 2006, 8, 5987–5990 CAS.
- A. Kishi, K. Moriyama and H. Togo, J. Org. Chem., 2018, 83, 11080–11088 CAS.
- K. Zhu, A. M. Shah, J. Berkow and A. Kiourti, Medicine and Biology, IEEE J. Electromagn. RF Microw. Med. Biol., 2020, 5, 124–131 Search PubMed.
- A. S. Karpov, P. Amiri, C. Bellamacina, M.-H. Bellance, W. Breitenstein, D. Daniel, R. Denay, D. Fabbro, C. Fernandez and I. Galuba, ACS Med. Chem. Lett., 2015, 6, 776–781 CAS.
- S. Pegoraro, M. Lang, T. Dreker, J. Kraus, S. Hamm, C. Meere, J. Feurle, S. Tasler, S. Prütting, Z. Kuras, V. Visan and S. Grissmer, Bioorg. Med. Chem. Lett., 2009, 19, 2299–2304 CAS.
- R. Rajagopalan, A. Bandyopadhyaya, D. R. Rajagopalan and P. Rajagopalan, Bioorg. Med. Chem. Lett., 2014, 24, 576–579 CAS.
- B. J. Zhang, M. F. Bao, C. X. Zeng, X. H. Zhong, L. Ni, Y. Zeng and X. H. Cai, Org. Lett., 2014, 16, 6400–6403 CAS.
- Z. Zuo, J. Liu, J. Nan, L. Fan, W. Sun, Y. Wang and X. Luan, Angew. Chem., Int. Ed., 2015, 54, 15385–15389 CAS.
- W. Hu, X. Wang, Y. Peng, S. Luo, J. Zhao and Q. Zhu, Org. Lett., 2022, 24, 3642–3646 CAS.
- P. Rodríguez-Salamanca, R. Martín-de la Calle, V. Rodríguez, P. Merino, R. Fernández, J. M. Lassaletta and V. Hornillos, Chem. Sci., 2021, 12, 15291–15297 Search PubMed.
- R.-G. Shi, X.-H. Wang, R. Liu and C.-G. Yan, Chem. Commun., 2016, 52, 6280–6283 CAS.
- M. P. Pollastri, Curr. Protoc. Pharmacol., 2010, 49, 9.12.11 Search PubMed.
- M. D. Hall, K. A. Telma, K.-E. Chang, T. D. Lee, J. P. Madigan, J. R. Lloyd, I. S. Goldlust, J. D. Hoeschele and M. M. Gottesman, Cancer Res., 2014, 74, 3913–3922 CAS.
- M. D. Astuti, T. Hara, N. Ichikuni and S. Shimazu, Green Chem., 2019, 21, 2307–2315 Search PubMed.
- J. Sherwood, J. H. Clark, I. J. Fairlamb and J. M. Slattery, Green Chem., 2019, 21, 2164–2213 CAS.
- P. G. Jessop, Green Chem., 2011, 13, 1391–1398 CAS.
- E. S. Onaivi, H. Ishiguro, J. P. Gong, S. Patel, A. Perchuk, P. A. Meozzi, L. Myers, Z. Mora, P. Tagliaferro and E. Gardner, Ann. N. Y. Acad. Sci., 2006, 1074, 514–536 CAS.
- C. Xing, Y. Zhuang, X. E. Zhou, M. Chen, L. Wang, X. Meng, Y. Xue and J. J. C. Wang, Cell, 2020, 180, 645–654 CAS .e613.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc06321f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |