
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrasonic reduction: an unconventional route to exsolute Ag from perovskite La(Ag)FeO3−δ for enhanced catalytic oxidation activity†
Haijun
Wu
,
Zhibin
Geng
 *,
Xu
Zhao
,
Qi
Wang
,
Ming
Ya
,
Taotao
Huang
,
Junzhi
Li
,
Liping
Li
* and
Guangshe
Li
*
*,
Xu
Zhao
,
Qi
Wang
,
Ming
Ya
,
Taotao
Huang
,
Junzhi
Li
,
Liping
Li
* and
Guangshe
Li
*
State Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry, Jilin University, Changchun 130012, P. R. China. E-mail: gengzb@jlu.edu.cn; lipingli@jlu.edu.cn; guangshe@jlu.edu.cn
First published on 30th January 2024
Abstract
We explore an uncommon ultrasonic reduction method to exsolute Ag from perovskite La0.87Ag0.03FeO3−δ, forming a composite with enhanced catalytic oxidation activity. Such a mild exsolution is based on the coupling effect of ultrasonic cavitation and reducible BH4−, and holds great potential in the fields of energy and environment catalysis.
Perovskite oxide-based composites are widely considered to be key catalysts in the fields of energy and environment due to their strong electron transfer between components.1–10 Many efforts have been made to develop synthetic methods for perovskite oxide-based composites with tight combination, strong interaction, and uniform particle distribution.5–19 Despite several advances in this regard, harsh conditions such as high-pressure hydrogen atmosphere and high temperature are still the primary exsolution methods for forming composites. For example, under conditions of H2 and 900 °C, Co3+ was exsoluted from bulk La0.3Sr0.7Co0.07Ti0.93O3−δ to the surface and formed nano-sized Co clusters that reduced the anode polarization resistance and improved the solid oxide fuel cell performance of La0.3Sr0.7TiO3−δ as well.11 Nano-alloy Fe–Ni can be exsoluted from Sr2Fe1.3Ni0.2Mo0.5O6–δ by H2 annealing, showing enhanced oxygen evolution activity.14 Kim's team constructed an Ir/LaFe0.99Ir0.01O3 composite by exsolution, and the composite showed good hydrogen production properties.17 It is noted that the classical thermal reduction exsolution method is conducted at a temperature above 500 °C and in H2 atmosphere,11–13 and moreover, the pristine oxides tend to undergo structural distortion and even structure collapse during the exsolution process.20 Nowadays, sustainable development requires environment-friendly exsolution methods without involving harsh conditions such as high temperature and hydrogen atmosphere, which remains highly challenging.
Ultrasonic reduction exsolution could be promising for meeting such challenges, due to the coupling effects of ultrasonic cavitation21–23 and strengthened reducibility of NaBH4 solution. High-intensity ultrasonic waves can generate a large number of tiny cavitation bubbles in the propagation medium,24 while the collapse of the cavitation bubbles can create a local environment of high temperature, pressure, and shockwaves, providing a special physicochemical environment for chemical reactions. The ultrasonic waves strengthened the reducibility of the NaBH4 solution, creating a reaction environment with local high temperature and pressure, and strong reducibility, which is similar to the classical high-pressure hydrogen atmosphere annealing exsolution. Even so, thermal reduction exsolution is carried out in a high temperature atmosphere, and thus the lattice oxygen could combine with active hydrogen to form water molecules and escape from the pristine oxide. In this case, a large number of cations and oxygen vacancies are generated in the reaction, which could lead to lattice collapse and even phase transformation.4,14,15 Fortunately, ultrasonic reduction exsolution is carried out in an aqueous solution at room temperature and pressure, and the solution environment prevents the escape of lattice oxygen after combining with active hydrogen, confirming the stability of the catalysts.25,26 Therefore, ultrasonic reduction exsolution could have potential for the fabrication of advanced composites.
In this work, we report an ultrasonic reduction exsolution that was successfully applied to the synthesis of composite Ag0.03−x/La0.87AgxHyFeO3−δ (LAFO-UR). Under ultrasonic waves, the active hydrogen was inserted into the lattice of perovskite LAFO, which then transfers electrons to Ag+ ions, leading to their reduction and exsolution from the lattice to form Ag nanoparticles on the surface of the perovskite, and construct a composite Ag0.03−x/La0.87AgxHyFeO3−δ (Fig. 1a). The crystal structures of the synthesized samples, including LaFeO3 (LFO), LAFO, and LAFO-UR, were verified by X-ray diffraction (XRD) patterns (Fig. 1b), which showed that all signal peaks of the synthesized LFO can be attributed to perovskite LaFeO3 (PDF#88-0641), similar to other two samples LAFO and LAFO-UR. Even so, from the enlarged XRD patterns in the interval of 31°–34° (Fig. 1c), one can see that the peaks of LAFO and LAFO-UR shifted towards lower angles relative to pure perovskite LFO. This demonstrates the lattice expansion after Ag doping in the perovskite due to the larger radius of Ag+ compared to La3+. Additionally, LAFO-UR exhibited a smaller shift towards higher angles relative to LAFO, indicating that some of the doped Ag+ ions were reduced and escaped from the perovskite lattice. The results of XRD refined data provide proof of the above conclusions (Fig. S3 and Table S1, ESI†).
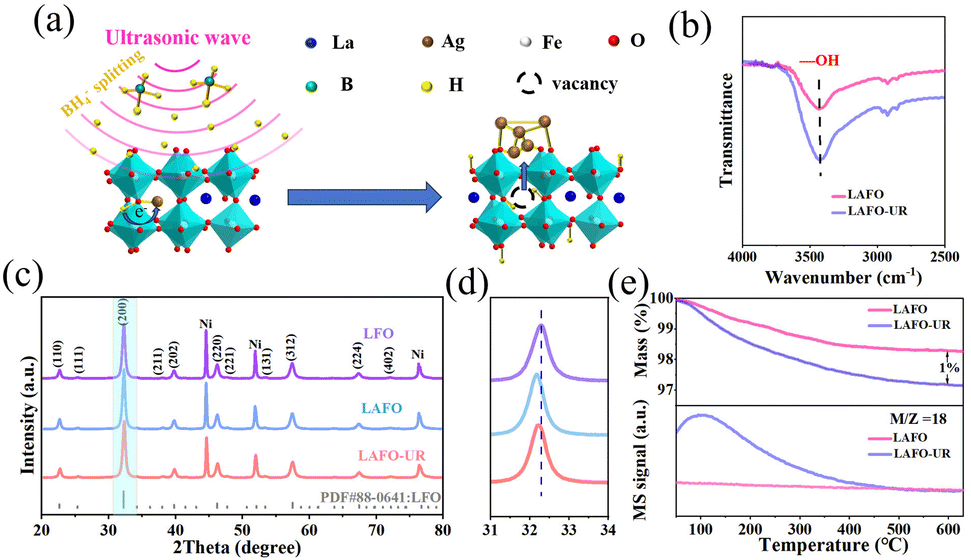 | ||
| Fig. 1 (a) Schematic illustration of LAFO ultrasound reduction, (b) FT-IR data of LAFO and LAFO-UR, (c) XRD patterns of LFO, LAFO, and LAFO-UR (Ni is the internal standard), (d) enlarged XRD patterns in the interval of 31–34°, and (e) TG-MS data of LAFO and LAFO-UR. | ||
The mechanism of ultrasonic reduction exsolution was investigated by Fourier Transform Infrared Spectroscopy (FT-IR) (Fig. 1e) and Thermogravimetric Mass Spectrometry (TG-MS) (Fig. 1d and Fig. S5, ESI†). The FT-IR analysis revealed that LAFO-UR contained more O–H species compared to LAFO, as evidenced by an observable characteristic peak at a wavelength of 3430 cm−1 (Fig. 1b). This conclusion is further supported by TG-MS data; the mass loss of LAFO-UR at 600 °C is 1% higher than that of LAFO (Fig. 1d), and this difference in mass loss is originated from the release of water from LAFO-UR, possibly formed by the condensation of O–H at high temperatures. The results suggest that hydrogen inserted could combine with lattice oxygen during the ultrasonic reduction process, leading to the formation of water, while lattice oxygen does not escape during the reaction process. The data of TG can indeed be used to calculate the amount of protons inserted into LAFO, and the molecular formula of LAFO-UR is Ag0.03–x/La0.87AgxH0.13FeO3−δ. Consequently, no oxygen vacancies were generated during the LAFO reduction process, as indicated by Electron Paramagnetic Resonance (EPR) (Fig. S6, ESI†).
The morphology and elemental distribution of the samples LFO, LAFO, and LAFO-UR were observed by Transmission Electron Microscope (TEM) images. Both LAFO and LAFO-UR exhibited particle sizes ranging from 15 to 50 nm, and no significant morphology change was observed after ultrasonic reduction. High Resolution Transmission Electron Microscope (HRTEM) analysis revealed lattice spacings of 0.275 and 0.35 nm for LAFO and LAFO-UR, corresponding to the crystal planes (002) and (111) of perovskite LaFeO3, respectively. Additionally, small nanoparticles with a lattice spacing of 0.235 nm were found in LAFO-UR, which was consistent with the (111) crystal plane of Ag metal (Fig. 2g), suggesting the formation of Ag nanoparticles on the surface of LAFO. Elemental mapping illustrated that La and Fe were uniformly distributed in both LAFO and LAFO-UR (Fig. 2b and f); meanwhile, in LAFO-UR, parts of Ag (Fig. 2f) were significantly aggregated compared to LAFO (Fig. 2b), with the sizes of the Ag aggregations ranged from 3 to 5 nm, consistent with particle size of Ag nanoparticles observed in TEM images (Fig. S7, ESI†). This result is further confirmed by linear scanning. In LAFO, the changing trend of Ag content is in the same way as that of La and O (Fig. 2d). Nevertheless, in LAFO-UR (Fig. 2h and Fig. S8, ESI†), Ag shows an aggregated distribution, and the surface nanoparticles mainly consist of Ag element, which provides direct evidence for the exsolution of Ag.
 | ||
| Fig. 2 (a) and (e) TEM images. (b) and (f) Elemental mapping. (c) and (g) HRTEM images. (d) and (h) Linear scan of LAFO and LAFO-UR. | ||
The surface composition and structure of the samples were obtained by X-ray Photoelectron Spectroscopy (XPS). From Ag 3d spectra of LAFO and LAFO-UR in Fig. 3a, one can see a larger Ag+ signal peak at 367.8 eV, in addition to the signal peak of Ag0 at 368.5 eV for LAFO-UR, which indicates the presence of Ag nanoparticles after ultrasonic reduction exsolution in LAFO-UR. The O 1s spectrum (Fig. 3c) shows that the peak at 529.3 eV corresponds to Fe–O–Fe in the perovskite,27 while the peak at 531.25 eV is attributed to O–H related species (Fe–O–H, H–O–H, etc.) on the surface of the perovskite. Obviously, the content of O–H on the surface of LAFO-UR is found to be significantly higher than that of LAFO. In addition, Fe 2p spectra indicate that the valence state of Fe is closer to +3 (Fig. 3b).
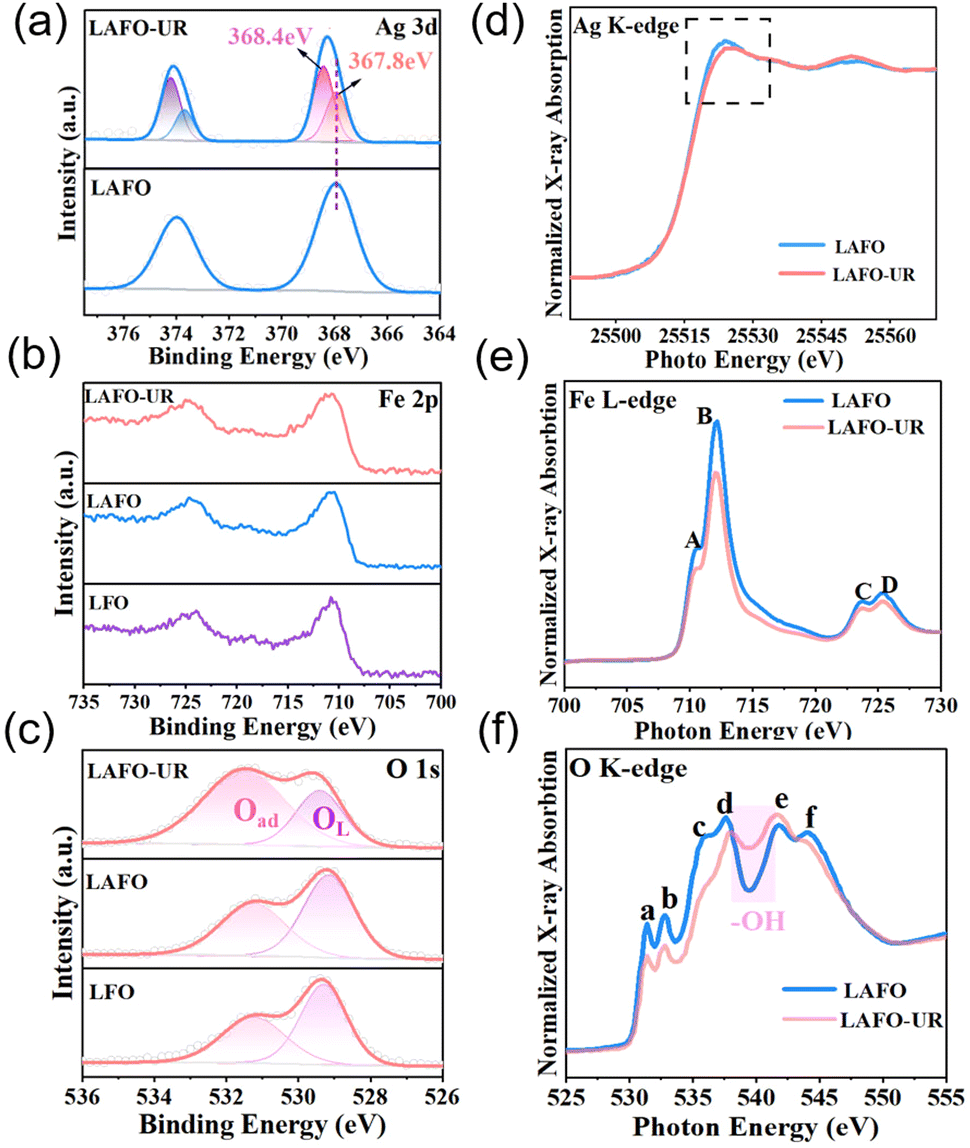 | ||
| Fig. 3 (a) Ag 3d XPS spectra of LAFO and LAFO-UR, (b) Fe 2p and (c) O 1s XPS spectra of LAFO and LAFO-UR, relative to LFO; (d) Ag K-edge, (e) Fe L-edge, and (f) O K-edge XAFS spectra of LAFO and LAFO-UR. | ||
Synchrotron X-ray Absorption Fine Structure (XAFS) was used to study the electronic states of the samples LAFO and LAFO-UR. In Fig. 3d, for Ag K-edge spectra, the intensity of the white line peak is related to the valence state.28 LAFO-UR exhibits a lower intensity of the white line peak in comparison with LAFO, indicating a significant decrease in valence state of Ag after ultrasonic reduction. Fig. 3e shows the normalized Fe L-edge spectra. Peaks A and C could be attributed to the Fe 3d–O 2p hybrid t2g orbital, while peaks B and D could be assigned to the Fe 3d–O2 p hybrid eg orbital.29 The peak intensity of LAFO-UR is slightly lower than that of LAFO, indicating that the density of electronic states of Fe in LAFO-UR is higher than that of Fe in LAFO. Meanwhile, the ratio of peak B to peak A was calculated (Table S4, ESI†), and the ratio values of LAFO and LAFO-UR were 2.13 and 2.04, respectively. This indicates that the probability of electron transfer from the t2g orbital to the eg orbital increases in LAFO-UR, as reported elsewhere.29 Therefore, the valence of Fe decreased after ultrasonic reduction. Fig. 3f is the normalized O K-edge spectra. Peaks a and b could be attributed to the orbitals t2g and eg in the Fe 3d–O 2p hybrid orbital. Peaks c and d could be associated with the La 5d–O 2p hybrid zone, while peaks e and f are derived from Fe 4sp and O 2p.30 To further discuss the electronic states, we calculated the intensity ratio of peak b to peak a (Table S5, ESI†). The ratio values for LAFO and LAFO-UR are 1.07 and 1.11, respectively, and this result suggests a transfer of eg orbital electrons in the Fe 3d-O 2p hybrid orbital to the t2g orbital, leading to a decrease in the valence of O, like the observation reported elsewhere.29 The main reason for the decrease of the O valence state is charge transfer from the Ag nanoparticles to the perovskite. The peak intensity of LAFO-UR at 538 ∼ 542 eV (Fig. 3f) was enhanced because of the production of hydroxyl groups, as reported elsewhere.31,32
The oxidation activity of the samples LFO, LAFO, LAFO-UR, and hydrogen-annealed LAFO (LAFO-H2-400) was investigated, as shown in Fig. 4a. LAFO-UR exhibits the best catalytic performance among all the samples. Additionally, the 10%, 50%, and 90% conversion temperatures of LAFO-UR are 150 °C, 175 °C, and 195 °C, respectively (Fig. 4b), all of which are lower than those of LAFO and LAFO-H2-400. A linear Arrhenius relationship between ln(rate) and the reciprocal of the temperature (1000/T) was obtained for LFO, LAFO and LAFO-UR (Fig. 4c). Activation energy indirectly reflects catalytic activity.33 The activation energy of LAFO-UR is calculated to be 61.97 KJ·mol−1, which is lower than those of LFO and LAFO, indicating that the catalytic CO oxidation of LAFO-UR is easier to occur at low temperatures.
 | ||
| Fig. 4 (a) CO catalytic oxidation curves and (b) 2D histograms of 10%, 50%, and 90% CO conversion versus reaction temperature for the given samples; (c) Arrhenius plots, (d) H2-TPR, and (e) O2-TPD for samples LFO, LAFO, and LAFO-UR. (d) H2-TPR and (e) O2-TPD of LFO, LAFO, and LAFO-UR. (f) In situ DRIFTS spectra of LAFO-UR in a reactive atmosphere (1% CO, 20% O2/He). | ||
H2-TPR and O2-TPD were used to characterize the change in the chemical nature of the samples. In the H2-TPR analysis (Fig. 4d), it is observed that the reduction temperature of Ag+ (135 °C) is below 300 °C, while the temperature range for reducing Fe3+ to Fe2+ is 300–500 °C. The reduction temperatures of Ag+ and Fe3+/Fe2+ in LAFO-UR have been significantly increased compared to LAFO. This result demonstrates that Ag+ and Fe3+/Fe2+ are activated during ultrasonic reduction, and are easily reduced at lower temperatures. O2-TPD is further used to understand the oxygen species desorption behavior of the samples (Fig. 4e). Low temperature desorption of physically adsorbed molecular O2 and adsorbed O2− (50–200 °C), and intermediate temperature desorption of O−, O2− and surface lattice oxygen (300–500 °C) are shown in Fig. 4e. The low and medium temperature ranges of LAFO-UR shifted towards lower temperatures. Moreover, the peak area in the low-temperature region for LAFO-UR is larger than those for LFO and LAFO, and a new peak appears at 297 °C. This observation is likely since exsolution of Ag activates the oxygen species on the surface of LAFO. In order to reveal the reaction mechanism for CO catalytic oxidation, in situ Diffuse Reflaxions Infrared Fourier Transformations Spectroscopy (DRIFTS) was examined (Fig. 4f). The peak positions of CO (at 2100–2200 cm−1), HCOO (at 1560 cm−1) and HCO3* (at 1437 cm−1 and 1339 cm−1) are observed, and the peak signal intensity of CO gradually decreases as the temperature increases, while the peaks for Ag adsorbing CO are not observed. Based on these observations, it can be deduced that with Ag as the absorption sites of O2, the mechanism of CO oxidation conforms to the Eley–Rideal (E–R) mechanism. The presence of Ag weakens the Fe–O bonds,34,35 and the activated –OH on the surface of the catalyst reacts with CO to produce bicarbonate and formate, which subsequently react with O2 adsorbed on the active site of Ag to produce CO2. The reaction path of LAFO is the same as that of LAFO-UR (Fig. S15, ESI†).
In summary, an unconventional exsolution method was successfully applied to prepare a Ag0.03−x/La0.87AgxHyFeO3−δ composite for CO oxidation. The formation of Ag nanoparticles on the surface was comfirmed by XPS and HRTEM. Hydrogen was found to be inserted into the perovskite lattice as indicated by FI-IR and TG-MS, which compensated for the cation losses caused by Ag exsolution and maintained the stability of the perovskite structure. The surface Ag nanoparticles increased the adsorption sites of O2, weakened the Fe–O bond, and activated the lattice oxygen on the sample surface, resulting in superior CO oxidation activity. The ultrasonic reduction exsolution has demonstrated its potential for synthesizing efficient composite catalysts, holding great potential in the fields of energy, environment, and catalysis.
This work was financially supported by the National Natural Science Foundation of China (Grant No. 22101097, 22293041, and 22175070), Jilin Provincial Science and Technology Department (SKL202302018). The authors thank beamlines MCD-A and MCD-B (Soochow Beamline for Energy Materials) at NSRL for providing beam time.
Conflicts of interest
There are no conflicts to declare.References
- D. Neagu, G. Tsekouras and D. N. Miller, et al. , Nat. Chem., 2013, 5, 916–923 CrossRef CAS PubMed.
- A. Opitz, A. Nenning, C. Rameshan and R. Rameshan, et al. , Angew. Chem., Int. Ed., 2015, 54, 2628–2632 CrossRef CAS PubMed.
- J. Zhu, S. Pang, T. Dittrich, Y. Gao and W. Nie, et al. , Nano Lett., 2017, 17, 6735–6741 CrossRef CAS PubMed.
- Y. Jiang, Z. Geng, Y. Sun and X. Wang, et al. , ACS Sustainable Chem. Eng., 2019, 8, 302–310 CrossRef.
- D. Neagu, T. S. Oh, D. N. Miller and H. Ménard, et al. , Nat. Commun., 2015, 6, 8120 CrossRef PubMed.
- S. Wang, Y. Gao, S. Miao and T. Liu, et al. , J. Am. Chem. Soc., 2017, 139, 11771–11778 CrossRef CAS PubMed.
- X. Wang, Z. Pan, X. Chu and K. Huang, et al. , Angew. Chem., Int. Ed., 2019, 58, 11720–11725 CrossRef CAS PubMed.
- F. Lasserre, S. Suarez and F. Mücklich, Mater. Res. Express, 2016, 3, 125007 CrossRef.
- M. Chu, C. Chen, Y. Wu, X. Yan and S. Jia, et al. , Green Energy Environ., 2022, 4, 792–798 CrossRef.
- Y. Cong, Z. Geng, Q. Zhu and H. Hou, et al. , Angew. Chem., Int. Ed., 2021, 133, 23568–23575 CrossRef.
- S. H. Cui, J. H. Li, X. W. Zhou and G. Y. Wang, et al. , J. Mater. Chem. A, 2013, 34, 9689–9696 RSC.
- J. H. Myung, D. Neagu, D. N. Miller and J. T. Irvine, Nature, 2016, 537, 528–531 CrossRef CAS PubMed.
- Y. Gao, D. Chen, M. Saccoccio, Z. Lu and F. Ciucci, Nano Energy, 2016, 27, 499–508 CrossRef CAS.
- K. Zhu, T. Wu, M. Li, R. Lu and X. Zhu, et al. , J. Mater. Chem. A, 2017, 5, 19836–19845 RSC.
- Y. Jin, W. Huo, L. Zhang and Y. Li, et al. , Chem. Commun., 2021, 57, 7168–7171 RSC.
- L. Yao, Z. Geng, W. Zhang and X. Wu, et al. , ACS Sustainable Chem. Eng., 2020, 8, 17194–17200 CrossRef CAS.
- Y. Kim, H. S. Kim, D. Kang, M. Kim and J. W. Lee, Chem. Eng. J., 2023, 468, 143662 CrossRef CAS.
- Z. Sun, W. Fan and Y. Bai, et al. , ACS Appl. Mater. Interfaces, 2021, 25, 29755–29763 CrossRef PubMed.
- J. Ma, Z. Geng, Y. Jiang and X. Hou, et al. , J. Alloys Compd., 2021, 854, 157154 CrossRef CAS.
- G. S. Pawar, A. Elikkottil, S. Seetha and K. S. Reddy, et al. , ACS Appl. Energy Mater., 2018, 1, 3449–3456 CrossRef CAS.
- B. Fu, J. Li, H. Jiang, X. He, Y. Ma and J. Wang, et al. , Nanoscale, 2022, 14, 14073–14081 RSC.
- R. Li, Z. Wei, T. Huang and A. Yu, Electrochim. Acta, 2011, 56, 6860–6865 CrossRef CAS.
- L. Yao, Z. Geng, W. Zhang and X. Wu, et al. , ACS Sustainable Chem. Eng., 2020, 8, 17194–17200 CrossRef CAS.
- F. Okejiri, Z. Zhang, J. Liu, M. Liu and S. Yang, et al. , ChemSusChem, 2020, 13, 111–115 CrossRef CAS PubMed.
- R. Huang, K. Kim and H. J. Kim, et al. , ACS Appl. Nano Mater., 2019, 1, 486–495 Search PubMed.
- J. X. Liang, X. F. Yang, A. Wang and T. Zhang, et al. , Catal. Sci. Technol., 2016, 18, 6886–6892 RSC.
- L. Lv, Z. Li, Y. Ruan and Y. Chang, et al. , Electrochim. Acta, 2018, 286, 172–178 CrossRef CAS.
- P. Wang, N. W. Menzies, P. G. Dennis and J. Guo, et al. , Environ. Sci. Technol., 2016, 15, 8274–8281 CrossRef PubMed.
- Y. Cong, Q. Tang, X. Wang and M. Liu, et al. , ACS Catal., 2019, 9, 11743–11752 CrossRef CAS.
- Y. Ma, N. Wassdahl, P. Skytt and J. Guo, et al. , Phys. Rev. Lett., 1992, 17, 2598–2601 CrossRef PubMed.
- H. Wang, L. Wu, J. Jiao and J. Zhou, et al. , J. Mater. Chem. A, 2015, 3(12), 6517–6525 RSC.
- X. Wu, S. Xiong, J. Zhu, J. Wang, J. Shen and P. K. Chu, Nano Lett., 2009, 9, 4053–4060 CrossRef CAS PubMed.
- X. Li, X. Wang, J. Ding and M. Ma, et al. , ACS Catal., 2023, 13, 6338–6350 CrossRef CAS.
- H. Ha, S. Yoon, K. An and H. Y. Kim, ACS Catal., 2018, 12, 11491–11501 CrossRef.
- J. Zhang, L. Li, X. Huang and G. Li, J. Mater. Chem., 2012, 22, 10480–10487 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc06113b |
| This journal is © The Royal Society of Chemistry 2024 |