
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of highly substituted 1,3-dienes through halonium promoted 1,2-sulfur migration of propargylic thioethers†
Clara
Martínez-Núñez
,
Noelia
Velasco
 ,
Roberto
Sanz
and
Samuel
Suárez-Pantiga
*
,
Roberto
Sanz
and
Samuel
Suárez-Pantiga
*
Departamento de Química, Facultad de Ciencias, Universidad de Burgos, Pza. Misael Bañuelos s/n, 09001-Burgos, Spain. E-mail: svsuarez@ubu.es
First published on 17th January 2024
Abstract
Conjugated 1-bromo or 1-iodo-1,3-dienes bearing a sulfide substituent have been synthesized via 1,2-sulfur migration from propargylic thioethers upon activation with NIS or NBS. The reaction generally proceeds with high control over the regio- and diastereoselectivity. Highly substituted thiophenes and selenophenes are easily obtained from the generated dienes.
Conjugated dienes are relevant structural motifs due to their applications in material science and organic synthesis,1 their prevalence in natural products,2 and broad versatility as building blocks in a plethora of reactions,3,4 enabling the rapid construction of molecular complexity. These features have caused a continuous interest in synthesizing 1,3-dienes.5 Recently, stereoselective strategies for preparing highly substituted 1,3-dienes have attracted much attention.6 Starting from alkynes, several reports have succeeded in producing conjugated dienes using palladium catalysts,7 or bimetallic catalytic systems.8 Migration of propargylic esters to the alkyne position followed by elimination afforded functionalized 1,3-dienes using Au- or Cu-catalysts (Scheme 1, eqn (1)),9 or by combining both metals with hypervalent iodonium salts.10 Also, a modular strategy for constructing 1-halo-1,3-dienes from alkynes has been described in two steps employing a copper catalyst and organolithiums (Scheme 1, eqn (2)).11 Activated 1,3-dienes bearing heteroatomic substituents are highly relevant building blocks and, thus, a common target in synthesis.1,12 Although less explored than their oxygenated or nitrogenated counterparts, sulfide-substituted conjugated dienes13,14 have been acquiring interest partly due to their diverse synthetic applications like in both Diels-Alder13 and inverse electron-demand Diels-Alder13b reactions after oxidation to sulfones.
 | ||
| Scheme 1 Background, previous work and proposed tetrahydrocarbazolone synthesis from 4-(indol-2-yl)-4-oxobutanals. | ||
In recent years, notable efforts in providing efficient routes of syntheses of these dienes have succeeded in accessing 3-substituted-(1,3-dien-2-yl)sulfanes via alkyne hydrothiolation followed by dehydration processes.15 Also, Ananikov, in collaboration with König, described an elegant photocatalyzed process to synthesize precursors of activated 1,3-dienes that, upon treatment with a suitable base, release a challenging trisubstituted sulfur-activated conjugated diene (Scheme 1, eqn (3)), which could be engaged in further transformations,16 circumventing the isolation and purification of reactive dienes.
On the other hand, sulfur migration involving alkenylation processes has experienced continuous growth,17 arising as a powerful strategy for accessing more complex compounds like heterocycles and alkenyl sulfides. In this sense, we have recently reported the synthesis of indenes through an alkyne iodothiolation of propargylic thioethers via 1,3-sulfur shift (Scheme 1, eqn (4)).18 Inspired by these findings, we envisioned that iodonium-induced sulfur migration of propargyl thioethers could provide access to tetrasubstituted alkenes with high control on the diastereoselectivity.19 A base could favor elimination reactions over the sulfonium intermediate 2, enabling of highly substituted conjugated dienes bearing easily tunable halide and sulfur-based substituents (Scheme 1d).
Initially, we subjected the readily available model thioether201a to reaction with I2 in the presence of an external base like K2CO3 (Table 1), observing the formation of conjugated diene 3a derived from alkyne activation followed by 1,2-sulfur migration only in minor amounts (entries 1 and 2). In the absence of the base starting material was recovered (entry 3). Considering the tendency of the I2/carbonate system to afford diiodinated products with similar starting materials,21 we tested NIS with the carbonate (entry 4). To our delight, diene 3a was obtained in high yields as a mixture of diastereoisomers (ca. 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), though isomer E was formed preferentially. A lower base amount achieved a high yield (entry 5). Similar results were provided with catalytic amounts of DBU (entry 6). In the absence of a base (entry 7), the reaction proceeded smoothly in 30 min without observing thiochromene byproducts.20 This result suggests that the succinimidate anion acts as a base in the elimination step. Other solvents needed higher reaction times to achieve full conversion (entries 8 and 9). Higher diastereoselectivity was obtained by lowering the temperature (entries 10 and 11). The transformation performed readily well without the base, affording almost exclusively the diene 3a as the isomer E after 2 h at 0 °C (entry 10). Isomerization of diene (E)-3a may occur upon exposure to sunlight or catalytic amounts of acid, so the reactions were performed in the dark to minimize it.
:1), though isomer E was formed preferentially. A lower base amount achieved a high yield (entry 5). Similar results were provided with catalytic amounts of DBU (entry 6). In the absence of a base (entry 7), the reaction proceeded smoothly in 30 min without observing thiochromene byproducts.20 This result suggests that the succinimidate anion acts as a base in the elimination step. Other solvents needed higher reaction times to achieve full conversion (entries 8 and 9). Higher diastereoselectivity was obtained by lowering the temperature (entries 10 and 11). The transformation performed readily well without the base, affording almost exclusively the diene 3a as the isomer E after 2 h at 0 °C (entry 10). Isomerization of diene (E)-3a may occur upon exposure to sunlight or catalytic amounts of acid, so the reactions were performed in the dark to minimize it.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | I+ source | Base | T | t (h) |
3a Yield (%)b (E:Z) |
| a Reaction conditions: thioether 1a (0.1 mmol), I+ source (0.15 mmol), base (0.1 mmol), solvent (1 mL), temperature (T), and reaction time (t). b Yield determined by 1H NMR using CH2Br2 as internal standard and referred to starting product 1a. c Unreacted starting material 1a was recovered. d The base was employed in catalytic amounts (10 mol%). e Yield after column chromatography referred to starting product 1a. f No full conversion (86%) was observed. | ||||||
| 1 | MeNO2 | I2 | K2CO3 | r.t. | 6 | 16 (4:1) |
| 2 | CH2Cl2 | I2 | K2CO3 | r.t. | 6 | 15 (>20:1) |
| 3 | CH2Cl2 | I2 | — | r.t. | 24 | —c |
| 4 | CH2Cl2 | NIS | K2CO3 | r.t. | 24 | 92 (4.4:1) |
| 5d | CH2Cl2 | NIS | K2CO3 | r.t. | 24 | 88 (3:1) |
| 6d | CH2Cl2 | NIS | DBU | r.t. | 24 | >95 (2:1) |
| 7 | CH2Cl2 | NIS | — | r.t. | 0.5 | 79 (2:1) |
| 8 | Toluene | NIS | — | r.t. | 24 | 77 (2:1) |
| 9 | THF | NIS | — | r.t. | 24 | 56 (4:1) |
| 10 | CH2Cl2 | NIS | — | 0 °C | 2 | >95 (90)e (20:1) |
| 11 | CH2Cl2 | NIS | K2CO3 | 0 °C | 24 | 81 (7:1)f |

Then, we investigated the scope of the reaction by applying the optimized reaction conditions to a diverse range of thioethers 1 (Table 2). Initially, we varied the substituent at R1 linked to the S-atom that experiences the migration. Substrates bearing phenyl or different p- or o-substituted aryl groups with halogens or methoxy groups were easily transformed into corresponding dienes (3b–f) in high yields. Also, elevated diastereoselectivities (>20:1) were observed except for products 3c and 3f, which required prolonged reaction times. When a naphthalene (1g) or different alkyl groups (1h–k) were installed at the S-atom, the dienes (E)-3g–k were formed with complete diastereoselectivity and good yields even when other functional groups were present in the alkyl chain (3j and 3k). Though, compound 3j was obtained in lower yields probably due to partial oxidation of the S-atom with the excess of NIS. Next, we investigated the behavior of substituents at the propargylic positions R2 and R3. Different cyclic, linear, or branched alkyl groups were well-tolerated, affording selectively dienes (E)-3l–o in high yields. Nevertheless, diene 3p, in which the configuration of the tetrasubstituted alkene is fixed, was obtained as a mixture 3:1 of two diastereoisomers where (3E,5E)-3p was the major compound. Thioethers bearing unsubstituted alkynes (1r and 1q) were also successfully employed. However, dienes bearing a methyl (3r) or a phenyl (3q) group at R2 position were highly reactive, resulting in lower yields and diastereoselectivities due to rapid isomerization. Other alkyl substituents at position R4 afforded dienes 3s and 3t in high yields. Notably, linear alkyl chains delivered diene 3t as a single isomer, whereas bulky groups at R4 like t-Bu (3s) provided lower diastereoselectivities.
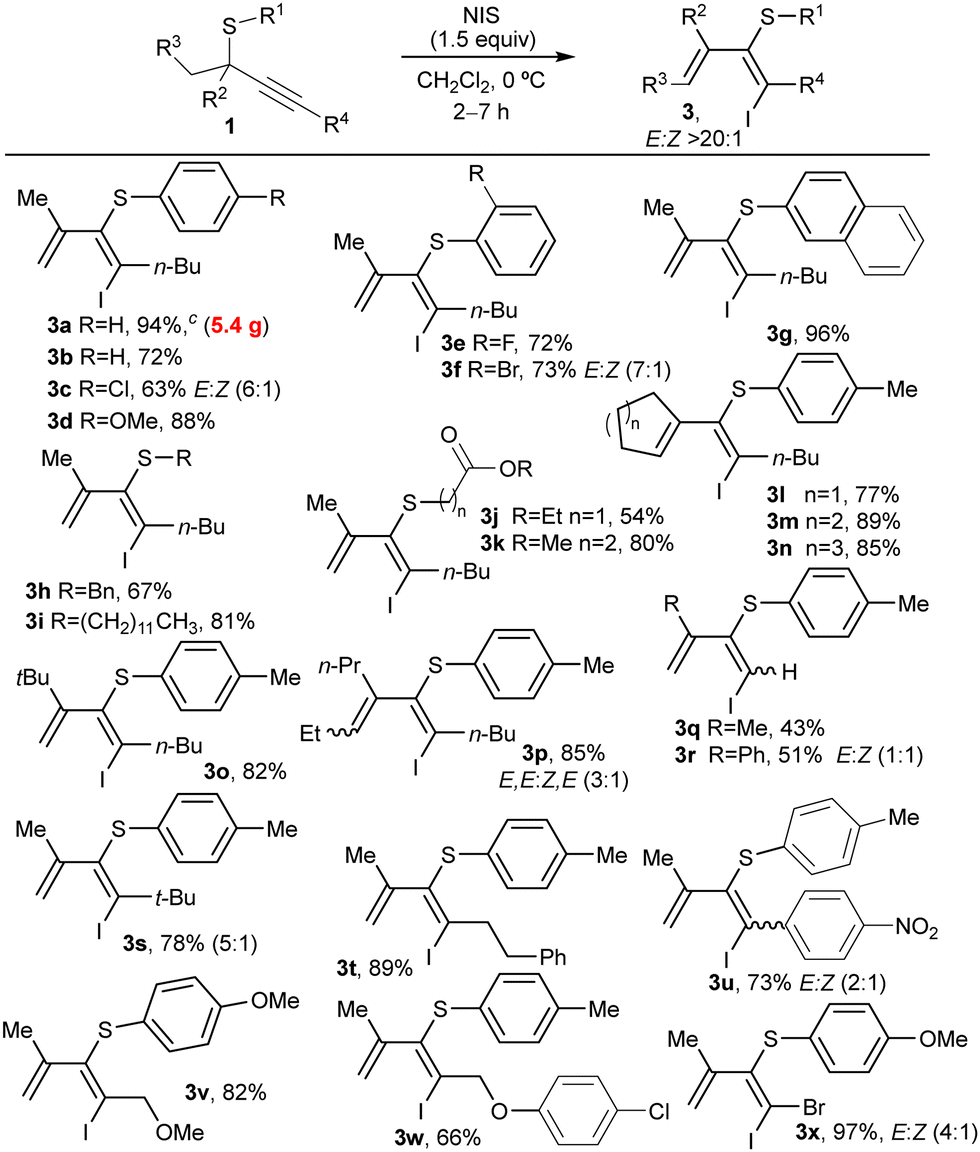
Propargylic thioethers bearing arenes directly attached at the alkyne produced the previously reported thiochromenes, except for those substrates bearing strong electron-withdrawing groups, like a p-nitro (3u), at the aromatic substituent placed at R4. Nevertheless, lower selectivities were obtained for 3u due to rapid isomerization in the reaction media. The reaction exhibited good compatibility with functional groups present in the alkyl chain, like, for example, methyl- or aryl ethers, that afforded the corresponding dienes 3v and 3w, respectively. Similarly, bromine-substituted alkyne 1x provided the diene 3x in almost quantitative yield as a mixture enriched in the diastereoisomer E. The reactions showed excellent performance at a multigram scale to obtain 5.4 g (94%) of diene 3a with complete selectivity. By contrast, S-aryl secondary propargyl thioethers (R2 = R3 = H) were found to be unreactive under optimized conditions.22
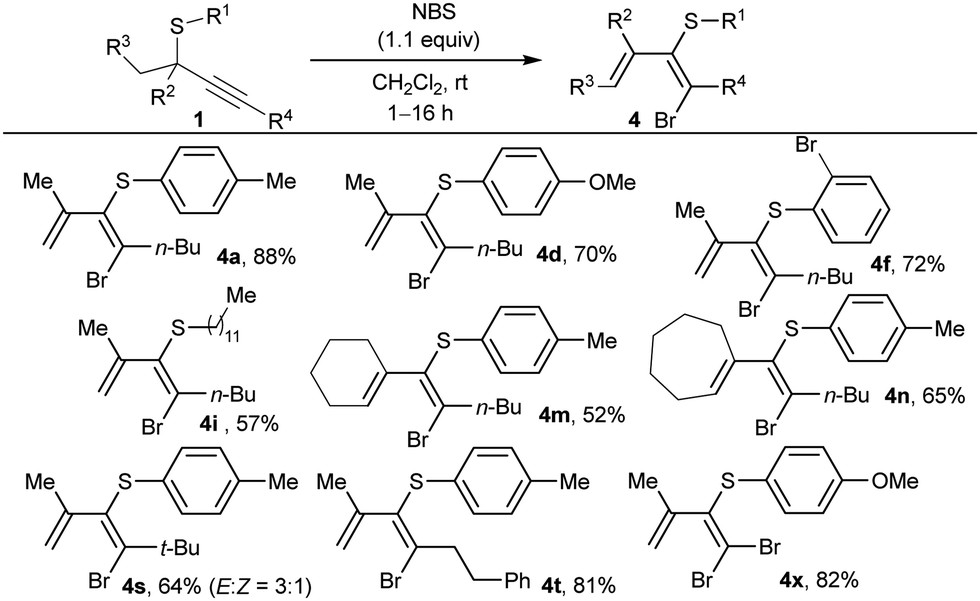
Next, we evaluated other monopositive halonium ion sources. By using N-bromosuccinimide (NBS) instead of NIS, alkyne activation of 1a followed by 1,2 sulfur migration also occurred, affording the corresponding 1-bromo-1,3-diene 4a (Table 3). By contrast, Selectfluor or N-chlorosuccinimide only originated the C–S bond cleavage at the propargylic position. The excess of NBS reagent was demonstrated to be detrimental as further reactions of the generated dienes with the additional amounts of halogenating agent were observed. So, the amount of NBS was reduced to 1.1 equivalents to achieve optimal results. Then, thioethers 1 substituted at the alkyne were tested (Table 3), accessing to 1-bromo-1,3-dienes 4 in slightly lower yields than the related iododienes. Remarkably, the reaction with NBS proceeded smoothly at r.t., affording only one diastereoisomer in almost all the cases except for bulky substituent at R4 (4s), with similar selectivity to that one observed with NIS to obtain 3s. Bromodienes 4 exhibited higher stability, and no isomerization was observed upon exposure to sunlight or acids.
To further illustrate the synthetic potential of the dienes, we decided to test them as precursors in synthesizing valuable heterocycles like 3-thiorganyl substituted thiophenes and selenophenes23 (Table 4). Conjugated iododienes 3 reacted with elemental sulfur or selenium with a copper iodide catalyst and a base, enabling a straightforward synthesis of highly substituted thiophenes and selenophenes.24 A variety of trisubstituted thiophenes (5a, 5i, 5o, and 5t) were obtained in high yields by reaction with elemental sulfur at 90 °C in DMF.
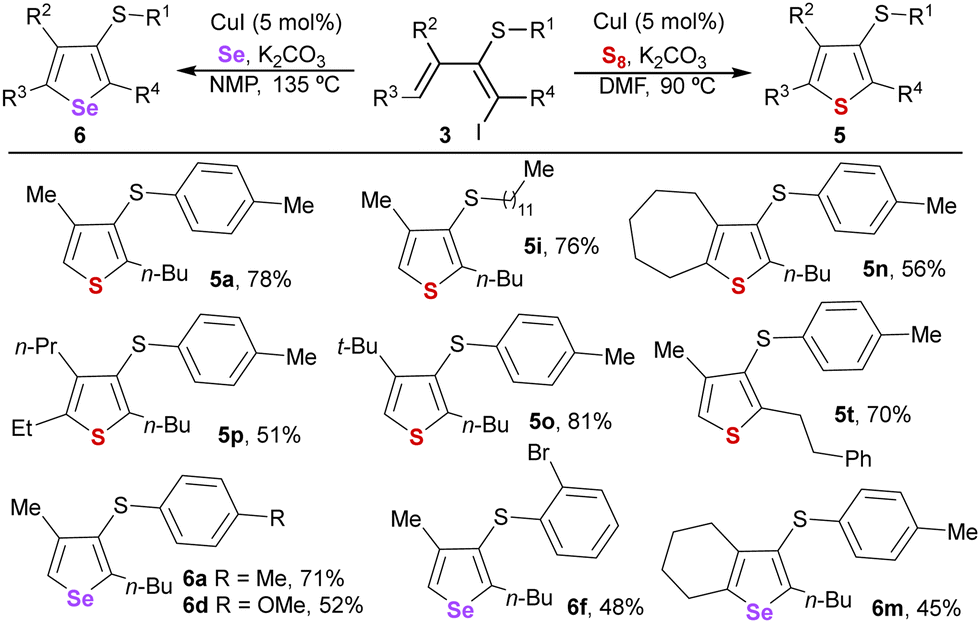
The reaction with pentasubstituted dienes 3n and 3p was demonstrated to be more challenging, although tetrasubstituted thiophenes 5n and 5p could also be synthesized in slightly lower yields. Regarding the synthesis of selenophenes, higher temperatures were required to accomplish an efficient heterocyclization reaction using the more stable NMP. Under these conditions, a selection of different representative selenophenes bearing three (6a, 6d, and 6f) or four substituents (6m) was afforded.
A plausible mechanism for the formation of conjugated 1-halo-1,3-dienes is depicted in Scheme 2. The interaction of thioether 1 with NIS generates cyclic iodonium intermediate 7. Then, an anti-nucleophilic attack of the S-atom located at the propargylic position produces a less studied methylenethiiranium intermediate 2.25 The nature of R4 substituent plays a crucial role in favoring diene formation. In this sense, alkyl, hydrogen, halogen, or electron-poor aromatics substituents prefer further evolution into sulfonium intermediate 2, whereas electron-rich arenes could cause slippage of the iodonium ion, delivering alternative competitive thiochromene formation. Finally, conjugated iododiene 3 is released after deprotonation of the intermediate 2 by the succinimidate anion derived from the NIS.
 | ||
| Scheme 2 Mechanistic proposal. | ||
In conclusion, a route to synthesize highly substituted conjugated dienes has been described by modulating the rich and diverse reactivity of propargylic thioethers towards alkynophilic reagents like NIS or NBS. The transformation proceeds under mild reaction conditions, enabling the formation of a variety of 1-halo-2-thio-1,3-dienes in high yields and, in general, with good control over the diastereoselectivity on the generated tetrasubstituted alkene. The process was robust enough to be performed under air and scalable. The high control over the regio- and diastereoselectivity observed could be reasoned by a key intermediate formed after an anti nucleophilic S-attack to the alkyne activated by the halonium source. The choice of substrate and reaction conditions enable this 1,2-sulfur migration followed by succinimidate anion-assisted elimination sequence to obtain conjugated dienes over other alternative reactivity patterns of propargylic thioethers. The generated halodienes demonstrated potential as valuable building blocks for subsequent transformation, like synthesizing highly substituted thiophenes and selenophenes upon reaction with elemental sulfur or selenium under copper catalysis.
We gratefully acknowledge MCIN (PID2020-115789GB-C21/AEI/10.13039/501100011033), and Junta de Castilla y León and FEDER (BU028P23) for financial support. C. M.-N. thanks Junta de Castilla y León and Fondo Social Europeo, for a predoctoral contract. S. S.-P. thanks a Ramón y Cajal (RYC2021-031533-I) contract by MCIN/AEI/10.13039/501100011033 and European Union “NextGenerationEU”/PRTR.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Z. Rappoport, PATAI’S Chemistry of Functional Groups: The Chemistry of Dienes and Polyenes, Wiley, 2000, vol. 2 Search PubMed.
- S. D. Rychnovsky, Chem. Rev., 1995, 95, 2021–2040 CrossRef CAS.
- K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS.
- (a) J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Chem. Rev., 2015, 115, 5301–5365 CrossRef CAS PubMed; (b) G. J. P. Perry, T. Jia and D. J. Procter, ACS Catal., 2020, 10, 1485–1499 CrossRef CAS.
- (a) M. De Paolis, I. Chataigner and J. Maddaluno, in Stereoselective Alkene Synthesis, ed. J. Wang, Springer Berlin Heidelberg, 2012, pp. 87–146 Search PubMed; (b) P. Hubert, E. Seibel, C. Beemelmanns, J.-M. Campagne and R. M. de Figueiredo, Adv. Synth. Catal., 2020, 362, 5532–5575 CrossRef CAS; (c) R. G. Soengas and H. Rodríguez-Solla, Molecules, 2021, 26, 249 CrossRef CAS PubMed.
- (a) S. Manna, S. Paul, W.-Y. Kong, D. Aich, R. Sahoo, D. J. Tantillo and S. Panda, Angew. Chem., Int. Ed., 2023, 62, e202309136 CrossRef CAS PubMed; (b) W.-J. Kong, S. N. Kessler, H. Wu and J.-E. Bäckvall, Org. Lett., 2023, 25, 120–124 CrossRef CAS PubMed; (c) G.-X. Liu, X.-T. Jie, X.-l Li, L.-S. Yang, H. Qiu and W.-H. Hu, ACS Catal., 2023, 13, 5307–5314 CrossRef CAS; (d) W.-S. Zhang, D.-W. Ji, Y. Li, X.-X. Zhang, Y.-K. Mei, B.-Z. Chen and Q.-A. Chen, Nat. Commun., 2023, 14, 651 CrossRef CAS PubMed; (e) R.-P. Li, J. Li, X. Chen, J. Liu, X. Wang and S. Tang, Adv. Synth. Catal., 2022, 364, 4260–4265 CrossRef CAS.
- (a) N. J. Green, A. C. Willis and M. S. Sherburn, Angew. Chem., Int. Ed., 2016, 55, 9244–9248 CrossRef CAS PubMed; (b) J. Liu, J. Yang, W. Baumann, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2019, 58, 10683–10687 CrossRef CAS PubMed; (c) X.-B. Xiang, S. Wang, T. Xu and S. Chen, Org. Lett., 2023, 25, 587–591 CrossRef CAS PubMed.
- (a) T. Long, C. Zhu, L. Li, L. Shao, S. Zhu, M. Rueping and L. Chu, Nat. Commun., 2023, 14, 55 CrossRef CAS PubMed; (b) C.-J. Hou, A. W. Schuppe, J. L. Knippel, A. Z. Ni and S. L. Buchwald, Org. Lett., 2021, 23, 8816–8821 CrossRef CAS PubMed.
- R. Kazem Shiroodi, A. S. Dudnik and V. Gevorgyan, J. Am. Chem. Soc., 2012, 134, 6928–6931 CrossRef CAS PubMed.
- Z. Sun, M. Dai, C. Ding, S. Chen and L.-A. Chen, J. Am. Chem. Soc., 2023, 145, 18115–18125 CrossRef CAS PubMed.
- E. Rivera-Chao and M. Fañanás-Mastral, Angew. Chem., Int. Ed., 2021, 60, 16922–16927 CrossRef CAS PubMed.
- A. Deagostino, C. Prandi, C. Zavattaro and P. Venturello, Eur. J. Org. Chem., 2006, 2463–2483 CrossRef CAS.
- (a) F. Peng, R. E. Grote, R. M. Wilson and S. J. Danishefsky, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10904–10909 CrossRef CAS PubMed; (b) J. Choi, H. Park, H. J. Yoo, S. Kim, E. J. Sorensen and C. Lee, J. Am. Chem. Soc., 2014, 136, 9918–9921 CrossRef CAS PubMed.
- (a) J.-E. Bäckvall and A. Ericsson, J. Org. Chem., 1994, 59, 5850–5851 CrossRef; (b) S. S. Zalesskiy, N. S. Shlapakov and V. P. Ananikov, Chem. Sci., 2016, 7, 6740–6745 RSC.
- E. S. Degtyareva, E. V. Borkovskaya and V. P. Ananikov, ACS Sustainable Chem. Eng., 2019, 7, 9680–9689 CrossRef CAS.
- J. V. Burykina, A. D. Kobelev, N. S. Shlapakov, A. Y. Kostyukovich, A. N. Fakhrutdinov, B. König and V. P. Ananikov, Angew. Chem., Int. Ed., 2022, 61, e202116888 CrossRef CAS PubMed.
- For 1,2 sulfur migration, see: (a) J. T. Kim, A. V. Kel'in and V. Gevorgyan, Angew. Chem., Int. Ed., 2003, 42, 98–101 CrossRef CAS PubMed; (b) A. S. Dudnik, A. W. Sromek, M. Rubina, J. T. Kim, A. V. Kel'in and V. Gevorgyan, J. Am. Chem. Soc., 2008, 130, 1440–1452 CrossRef CAS PubMed; (c) L. Peng, X. Zhang, S. Zhang and J. Wang, J. Org. Chem., 2007, 72, 1192–1197 CrossRef CAS PubMed; (d) T. Yuan, B. Ryckaert, K. Van Hecke, J. Hullaert and J. M. Winne, Angew. Chem., Int. Ed., 2021, 60, 4070–4074 CrossRef CAS PubMed . For 1,3 sulfur migration, see: ; (e) S. B. Wagh, R. R. Singh, R. L. Sahani and R.-S. Liu, Org. Lett., 2019, 21, 2755–2758 CrossRef CAS PubMed . For 1,4 sulfur migration, see: ; (f) J. A. Kaplan, A. Issaian, M. Stang, D. Gorial and S. A. Blum, Angew. Chem., Int. Ed., 2021, 60, 25776–25780 CrossRef CAS PubMed; (g) F. Martínez-Lara, A. Suárez, N. Velasco, S. Suárez-Pantiga and R. Sanz, Adv. Synth. Catal., 2022, 364, 132–138 CrossRef . For related halogen-induced 1,2-migrations of heteroatoms, see also: ; (h) D.-H. Tan, Z.-H. Chen, L. Yang, C.-T. Li, F.-H. Tu, Q. Li and H. Wang, Sci. China Chem., 2022, 65, 746–752 CrossRef CAS; (i) F.-H. Tu, Y. Wang, Z. Li, S. Yang, Y. Li, D.-H. Tan, Q. Li, H. Gao and H. Wang, Cell Rep. Phys. Sci., 2022, 3, 101194 CrossRef CAS.
- N. Velasco, C. Martínez-Núñez, M. A. Fernández-Rodríguez, R. Sanz and S. Suárez-Pantiga, Adv. Synth. Catal., 2022, 364, 2932–2938 CrossRef CAS.
- A. N. V. Satyanarayana, N. Mukherjee and T. Chatterjee, Green Chem., 2023, 25, 779–788 RSC.
- N. Velasco, A. Suárez, F. Martínez-Lara, M. Á. Fernández-Rodríguez, R. Sanz and S. Suárez-Pantiga, J. Org. Chem., 2021, 86, 7078–7091 CrossRef CAS PubMed.
- F. Yang, T.-N. Jin, M. Bao and Y. Yamamoto, Tetrahedron Lett., 2011, 52, 936–938 CrossRef CAS.
- For further details, see ESI†.
- (a) M. Wang, Q. Fan and X. Jiang, Org. Lett., 2016, 18, 5756–5759 CrossRef CAS PubMed; (b) M. Wang, J. Wei, Q. Fan and X. Jiang, Chem. Commun., 2017, 53, 2918–2921 RSC . For some applications of 3-thiorganyl thiophenes and selenophenes in material science, see: ; (c) R. Cagnoli, M. Lanzi, E. Libertini, A. Mucci, L. Paganin, F. Parenti, L. Preti and L. Schenetti, Macromolecules, 2008, 41, 3785–3792 CrossRef CAS; (d) Y. H. Wijsboom, Y. Sheynin, A. Patra, N. Zamoshchik, R. Vardimon, G. Leitus and M. Bendikov, J. Mat. Chem., 2011, 21, 1368–1372 RSC . For application in drugs, see: ; (e) D. Tondi, R. A. Powers, E. Caselli, M. a-C. Negri, J. Blázquez, M. P. Costi and B. K. Shoichet, Chem. Biol., 2001, 8, 593–610 CrossRef CAS PubMed.
- (a) B. Wu and N. Yoshikai, Angew. Chem., Int. Ed., 2013, 52, 10496–10499 CrossRef CAS PubMed; (b) J. Wu and N. Yoshikai, Angew. Chem., Int. Ed., 2016, 55, 336–340 CrossRef CAS PubMed.
- For related intermediates employing gold catalysts, see also: ref. 17c and d.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, and NMR spectra. See DOI: https://doi.org/10.1039/d3cc06194a |
| This journal is © The Royal Society of Chemistry 2024 |