
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Activation of non-polar bonds by an electron-rich gallagermylene†
Anna
Bücker
a,
Alexander
Gehlhaar
a,
Christoph
Wölper
a and
Stephan
Schulz
 *ab
*ab
aInstitute of Inorganic Chemistry, University of Duisburg-Essen, Essen 45117, Germany. E-mail: stephan.schulz@uni-due.de
bCenter for Nanointegration Duisburg-Essen (CENIDE), University of Duisburg-Essen, Duisburg 47057, Germany
First published on 8th February 2024
Abstract
The electron-rich germylene LGa(μ–Cl)GeArMes (1) (L = CH[C(Me)N(Dipp)]2, Dipp = 2,6-iPr2C6H3, ArMes = 2,6-Mes2C6H3, Mes = 2,4,6-Me3C6H2) shows promising potential in the σ-bond activation of unpolar molecules as is shown in oxidative addition reactions with H2 and P4, yielding L(Cl)GaGe(H)2ArMes (2) and L(Cl)Ga(P4)GeArMes (3). Compounds 2 and 3 were characterised spectroscopically (1H, 13C{1H}, (31P{1H}), IR) and by single-crystal X-ray diffraction (sc-XRD).
The activation of small molecules, i.e., H2, CO, CO2, NH3, P4 or ethylene, and their utilization for synthetic purposes including catalytic reactions is of fundamental importance in chemistry. While such reactions have been dominated by transition metal complexes for decades, the use of low valent main group complexes, whose electronic nature mimics that of transition metal complexes, has received increasing interest in recent years.1 In particular the activation and cleavage of strong non-polar σ-bonds, i.e., the H–H bond of H2, has received increasing attention. The reaction of the digermyne ArDippGeGeArDipp (ArDipp = 2,6-Dipp2C6H3; Dipp = 2,6-iPr2C6H3) with H2, which was reported by Power et al., represents a milestone since the digermene ArDipp(H)Ge
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ge(H)ArDipp and the germanes ArDipp(H)2GeGe(H)2ArDipp and ArDippGe(H)3 were formed under very mild reaction conditions.2 Later on, the amido-substituted digermynes [L1,2Ge]2 (L1 = N(SiMe3)(Ar1), Ar1 = 2,6-(CHPh2)2-4-MeC6H2; L2 = N(SiiPr3)(Ar2), Ar2 = 2,6-(CHPh2)2-4-iPrC6H2)3 were found to be active in H2 activation, yielding L1Ge(H)2GeL1,3a and L2(H)GeGe(H)L2,3b respectively. Since then, reactions of H2 with digermynes RGeGeR,1a digermavinylidene (L3B)2GeGe (L3B = [HCN(Dipp)]2B), which reacted with H2 (4 atm) to the symmetric digermane [L3BGe(H)2]2,4a and with a digermynyl aluminum complex, which slowly activated H2 (1 atm) at ambient temperature,4b have been reported.
Ge(H)ArDipp and the germanes ArDipp(H)2GeGe(H)2ArDipp and ArDippGe(H)3 were formed under very mild reaction conditions.2 Later on, the amido-substituted digermynes [L1,2Ge]2 (L1 = N(SiMe3)(Ar1), Ar1 = 2,6-(CHPh2)2-4-MeC6H2; L2 = N(SiiPr3)(Ar2), Ar2 = 2,6-(CHPh2)2-4-iPrC6H2)3 were found to be active in H2 activation, yielding L1Ge(H)2GeL1,3a and L2(H)GeGe(H)L2,3b respectively. Since then, reactions of H2 with digermynes RGeGeR,1a digermavinylidene (L3B)2GeGe (L3B = [HCN(Dipp)]2B), which reacted with H2 (4 atm) to the symmetric digermane [L3BGe(H)2]2,4a and with a digermynyl aluminum complex, which slowly activated H2 (1 atm) at ambient temperature,4b have been reported.
In marked contrast, H2 activation reactions of germylenes R2Ge have been reported only rarely.5 H2 activation by aryl-substituted germylenes ArMes2Ge (A) (ArMes = 2,6-Mes2-C6H3, Mes = 2,4,6-Me3-C6H2) and ArDipp2Ge proceeded either via the formation of the corresponding germane ArMes2GeH2 or with the release of the ligand ArDippH and ArDippGeH3.6a The crucial influence of the organic ligand on the germylene reactivity was demonstrated by Aldridge et al. by comparing different acyclic germylenes ArMesGeR′ (R′ = N(Dipp)H, CH(SiMe3)2, P(SiMe3)2 and Si(SiMe3)3 (B)).6b Electropositive ligands lead to smaller HOMO–LUMO gaps, resulting in an increased reactivity. This was further demonstrated by Jones et al. for the acyclic zincagermylene (TBoN)(L4Zn)Ge (TBoN = N(SiMe3){B[N(Dipp)CH]2}; L4 = N(SiiPr3)(Ar1)) (C), which reacted in toluene solution with H2 at r.t. within five seconds to the corresponding germanium(IV) dihydride.6c In contrast, the cationic tungstagermylene [Cp*(CO)3WGe(IDipp)](BAr4F) (IDipp = [HCN(Dipp)]2C; ArF = 3,5-(CF3)2C6H3) (1 atm, 60 °C, 24 h)6d and a PMe3-coordinated cyclic (alkyl)(boryl)germylene (1 atm, 50 °C, 12 h) reacted with H2 only at elevated temperatures to the corresponding germanes.6e
Due to its fundamental interest, the activation of H2 in reactions with metallylenes was further investigated by use of relativistic density functional theory (DFT), showing that the decreasing reactivity of tetrylenes from carbenes to stannylenes mainly results from a worsening of the back-donation from the tetrylene lone-pair orbital and the H2 σ*-orbital, despite an increase in interaction energy of the LUMO of the terylene and the HOMO of H2. However, decreasing the electronegativity of the tetrylene ligand resulted in significantly lower reaction barriers due to a reduced Pauli repulsion, which was identified as a main hindrance.7
In addition, the activation of non-polar P–P σ-bonds of white phosphorus (P4) is also of broad interest.8 Among several pathways, oxidative addition reactions of P4 to tetrylenes, in particular silylenes,9 have been reported. In contrast, reactions of heavier tetrylenes with P4 are rather scarce.10 The acyclic germylene ArMes2Ge was found to reversibly activate P4,10a while we demonstrated the beneficial effect of electropositive metal-based ligands in L(Cl)M-SiL5 (L = HC[C(Me)N(Dipp)]2, L5 = PhC[N(tBu)]2, M = Al, Ga), which reacted in an unprecedented [2+1+1] fragmentation reactions with P4.9f In addition, the reaction of L(Cl)Ga-SiL5 with Cp*Fe(η5-Pn5) (Pn = P, As) resulted in Pn–Pn and Si–Ga bond cleavage, which is caused by the insertion of the silylene into the cyclo-Pn5 rings.11
We recently reported the synthesis of the unusual Cl-bridged gallagermylene LGa(μ–Cl)GeArMes (ArMes = 2,6-Mes2C6H3, Mes = 2,4,6-Me3C6H2) (1),12a which reacted with CO2 with activation of the polar C–O double bond (decarbonylation) to the germylene ether, whereas reactions with isocyanates and carbodiimides proceeded with insertion into the Ga–Ge bond.12b In addition, 1 reacted with ethylene with insertion into the Ga–Ge bond followed by dimerization of the as-formed germylene to the corresponding digermene, which then reacted with ethylene in a [2+2] cycloaddition to the 1,2-digermacyclobutane.12c
These promising reactivity studies prompted us to analyse the electronic structure of 1 in more detail by use of ORCA 5.0.413a and the NBO program package (version 7.0.10)13b at the def2-TZVPP level of theory (def2-QZVP for E > Ne)13c using the atom-pairwise dispersion correction based on tight binding partial charges (D4)13d,e with the PBE013f,g functional and to compare the electronic structure of germylene 1 with that the acyclic germylenes A, B and C, respectively. As we reported previously,12a the geometry optimization resulted in a shift of the chlorine atom to the Ge atom (structure 1_opt). Our computations (Table 1) indicate a decreasing natural partial charge of the Ge atom going from A to 1_opt/C, which agrees with the increasing electropositive character of the respective ligands.
| Q(Ge) | E HOMO | E LUMO | ΔEHOMO–LUMO | |
|---|---|---|---|---|
| A | +1.13 | –5.25 | –1.96 | 3.29 |
| B | +0.78 | –5.26 | –2.13 | 3.13 |
| C | +0.41 | –5.15 | –1.64 | 3.51 |
| 1_opt | +0.45 | –4.98 | –1.32 | 3.67 |
Although the electronic structures of the acyclic germylenes including the HOMO–LUMO gaps are comparable, an increase of the energy gap (ΔEHOMO–LUMO) from A to 1_opt is observed (Table 1). However, the relative energetic locations of the frontier orbitals indicate more destabilized orbitals of 1_opt, which might enhance the relevant orbital interaction between the germylene lone-pair and the H2 σ*-orbital. The activation of non-polar σ-bonds by use of germylene 1_opt therefore seemed reasonable and we studied its reactions with H2 and P4, respectively.
The dark red solution solution of 1 in benzene-d6 immediately turned colourless upon expose to a H2 atmosphere at ambient temperature (Scheme 1). According to in situ1H NMR spectroscopic studies, the formation of L(Cl)GaGe(H)2ArMes (2) is immediately completed (ESI,† Fig. S9) and the activation of H2 proceeded much faster and under milder conditions than observed with germylenes A and B and almost as fast as C (Fig. 1). Even though the computed electronic structures of the germylenes 1 and C are hardly comparable, the significant faster reaction rate observed with germylene 1 compared to A and B can be attributed to a combination of reduced positive charge of the Ge atom and of destabilized frontier orbitals.
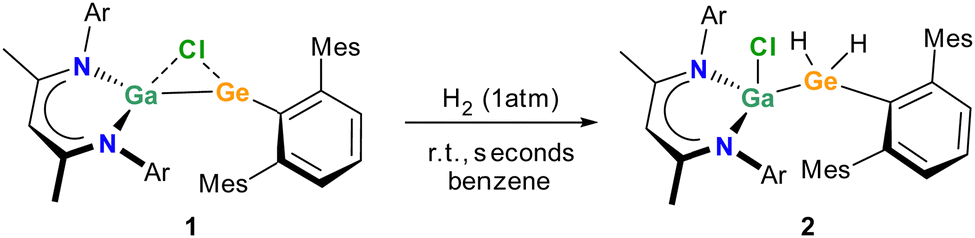 | ||
| Scheme 1 Synthesis of 2 by reaction of germylene 1 with H2. | ||
 | ||
| Fig. 1 Selected acyclic germylenes, which are active in H2 activation, including computed natural charges. The natural charge of 1 refers to the charge of 1_opt (vide infra). Mes = 2,4,6-Me3C6H2, Ar = 2,6-iPr2C6H3, Ar1 = 2,6-(CHPh2)2-4-MeC6H2. | ||
Germane 2 is soluble in benzene, toluene and n-hexane, and its 1H and 13C{1H} NMR spectra show the expected resonances of the β-ketiminate and the terphenyl ligands. The characteristic Ge–H resonance was detected at 3.33 ppm, which is shifted to higher field compared to previously reported germanes (ArMes2GeH2: 4.61 ppm6a (ArMes)(Si(SiMe3)3)GeH2: 3.90 ppm6b (TBoN)(L2Zn)GeH2: 3.88 ppm6c). The FT-IR spectrum shows Ge–H stretching bands at ν = 2019 and 1942 cm−1, respectively, which is in a comparable range reported for (TBoN)(L3Zn)GeH2 (2054, 1995 cm−1)6c and ArMes2GeH2 (2113, 1731 cm−1).6a
Crystals of 2 suitable for a single crystal X-ray diffraction (sc-XRD) study were grown from a saturated benzene solution at 6 °C. 2 crystallises in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) as colourless blocks (Fig. 2).
as colourless blocks (Fig. 2).
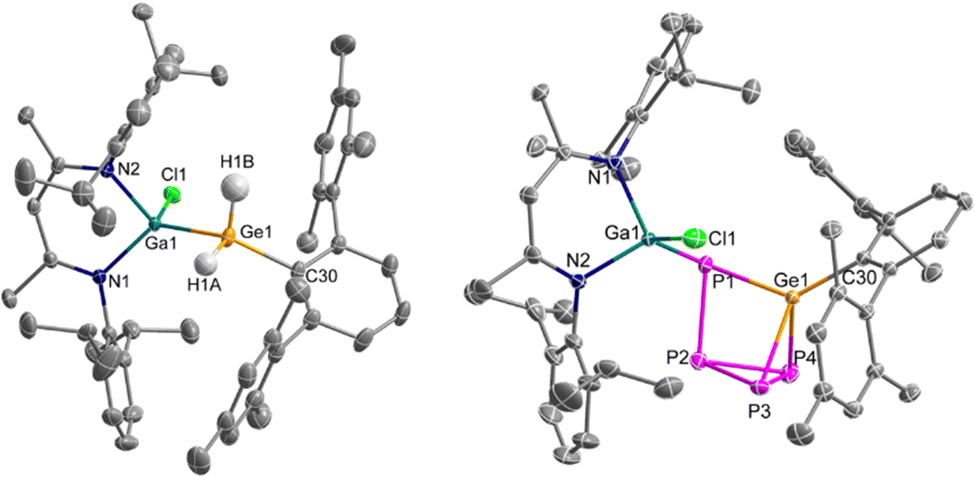 | ||
| Fig. 2 Crystal structures of compounds 2 (left) and 3 (right) with thermal ellipsoid (50%); H atoms except for GeH2, disorder and solvent molecules are omitted for clarity. Selected bond lengths [Å] and angles [°]: 2: Ga1–Ge1: 2.4381(3), Ge1–C30: 1.9725(16), Ga1–Cl1: 2.2341(5); Ga1–Ge1–C30: 129.19(5); 3: Ga1–P1: 2.3486(3), Ga1–Cl1: 2.2018(4), P1–P2: 2.2557(5), P2–P3: 2.2154(5), P3–P4: 2.2694(6), P2–P4: 2.208.79(5), P1–Ge1: 2.3165(3), P3–Ge1: 2.3001(4), P4–Ge1: 2.3344(4), Ge1–C30: 1.9600(11), Ga1–P1–Ge1: 107.142(14), P1–Ge1–P3: 96.355(13), P1–Ge1–P4: 89.406(14), P3–Ge1–P4: 58.633(15), P1–Ge1–C30: 132.63(3), P1–P2–P3: 100.612(18), P1–P2–P4: 94.237(19), P3–P2–P4: 61.720(17), P2–P3–P4: 59.000(17), P2–P4–P3: 59.280(16). | ||
The Ga–Ge (2.4381(3) Å) and Ge–C bond lengths (1.9725(16) Å) of 2 are slightly shortened compared to those of 1 (Ga–Ge: 2.4678(4) Å, Ge–C: 2.022(2) Å),12a while the Ge–C bond lengths reported for (ArMes)(Si(SiMe3)3)GeH2 (1.973(6) Å) and ArMes2GeH2 (1.973(3) Å) are almost identical. The Ga–Cl bond length (2.2341(5) Å) of 2 is within the known range for L(Cl)Ga ligands but much shorter compared to the Cl-bridged complex 1 (Ga–Cl: 2.6076(6) Å).12a The Ga–Ge–C bond angle is widened from 113.86(6)° in 1 to 129.19(5)° in 2 as was also observed for germanes (ArMes)2GeH2 (C–Ge–C: 127.9(2)°), [(Me3Si)3Si]Ge(H)2(ArMes) (Si–Ge–C: 125.8(2)°) and (TBoN)(L2Zn)GeH2 (Zn–Ge–N: 119.04(9)°), synthesised by oxidative addition of H2 to the corresponding germylenes A (C–Ge–C: 114.4(2)°), B (Si–Ge–C: 112.7(1)°) and C (Zn–Ge–N: 107.8(1)°), respectively.
To further evaluate the reactivity of the electron-rich germylene 1 toward unpolar compounds, we reacted 1 with white phosphorus. The reaction of 1 with P4 in benzene-d6 solution is completed after one minute at r.t. according to in situ1H NMR spectroscopy (ESI,† Fig. S10) to yield compound 3 (Scheme 2). Compound 3 contains a P42− moiety, which is formed by a consecutive activation/functionalization reaction of P4, which occurred with the cleavage of two P–P bonds and the regioselective formation of three new Ge–P and one Ga–P bonds, respectively. A comparable 1,2-silyl migration was only once reported for the reaction of vinyl(silyl)silylene with P4, which also occurred with formation of a P42− unit.9c However, in contrast to silylenes, which have been frequently reported to activate P4,91 belongs to the very short list of heavier tetrylenes capable for P4 activation. Only the acyclic germylene A10a and the acyclic stannylene ArMesSnSitBu310b were found to react with white phosphorus with insertion into one P–P bond. However, both complexes were found to release P4 under UV light irradiation. In addition, the distannyne {[(Dipp)NC(CH3)]2C6H3Sn}210c has been reported to react with P4.
 | ||
| Scheme 2 Synthesis of 3 by reaction of germylene 1 with P4. | ||
3 is poorly soluble in n-hexane but well soluble in toluene and benzene. Its 1H NMR and 13C{1H} spectra show the expected resonances of the β-diketiminate and terphenyl ligands. The 31P NMR spectra of 3 shows four resonances (198.5, −72.3, −378.6, −434.5 ppm) due to the magnetic inequivalency of the four P atoms, whereas three resonances (120.0, −181.0, −316.7 ppm) were reported for [MeIDippC(H)]Si(P4)[Si(SiMe3)3]9c (MeIDipp = [(Me)CN(Dipp)]2C).
Crystals of 3 suitable for sc-XRD analysis were obtained from a solution in hot n-hexane. 3 crystallises in the monoclinic space group C2/c as light-yellow platelets (Fig. 2). The Ge–P bond lengths are almost identical (2.3001(4) Å, 2.3165(3) Å, 2.3344(4) Å) and comparable to the Ge–P bond lengths reported for (ArMes)2Ge(P4) (2.3433(7) Å, 2.3509(9) Å). The Ge–C bond length of 1.9600(11) Å in 3 is slightly shorter compared to those in (ArMes)2Ge(P4) (Ge–C: 1.9975(12) Å, 1.9932(12) Å),10a and the P–P bond lengths (2.2557(5) Å, 2.2089(5) Å, 2.154(5) Å, 2.2694(6) Å) vary in a slightly larger range compared to the P–P bond lengths [MeIDippC(H)]Si(P4)[Si(SiMe3)3] (P–P: 2.2555(12) Å, 2.2262(12) Å, 2.2057(13) Å, 2.2615(10) Å).9c The Ga–P bond length (2.3486(3) Å) is within the typical range of gallandiyle coordinated compounds (2.343(9)–2.405(8) Å) obtained from the reaction of LGa with P4.14 In contrast, the Ga–P bond length (2.2510(3) Å) in [L(Cl)GaPSi(L5)P]2, which formed in the [2+1+1] fragmentation reaction of P4 with L(Cl)Ga-SiL5, is significantly shorter.9f The P2–P3–P4 bond angle (59.000(17)°) is almost identical to the corresponding angle in [MeIDippC(H)]Si(P4)[Si(SiMe3)3] (58.96(4)°).9c
The marked reactivity differences between the gallagermylene 1 and (ArMes)2Ge A in the P4 activation reaction point to a benefitial effect of the electropositive L(Cl)Ga substituent, which results in a lower formal charge of the Ge atom in 1 (vide supra). The oxidative addition of one P–P bond of P4 to the germylene centre in 1 proceeded much faster than observed with germylene A, which required four days at r. t. to achieve 75% yield.10a Moreover, gallagermylene 1 not only reacted with insertion of the germylene unit into one P–P bond, which is typically observed in reactions of tetrylenes with P4, but with additional 1,2-migration of the L(Cl)Ga substituent from the Ge to the P atom. The migration of the L(Cl)Ga substituent was also recently observed in the P–P bond activation of the P5 ring in Cp*Fe(η5-P5) upon reaction with L(Cl)Ga-SiL5,11 whereas heavier analogue L(Cl)Ga-GeL5 failed to activate Cp*Fe(η5-P5), clearly demonstrating the higher reactivity of Cl-bridged gallagermylene 1 compared to L(Cl)Ga-GeL5.
To conclude, the gallagermylene 1 shows promising potential in the activation of stable, non-polar σ-bonds of small molecules as was exemplarily demonstrated in the σ-bond activation reaction of H2, which is completed at ambient conditions within seconds. The high reactivity of 1 most likely results from the destabilisation of the HOMO and the benefitial effect of the electropositive L(Cl)Ga ligand. In addition, 1 represents a very rare example of a germylene that activates P4 in an unusual activation/functionalization manner, resulting in formation of a P42− unit as was previously only observed with an electron-rich vinyl(silyl)silylene.
Financial support by the DFG (SCHU1069/26-1; INST 20876/282-1 FUGG) and the University of Duisburg-Essen is acknowledged. We also acknowledge support by the Open Access Publication Fund of the University of Duisburg-Essen.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) F. Hanusch, L. Groll and S. Inoue, Chem. Sci., 2021, 12, 2001 RSC; (b) K. Oberdorf and C. Lichtenberg, Chem. Commun., 2023, 59, 8043–8058 RSC; (c) P. P. Power, Nature, 2010, 463, 171 CrossRef CAS PubMed; (d) T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608 CrossRef CAS PubMed; (e) T. J. Hadlington, M. Driess and C. Jones, Chem. Soc. Rev., 2018, 47, 4176 RSC; (f) C. Weetman and S. Inoue, ChemCatChem, 2018, 10, 4213 CrossRef CAS.
- G. H. Spikes, J. C. Fettinger and P. P. Power, J. Am. Chem. Soc., 2005, 127, 12232 CrossRef CAS PubMed.
- (a) J. Li, C. Schenk, C. Goedecke, G. Frenking and C. Jones, J. Am. Chem. Soc., 2011, 133, 18622 CrossRef CAS PubMed; (b) T. J. Hadlington, M. Hermann, J. Li, G. Frenking and C. Jones, Angew. Chem., Int. Ed., 2013, 52, 10199 CrossRef CAS PubMed.
- (a) A. Rit, J. Campos, H. Niu and S. Aldridge, Nat. Chem., 2016, 8, 1022 CrossRef CAS PubMed; (b) M. Chen, B. Lei, X. Wang, H. Rong, H. Song and Z. Mo, Angew. Chem., Int. Ed., 2022, 61, e202204495 CrossRef CAS.
- T. J. Hadlington, M. Driess and C. Jones, Chem. Soc. Rev., 2018, 47, 4176 RSC.
- (a) Y. Peng, J.-D. Guo, B. D. Ellis, Z. Zhu, J. C. Fettinger, S. Nagase and P. P. Power, J. Am. Chem. Soc., 2009, 131, 16272 CrossRef CAS; (b) M. Usher, A. V. Protchenko, A. Rit, J. Campos, E. L. Kolychev, R. Tirfoin and S. Aldridge, Chem. – Eur. J., 2016, 22, 11685 CrossRef CAS PubMed; (c) M. M. Juckel, J. Hicks, D. Jiang, L. Zhao, G. Frenking and C. Jones, Chem. Commun., 2017, 53, 12692 RSC; (d) K. Inomata, T. Watanabe, Y. Miyazaki and H. Tobita, J. Am. Chem. Soc., 2015, 137, 11935 CrossRef CAS; (e) B. Rao and R. Kinjo, Angew. Chem., Int. Ed., 2019, 58, 18150 CrossRef CAS.
- P. Vermeeren, M. T. Doppert, F. M. Bickelhaupt and T. A. Hamlin, Chem. Sci., 2021, 12, 4526 RSC.
- D. J. Scott, Angew. Chem., Int. Ed., 2022, 61, e202205019 CrossRef CAS PubMed.
- (a) Y. Xiong, S. Yao, M. Brym and M. Driess, Angew. Chem., Int. Ed., 2007, 46, 4511 CrossRef CAS; (b) S. S. Sen, S. Khan, H. W. Roesky, D. Kratzert, K. Meindl, J. Henn, D. Stalke, J.-P. Demers and A. Lange, Angew. Chem., Int. Ed., 2011, 50, 2322 CrossRef CAS; (c) M. M. D. Roy, M. J. Ferguson, R. McDonald, Y. Zhou and E. Rivard, Chem. Sci., 2019, 10, 6476 RSC; (d) D. Reiter, P. Frisch, D. Wendel, F. M. Hörmann and S. Inoue, Dalton Trans., 2020, 49, 7060 RSC; (e) Y. Wang, T. Szilvási, S. Yao and M. Driess, Nat. Chem., 2020, 12, 801 CrossRef CAS PubMed; (f) J. Schoening, A. Gehlhaar, C. Wölper and S. Schulz, Chem. – Eur. J., 2022, 28, e202201031 CrossRef CAS PubMed.
- (a) J. W. Dube, C. M. E. Graham, C. L. B. Macdonald, Z. D. Brown, P. P. Power and P. J. Ragogna, Chem. – Eur. J., 2014, 20, 6739 CrossRef CAS PubMed; (b) D. Sarkar, C. Weetman, D. Munz and S. Inoue, Angew. Chem., Int. Ed., 2021, 60, 3519 CrossRef CAS PubMed; (c) S. Khan, R. Michel, J. M. Dieterich, R. A. Mata, H. W. Roesky, J. Demers, A. Lange and D. Stalke, J. Am. Chem. Soc., 2011, 133, 17889 CrossRef CAS PubMed.
- X. Sun, A. Hinz, S. Schulz, L. Zimmermann, M. Scheer and P. W. Roesky, Chem. Sci., 2023, 14, 4769 RSC.
- (a) A. Bücker, C. Wölper, G. Haberhauer and S. Schulz, Chem. Commun., 2022, 58, 9758 RSC; (b) A. Bücker, C. Wölper and S. Schulz, Polyhedron, 2024, 247, 116702 CrossRef; (c) A. Bücker, C. Wölper, H. Siera, G. Haberhauer and S. Schulz, Dalton Trans., 2024, 53, 640 RSC.
- (a) F. Neese, WIREs Comput. Mol. Sci., 2022, 12, e1606 CrossRef; (b) E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0., Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2018 Search PubMed; (c) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC; (d) E. Caldeweyher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2017, 147, 34112 CrossRef; (e) E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2019, 150, 154122 CrossRef PubMed; (f) J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982 CrossRef CAS; (g) C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- F. Hennersdorf, J. Frötschel and J. J. Weigand, J. Am. Chem. Soc., 2017, 139, 14592 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and analytical data (NMR, IR spectra, crystallographic data) and details from DFT calculations. CCDC 2321151 (2) and 2321152 (3) contain the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc06223f |
| This journal is © The Royal Society of Chemistry 2024 |