
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Remote control of anion binding by CH-based receptors†
Paulina
Jurek
 ,
Marek P.
Szymański
and
Agnieszka
Szumna
*
,
Marek P.
Szymański
and
Agnieszka
Szumna
*
Institute of Organic Chemistry Polish Academy of Sciences, Kasprzaka 44/52, Warsaw 01-224, Poland. E-mail: agnieszka.szumna@icho.edu.pl
First published on 4th March 2024
Abstract
We show that the substitution of tetra(benzimidazole)resorcin[4]arenes with electron withdrawing groups on the upper rim enhances anion binding at the opposite edge by more than three orders of magnitude. Moreover, selective anion binding at either the OH/NH or CH binding sites is demonstrated.
Organic and inorganic anions of various shapes and properties are inherently present in biotic and abiotic environments. Their selective detection and capture by anion receptors constitute an important subfield of supramolecular chemistry1 and find applications in extraction,2 sensing,3 catalysis,4 materials science,5 and drug discovery.6 Classical hydrogen-bond-forming anion receptors are based on NH or OH groups.7 Recently, there has been a burgeoning interest in CH-based anion receptors due to their resistance to deprotonation.8,9 Although the CH⋯anion hydrogen bond is weaker than the classical hydrogen bond,10 high-density multipoint interactions and polarisation effects significantly amplify the binding affinity of CH-based receptors for anions. Polarisation effects induced by electron withdrawing groups (EWGs) are typically observed for CH donors that constitute a part of the aromatic rings (e.g. 1,2,3-trifluorobenzenes or 1,2,3-triazoles)11 or acyclic systems (e.g. cyanostilbenes12 or nitrones13). In this paper, we demonstrate that (1) a CH donor can also be located at an π-electron-rich aromatic ring, and (2) its polarisation can originate from remote unconjugated positions.14 These discoveries significantly expand the design principles for constructing and modulating the properties of CH-based anion receptors.
Resorcin[4]arenes are π-electron-rich macrocycles known to interact with cationic guests.15 However, recently, we16,17 and others18 demonstrated that resorcin[4]arenes can also interact with anions in numerous different ways. Those possessing OH groups, e.g.1, bind anions via classical OH···anion hydrogen bonds (Fig. 1a).16 Those with OAlk groups, e.g.2 and 3, interact with anions at the lower rim via CH···anion hydrogen bonds (Fig. 1b and c).17 The interactions of 2 with anions are weak (Ka(Cl−) = 144 M−1 in THF), but, after substitution with EWG groups (–NO2), as for 3, the interactions with anions become remarkably efficient (Ka(Cl−) >105 M−1 in THF). Both observations were non-trivial because 2 and 3 belong to π-electron-rich rings, which have not been considered before as capable of CH···anion interactions.
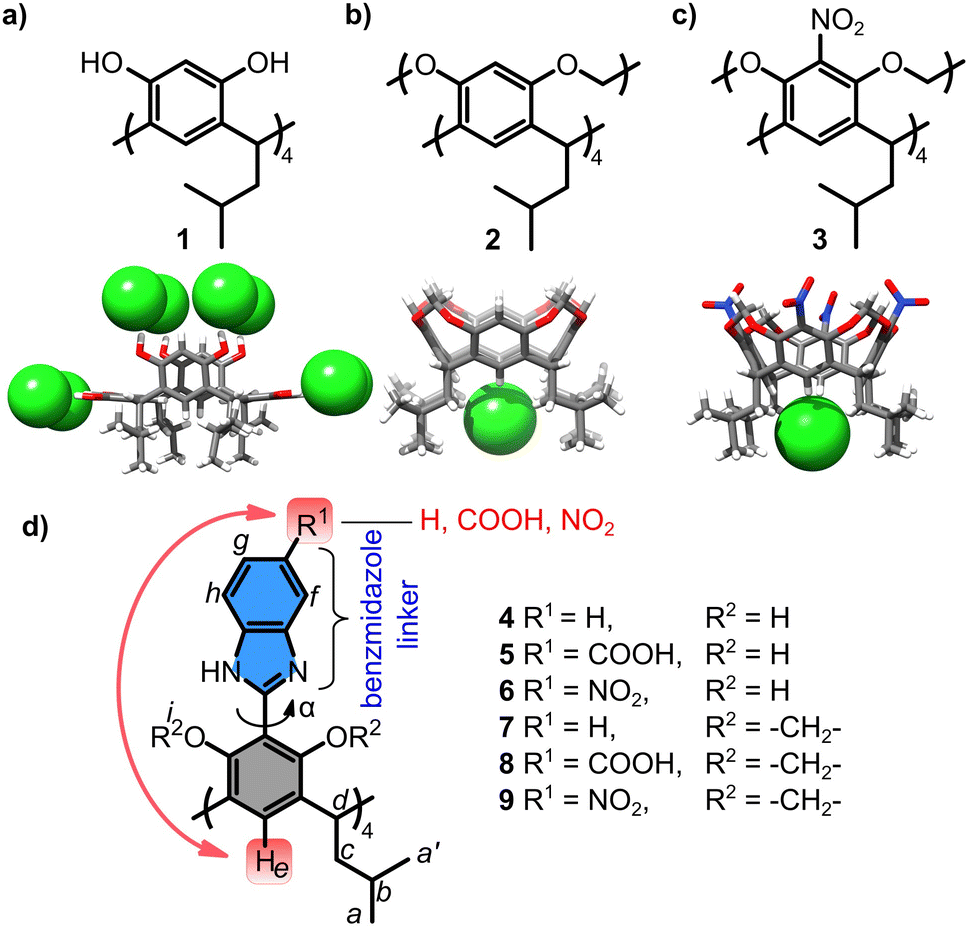 | ||
| Fig. 1 Anion binding by resorcin[4]arenes based on: (a) OH···anion interactions [ref. 16]; (b) and (c) CH···anion interactions [ref. 17]; (d) receptors studied in this work along with the notation of signals for NMR and definition of torsion α angle. | ||
In this work we evaluate the possibility of remote modulation of anion binding properties using a series of resorcin[4]arenes 4–9 (Fig. 1d). Their cores are substituted with linkers (benzimidazoles) and their properties are modulated by increasing the EWG character of the substituent at the linker (R1 = –H, –COOH, –NO2). It should be noted that the linker is attached to resorcinol rings via a single aryl–aryl bond; therefore, it is not conjugated and can adopt various dihedral angles between the aromatic units (α = 0–90°). Additional modulation comes from modifications of hydroxyl groups (R2 = H or –CH2−) that alter the electronic properties of the basic core, modulate the α angle, and can also form a competitive OH/NH-type binding site. Compounds 4, 5, 7, and 8 were synthesized previously for a different purpose.19 The synthesis of 6 and 9 is reported in the ESI.†
For the initial assessment of the properties, DFT calculations20 were performed for 3a–9a (analogues of 3–9 devoid of lower rim alkyl substituents, which have a negligible influence on the relevant parameters, Fig. S36, ESI†), 10 (monomer) and 11 (a derivative with nitrophenylene substituent). The results show a surprisingly large variation of the electrostatic surface potentials (ESPs, Fig. 2) at the lower rim depending on (a) remote substitution at the upper rim, and (b) substitution of OH groups. Derivatives 4a–6a, which contain free OH groups, form 12 intramolecular hydrogen bonds, which stabilize conformations with coplanar phenolic and benzimidazole rings (α = 1° ± 1°). The ESP potential at the lower rim becomes more positive with the increase of the electron-withdrawing character of the substituent at the upper rim (–H < –COOH < –NO2) and reaches 200 kJ mol−1 for 6a. For O-bridged derivatives 7a–9a, due to a lack of intramolecular hydrogen bonds, the benzimidazole and resorcinol rings are twisted (α = 35° ± 1°). Despite this twist, which reduces the resonance effects, the ESPs at the lower rim are also largely affected by the upper rim EWG substituents and reach even 221 kJ mol−1 for 9a. It should be noted that the ESPs reach higher values for O-bridged derivatives than for their analogues with free OH groups (e.g., compare 6a and 9a). Furthermore, the crucial role of the concentration of ESP in the apex area is clearly seen by the comparison of 9a with monomer 10 (87 kJ mol−1, α = 26°). In summary, theoretical calculations suggest that substituent effects transfer over large distances and even through a system of twisted biaryl connections. These remote modulations result in ESP values for 9a (221 kJ mol−1) that are only slightly lower than for resorcinarene 3a (247 kJ mol−1),17 in which the EWG substituent is directly attached to the core.
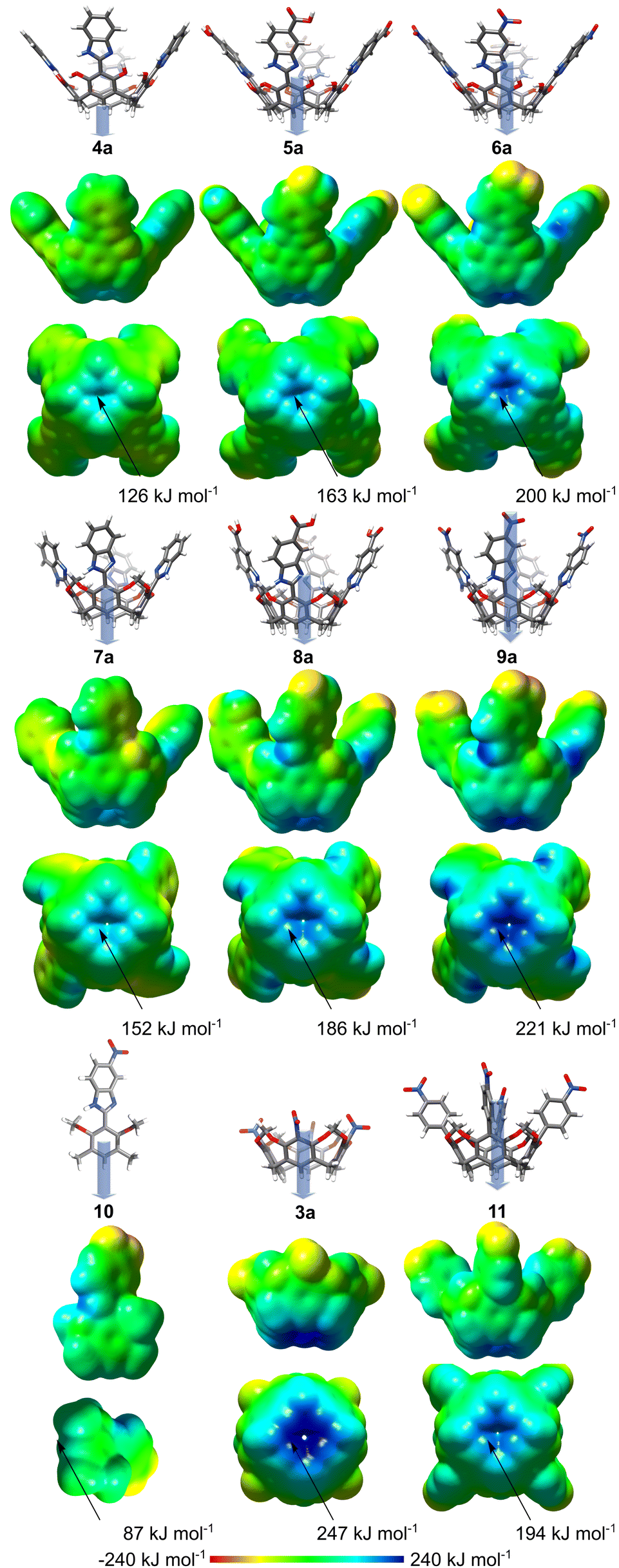 | ||
| Fig. 2 Theoretical calculations of molecular properties by DFT B3LYP 6-31+G(D,P) using the SMD solvent model (THF). ESPs are mapped on electron density isosurfaces (0.0004 e au−3), and relative dipole moment values (μ, D) are depicted as blue-grey arrows. | ||
To experimentally support the above hypotheses, anion binding properties were evaluated by 1H NMR titration of 4–6 with But4NBr in THF-d8 and 6, 8, and 9 in mixtures of THF-d8:DMSO-d6 (for solubility reasons). We were unable to obtain 7 of purity suitable for titrations (due to co-eluting partially substituted derivatives); therefore, 7 was excluded from the quantitative determination of Ka, but qualitative experiments show that it follows the trends observed for other derivatives.
During the 1H NMR titrations of 4–9 with But4NBr, the most pronounced changes were observed for the He signals (+0.8 ppm), and Hc (+0.5 ppm) indicating that anion binding occurs in the apex area of the lower rim (Fig. 3). The Ka(Br−) values increase in the order 4 < 5 < 6 < 8 < 9 (Table 1). The results show that the Br− binding affinity increases by two orders of magnitude (68 M−1 for 4 and 6000 M−1 for 6). A further increase in Br− binding affinity by more than an order of magnitude was observed upon O-bridging – Ka(9, Br−) in THF-d8:DMSO-d6, 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, v:v, is almost 30 times higher than Ka(6, Br−) in the same solvent. This trend agrees with the calculated ESPs and proves experimentally that the effect of remote EWG substitution on anion binding at the binding site is remarkable (reaching more than three orders of magnitude).
:10, v:v, is almost 30 times higher than Ka(6, Br−) in the same solvent. This trend agrees with the calculated ESPs and proves experimentally that the effect of remote EWG substitution on anion binding at the binding site is remarkable (reaching more than three orders of magnitude).
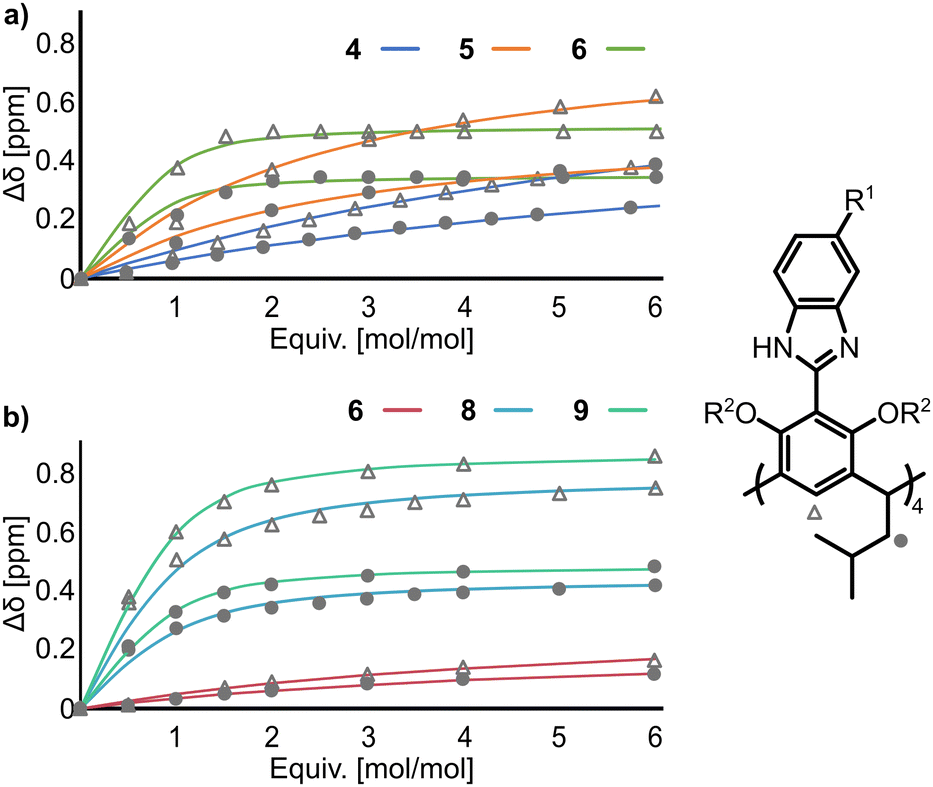 | ||
| Fig. 3
1H NMR titrations of receptors 4–9 (2 mM) with But4NBr (a) in THF-d8 and (b) in THF-d8:DMSO-d6 (9:1, v:v). | ||
:1 model, determined by 1H NMR titrations with But4NX)
| Receptor | Anion | THF | THF:DMSO 98:2, v:v |
THF:DMSO 90:10, v:v |
|---|---|---|---|---|
| a Determination not possible due to low solubility. b Titrations were not performed. | ||||
| 4 | Br− | 68 (±1%) | 34 (±1%) | <10 |
| 5 | Br− | 270 (±4%) | <10 | <10 |
| 6 | Br− | 6000 (±25%) | 800 (±24%) | 124 (±3%) |
| 8 | Br− | —a | —a | 1800 (±6%) |
| 9 | Br− | —a | 7000 (±14%) | 3400 (±6%) |
| 9 | Cl− | —a | 1900 (±11%) | 760 (±6%) |
| 9 | I− | —a | 2900 (±23%) | —b |
| 9 | ClO4− | —a | 450 (±2%) | —b |
The mechanism by which the effect of the EWG group is transferred over such a large distance and through twisted aryl–aryl bonds is not fully clear to us, and, most likely, many factors are correlated. Apparently, the resonance effect is not determining, because of the insensitivity of the effects to the aryl–aryl dihedral angle (compare 6a and 9a). Inductive effects and the polarisability seem to play a crucial role. This is illustrated by the comparison of the ESP values for 9a, which possesses long polarisable benzimidazole linkers (He⋯![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif) O2 11.1 Å), with 11, which possesses a shorter but less polarisable phenylene linker (He⋯O2 = 9.2 Å). As a result of these two factors, large dipole moments of the receptors are induced, which seem to be correlated with anion binding affinity.
O2 11.1 Å), with 11, which possesses a shorter but less polarisable phenylene linker (He⋯O2 = 9.2 Å). As a result of these two factors, large dipole moments of the receptors are induced, which seem to be correlated with anion binding affinity.
The selectivity of the receptors is determined by several factors, including spatial fit and the strength of the interactions. Receptor 9 favours Br− over Cl− by a factor of 3.7 (Table 1), despite Cl− forming stronger hydrogen bonds, indicating that the binding site may have geometric preference for Br−. Unprecedented site-selective binding is demonstrated for receptor 6, which possesses free OH groups. The comparison of the titrations of 6 with But4NCl and But4NBr in THF-d8 (Fig. 4) suggests that receptor 6 binds to Br− on the lower rim while Cl− on the upper rim. Thus, during Br− binding, the He signal is downfield shifted, indicating binding in the lower rim (Fig. 4b and d). On the contrary, during Cl− binding all signals of 6 substantially broaden and, after lowering of the temperature, substantial downfield shifts of its OH/NH signals are observed (Fig. 4c and Fig. S34, ESI†). The Hd signal splits into two, of which only one shifts substantially downfield and the changes saturate after 2 equiv. of Cl− are added (Fig. S33, ESI†). This is in agreement with the binding of two Cl− in the upper rim, between the arms in the opposite corners of the macrocycle, using OH/NH···Cl− hydrogen bonds (Fig. 4e). The geometry-optimized structure of 6a·(Cl−)2 has C2v symmetry, which is in agreement with the observed doubling of the signals, downfield shift of eight out of 12 NH/OH signals and two of the four Hd signals. The hypothetical geometry-optimized structure 6a·(Br−)2 has less favourable geometry (e.g. less hydrogen bonds, ESI†). This site-selective Cl−/Br− binding can result from geometrical reasons. It can also originate from the fact that Cl− is a ‘hard’ anion and therefore prefers a ‘hard’ binding site, whereas Br− has a more diffuse charge and therefore a higher affinity toward a more surface-diffused CH-based binding site.
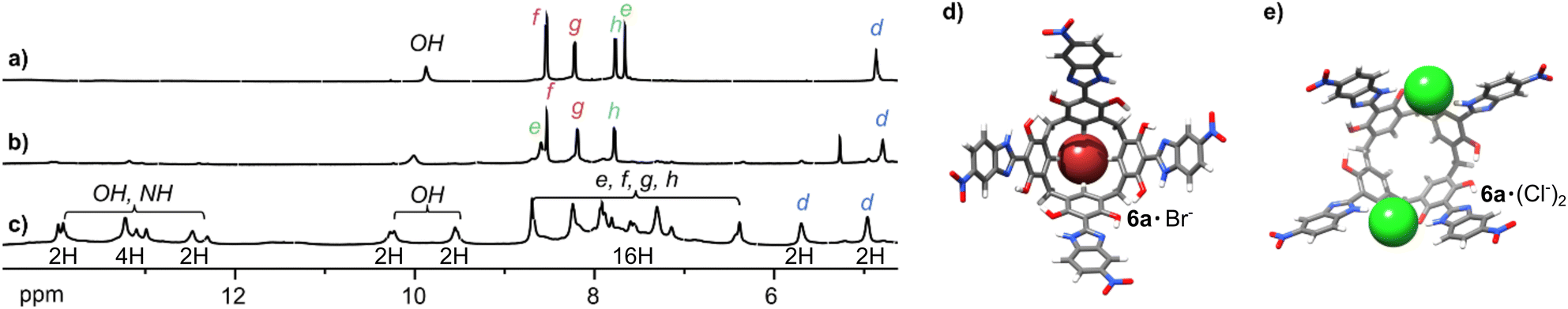 | ||
| Fig. 4 Anion binding modes for receptor 6. 1H NMR spectra of: (a) 6; (b) 6 + But4NBr; (c) 6 + But4NCl (2 mM each, THF-d8 at 233 K). Suggested structures of: (d) 6a·Br−; (e) 6a·(Cl−)2. | ||
In summary, we have introduced novel macrocyclic anion receptors derived from benzimidazole functionalized resorcin[4]arenes. Our findings demonstrate that substituting an electron-withdrawing group at the upper rim can remarkably enhance anion binding at remote lower rim positions by more than three orders of magnitude. Specifically, Br− is bound at the lower rim apex of the cone through CH⋯anion hydrogen bonds. The cone geometry plays a pivotal role in generating a positive electrostatic potential and a large vertical dipole moment for the receptor. Additionally, we have unveiled an unexpected site-selective differentiation in the binding of Cl− and Br−. The Cl− anions exhibit a preference for OH/NH-based binding sites, whereas the Br− anions favour the CH-based binding site within the same receptor.
This work was supported by the National Science Centre, Poland (OPUS 2021/41/B/ST4/01650). The calculations were performed at the Wroclaw Centre for Networking and Supercomputing (grant no. 299).
Conflicts of interest
There are no conflicts to declare.Notes and references
- P. Molina, F. Zapata and A. Caballero, Chem. Rev., 2017, 117, 9907 CrossRef CAS PubMed; L. Chen, S. N. Berry, X. Wu, E. N. W. Howe and P. A. Gale, Chemistry, 2020, 6, 61 CrossRef; N. H. Evans and P. D. Beer, Angew. Chem., Int. Ed., 2014, 53, 11716 CrossRef PubMed; L. K. Macreadie, A. M. Gilchrist, D. A. McNaughton, W. G. Ryder, M. Fares and P. A. Gale, Chemistry, 2022, 8, 46 CrossRef; J. Zhao, D. Yang, X.-J. Yang and B. Wu, Coord. Chem. Rev., 2019, 378, 415 CrossRef.
- W. Liu, A. G. Oliver and B. D. Smith, J. Org. Chem., 2019, 84, 4050 CrossRef CAS PubMed; S.-Q. Chen, W. Zhao and B. Wu, Front. Chem., 2022, 10, 905563 CrossRef PubMed; S. K. Kim, J. Lee, N. J. Williams, V. M. Lynch, B. P. Hay, B. A. Moyer and J. L. Sessler, J. Am. Chem. Soc., 2014, 136, 15079 CrossRef PubMed; K. Gloe, H. Stephan and M. Grotjahn, Chem. Eng. Technol., 2003, 26, 1107 CrossRef.
- R. M. Duke, E. B. Veale, F. M. Pfeffer, P. E. Kruger and T. Gunnlaugsson, Chem. Soc. Rev., 2010, 39, 3936 RSC; N. Busschaert, C. Caltagirone, W. V. Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038 CrossRef CAS PubMed.
- D. D. Ford, D. Lehnherr, C. R. Kennedy and E. N. Jacobsen, ACS Catal., 2016, 6, 4616 CrossRef CAS PubMed; A. Kerckhoffs, I. Moss and M. J. Langton, Chem. Commun., 2023, 59, 51 RSC; M. Li, Z. Zhang, Y. Yan, W. Lv, Z. Li, X. Wang and Y. Tao, Nat. Synth., 2022, 1, 815 CrossRef; G. Hirata and H. Maeda, Org. Lett., 2018, 20, 2853 CrossRef PubMed.
- W. Zhao, J. Tropp, B. Qiao, M. Pink, J. D. Azoulay and A. H. Flood, J. Am. Chem. Soc., 2020, 142, 2579 CrossRef CAS PubMed; N. G. White and M. J. MacLachlan, Chem. Sci., 2015, 6, 6245 RSC.
- N. Busschaert and P. A. Gale, Angew. Chem., Int. Ed., 2013, 52, 1374 CrossRef CAS PubMed; J. de Jong, J. E. Bos and S. J. Wezenberg, Chem. Rev., 2023, 123, 8530 CrossRef PubMed; L. Martínez-Crespo and H. Valkenier, ChemPlusChem., 2022, 87, e202200266 CrossRef PubMed; H. Li, H. Valkenier, A. G. Thorne, C. M. Dias, J. A. Cooper, M. Kieffer, N. Busschaert, P. A. Gale, D. N. Sheppard and A. P. Davis, Chem. Sci., 2019, 10, 9663 RSC.
- G. I. Vargas-Zúñiga and J. L. Sessler, Coord. Chem. Rev., 2017, 345, 281 CrossRef CAS PubMed; J. Gomez-Vega, J. M. Soto-Cruz, O. Juárez-Sánchez, H. Santacruz-Ortega, J. C. Gálvez-Ruiz, D. O. Corona-Martínez, R. Pérez-González and K. O. Lara, ACS Omega, 2022, 7, 22244 CrossRef PubMed; S. J. Wezenberg and B. L. Feringa, Org. Lett., 2017, 19, 324 CrossRef PubMed; S. Kundu, T. K. Egboluche and M. A. Hossain, Acc. Chem. Res., 2023, 56, 1320 CrossRef PubMed; P. R. Brotherhood and A. P. Davis, Chem. Soc. Rev., 2010, 39, 3633 RSC; F. M. Pfeffer, K. F. Lima and K. J. Sedgwicka, Org. Biomol. Chem., 2007, 5, 1795 RSC; P. A. Gale, Chem. Commun., 2008, 4525 RSC; P. A. Gale, J. R. Hiscock, N. Lalaoui, M. E. Light, N. J. Wellsa and M. Wenzela, Org. Biomol. Chem., 2012, 10, 5909 RSC; M. J. Chmielewski, M. Charon and J. Jurczak, Org. Lett., 2004, 6, 3501 CrossRef; K. M. Bąk, K. Chabuda, H. Montes, R. Quesada and M. J. Chmielewski, Org. Biomol. Chem., 2018, 16, 5188 RSC; L. S. Yakimova, D. N. Shurpika and I. I. Stoikov, Chem. Commun., 2016, 52, 12462 RSC; S. O. Kang, R. A. Begum and K. Bowman-James, Angew. Chem., Int. Ed., 2006, 45, 7882 CrossRef; M. J. Chmielewski, A. Szumna and J. Jurczak, Tetrahedron Lett., 2004, 45, 8699 CrossRef; S. A. Boer, E. M. Foyle, C. M. Thomas and N. G. White, Chem. Soc. Rev., 2019, 48, 2596 RSC.
- M. J. Langton, C. J. Serpell and P. D. Beer, Angew. Chem., Int. Ed., 2016, 55, 1974 CrossRef CAS; L. M. Eytel, H. A. Fargher, M. M. Haley and D. W. Johnson, Chem. Commun., 2019, 55, 5195 RSC; J. Cai and J. L. Sessler, Chem. Soc. Rev., 2014, 43, 6198 RSC; M. A. Majewski, Y. Hong, T. Lis, J. Gregoliński, P. J. Chmielewski, J. Cybińska, D. Kim and M. Stępień, Angew. Chem., Int. Ed., 2016, 55, 14072 CrossRef; H. Zhu, B. Shi, K. Chen, P. Wei, D. Xia, J. H. Mondal and F. Huang, Org. Lett., 2016, 18, 5054 CrossRef; H. Valkenier, O. Akrawi, P. Jurček, K. Sleziaková, T. Lízal, K. Bartik and V. Šindelář, Chemistry, 2019, 5, 429 CrossRef; C. Rando, J. Vázquez, J. Sokolov, Z. Kokan, M. Nečas and V. Šindelář, Angew. Chem., Int. Ed., 2022, 61, e202210184 Search PubMed; S. Kaabel, J. Adamson, F. Topić, A. Kiesilä, E. Kalenius, M. Öeren, M. Reimund, E. Prigorchenko, A. Lõokene, H. J. Reich, K. Rissanen and R. Aav, Chem. Sci., 2017, 8, 2184 RSC.
- D. Mondal, M. Ahmad, P. Panwaria, A. Upadhyay and P. Talukdar, J. Org. Chem., 2022, 87, 10 CrossRef CAS.
- V. S. Bryantsev and B. P. Hay, J. Am. Chem. Soc., 2005, 127, 82823 Search PubMed.
- I. Bandyopadhyay, K. Raghavachari and A. H. Flood, Chem. Phys. Chem., 2009, 10, 2535 CrossRef CAS; V. Haridas, S. Sahu, P. P. Praveen Kumar and A. R. Sapala, RSC Adv., 2012, 2, 12594 RSC; Y. Liu, W. Zhao, C. H. Chen and A. H. Flood, Science, 2019, 365, 159 CrossRef; S. Mirzaei, V. M. Espinoza Castro and R. Hernandez Sanchez, Chem. Sci., 2022, 13, 2026 RSC.
- S. Lee, C.-H. Chen and A. H. Flood, Nat. Chem., 2013, 5, 704 CrossRef CAS.
- X. He, R. R. Thompson, S. A. Clawson, F. R. Fronczek and S. Lee, Chem. Commun., 2023, 59, 4624 RSC.
- When this paper was under review, a paper showing an influence of remote inductive effects on anion binding was released, see: M. Chvojka, D. Madea, H. Valkenier and V. Šindelář, Angew. Chem., Int. Ed., 2023, e202318261 Search PubMed.
- N. K. Beyeh, A. Valkonen and K. Rissanen, Supramol. Chem., 2009, 21, 142 CrossRef CAS; N. K. Beyeh and K. Rissanen, Isr. J. Chem., 2011, 51, 769 CrossRef; H. Mansikkamäki, M. Nissinen and K. Rissanen, Chem. Commun., 2002, 1902 RSC; H. Mansikkamäki, M. Nissinen, C. A. Schalley and K. Rissanen, New J. Chem., 2003, 27, 88 RSC; N. K. Beyeh, D. P. Weimann, L. Kaufmann, C. A. Schalley and K. Rissanen, Chem. – Eur. J., 2012, 18, 5552 CrossRef; N. K. Beyeh, M. Göth, L. Kaufmann, C. A. Schalley and K. Rissanen, Eur. J. Org. Chem., 2014, 80 CrossRef.
- M. Chwastek, P. Cmoch and A. Szumna, Angew. Chem., Int. Ed., 2021, 60, 4540 CrossRef CAS; M. Chwastek, P. Cmoch and A. Szumna, J. Am. Chem. Soc., 2022, 144, 5350 CrossRef; E. R. Abdurakhmanova, P. Cmoch and A. Szumna, Org. Biomol. Chem., 2022, 20, 5095 RSC.
- E. R. Abdurakhmanova, D. Mondal, H. Jędrzejewska, P. Cmoch, O. Danylyuk, M. J. Chmielewski and A. Szumna, ChemRxiv, 2023, preprint DOI:10.26434/chemrxiv-2023-0f7tx.
- S. S. Zhu, H. Staats, K. Brandhorst, J. Grunenberg, F. Gruppi, E. Dalcanale, A. Lützen, K. Rissanen and C. A. Schalley, Angew. Chem., Int. Ed., 2008, 47, 788 CrossRef CAS.
- P. Jurek, H. Jędrzejewska, M. F. Rode and A. Szumna, Chem. – Eur. J., 2023, 29, e2022031 CrossRef CAS; P. Jurek, M. F. Rode, M. P. Szymański, M. Banasiewicz and A. Szumna, J. Mater. Chem. C, 2023, 11, 10642 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 (Revision C.01), Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR spectra, details of titrations and theoretical calculations, and coordinates of calculated structures. See DOI: https://doi.org/10.1039/d3cc06038a |
| This journal is © The Royal Society of Chemistry 2024 |