
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultiplicity-driven photochromism controls three-state fulgimide photoswitches†
Jakub
Copko
 and
Tomáš
Slanina
*
and
Tomáš
Slanina
*
Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo náměstí 542/2, Prague 6, 160 00, Czech Republic. E-mail: tomas.slanina@uochb.cas.cz
First published on 13th February 2024
Abstract
Fulgimide photoswitches display three-state photoswitching between isomeric forms Z, E and C. Fulgimides have therefore access to both large steric change of double bond isomerization and the large spectral change induced by electrocyclization. By controlling the multiplicity and photoisomerization conditions, we achieved precise and near-quantitative control over both isomerization modes.
Photochromic organic motifs are increasingly found in various applications,1 including optically active smart materials,2 catalysis,3 and photopharmacology.4 However, these applications are often restricted by properties of the materials, such as limited thermal and photostability, low quantum yields, and low conversions between isomers.5 Among the photochromic organic motifs that effectively address these challenges, fulgides stand out for their excellent photophysical properties and high thermal and photostability.6 Yet, despite outperforming commonly used photochromic motifs, including azobenzenes and stilbenes, in terms of PSS ratios,7 separation of absorption maxima,8 quantum yields,5 and thermal stability,5,9 their development have been recently stagnating. That could most likely be attributed to their (i) limited synthetic availability, (ii) complex three-state photoswitching (E, Z and C) and (iii) small geometrical change between E and C.10–12
The three-state photoswitching of fulgides derives from the dual photoreactivity of 1,3,5-hexatriene moiety in E form (Scheme 1).10 Upon irradiation, E undergoes conrotatory 6π-electrocyclization, yielding the closed form (C). Photoinduced fulgide cyclization is accompanied by a planarization of the central ring. This planarization markedly changes π-electron conjugation, bathochromically shifting absorption maxima, typically by 150 nm.10 Concomitantly, light can also trigger E–Z isomerization of the central double bond, affording an non-cyclizable form (Z) and vastly altering the steric demands of the molecule. E–Z isomerization was historically perceived as an unwanted side process.10 This has been consistently suppressed by introducing a bulky substituent R1, thereby limiting the photoreactivity of fulgides to their photoinduced cyclization.13
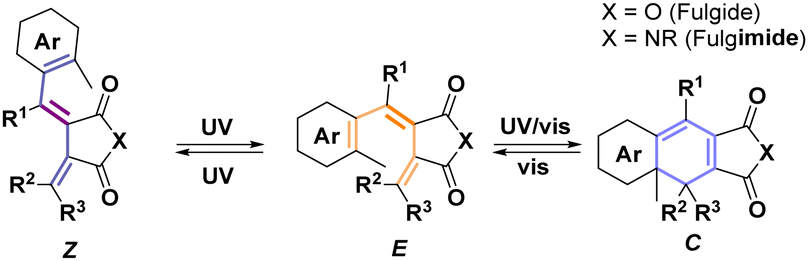 | ||
| Scheme 1 General structures and photoisomerization of fulgide. | ||
Although hindering photoisomerization simplifies the complex three-state photoswitching to a single process, it does so at the expense of the utility of fulgides. Rather than blocking, precisely controlling the dual isomerization may broaden fulgide's applications because a single monochromophoric unit may enable us to choose between both the color change induced by electrocyclization and the large steric change induced by double bond isomerization. This controlled dual isomerization could expand the utility of photochromic materials as the best strategy to access both of these advantageous properties currently requires either mixing, covalently linking two different photoswitches, forming multichromophoric systems, or combining monochromophoric system with chemical transformation.14–16 A solution for controlling dual monochromophoric photoswitching might be offered by multiplicity-based photochemistry. As shown recently, commonly used azobenzenes can undergo isomerization through triplet sensitization.17 Moreover, azobenzene photoswitching does not strictly proceed from the singlet excited state, but instead may be a multistate process, involving both singlet and triplet excited states.18 In fulgides, double bond isomerization mirrors that of azobenzenes and may also be triggered via the triplet excited state. However, the ultra-fast singlet excited state relaxation of fulgides prevents any intersystem crossing to triplet state, which can only be accessed by sensitization.
Thus, together with a properly designed molecule, excited-state multiplicity could help to control the outcome of photoinduced isomerization. Herein, we aim at tackling the key problems of fulgides. First, we developed a one-pot procedure to prepare several fulgimide photoswitches. Second, we precisely and near-quantitively tamed photoswitching between all three isomers by controlling excited-state multiplicity, thereby switching between photochromic motifs (E–Z isomerization and electrocyclization). As a result, we were able to control the color change induced by electrocyclization and the large geometrical change induced by double bond isomerization through a rational selection of functional groups. For this purpose, we chose indole as the aryl unit, trifluoromethyl as R1 (Scheme 1) and geminal cyclopropyls as R2 and R3 to enhance chemical stability and fatigue resistance and to ensure full operability in the visible spectrum.9,19,20 In addition to simple irradiation, we combined dual wavelength irradiation with triplet sensitization. By doing so, we precisely controlled the outcome of photoisomerization, and exploited orthogonal photochromism of monochromophoric fulgides.
To overcome problems associated with classical fulgide synthesis, we developed an alternative method. The anhydride function of fulgides decreases their thermal stability and requires purification. So, we prepared the target fulgimides avoiding the anhydride function by base-promoted cyclization of amide ester precursors rather than dehydrating the amide acid precursors (Scheme 2B).
 | ||
| Scheme 2 (A) Revised one-pot procedure towards 1a–h (B) classical approach (C) fulgimides 1a–h (D) on left absorption profiles of isomers of 1b (Z – purple, E – orange, C – blue); on right relative abundance of all 1d isomers during activation, 10 cycles of opening and closing, and deactivation. | ||
Cyclic lactone 2 underwent base-promoted ring opening, affording intermediate i1. The amide coupling reagent hexafluorophosphate azabenzotriazole tetramethyl uronium (HATU) facilitated conversion into precursor i2 for the final cyclization, which was eventually triggered using sodium hydride as a strong base (Scheme 2A). Optimized steps were then applied in a sequential, one-pot reaction, yielding the target imide 1 and a non-dehydrated acid amide by-product (i3). Compound i3 was then easily transformed to the target fulgimide by further HATU addition. Because this approach does not require purifying intermediates, the reaction is shortened from days to typically 4–6 hours and has a fourfold higher yield than the classical method (increasing from 15 to 60% for 1a, calculated from lactone 2). Given the stereochemistry of the elimination of lactone 2, this alternative approach leads solely to the Z isomer of fulgimide (Scheme S1, ESI†), which is inactive towards photoinduced electrocyclization. Previously, repetitive recrystallizations have been used to thermally trigger double bond isomerization, affording the active E isomer.20
Using our sequential, one-pot method, we prepared several new fulgimides, 1a–f, bearing an aliphatic amine (Scheme 2C). To broaden the scope of this method, protected glycine was introduced in 1g, showing potential for further functionalization with other amino acids. However, attaching aniline-like amines proved difficult because they are not as nucleophilic as their aliphatic counterparts. Only activated p-anisidine yielded 1h, albeit requiring harsher conditions. Most fulgimides had a functional group, enabling further modifications, such as nitrogen quaternization (1b, 1c), copper-catalyzed azide–alkyne cycloaddition (1d, 1e), and cross-coupling chemistry (1f), as shown by quantitatively coupling fulgimide 1d to benzyl azide with copper-mediated click chemistry (Scheme S3, ESI†).
Fulgide photoswitches exist in three isomeric forms – two open isomers (historically denoted E and Z) and a closed isomer (C) (Scheme 1). Because of its geometry, Z cannot undergo photoinduced ring closing and is, therefore, denoted as an inactive form. Upon double bond isomerization in inactive form Z, the resulting active form E readily undergoes photoinduced electrocyclization, affording the closed form C of the fulgimide. Photoinduced isomerization between active and closed forms is accompanied by a distinctive color change.6,10 As such, photoswitching between E and C has been historically regarded as the key mode of action and double bond isomerization as an unwanted side reaction. But double bond isomerization, accompanied by a large structural change, may act as a lock, blocking photocyclization between E and C. By controlling these processes – initial Z to E double bond isomerization (activation), closing and opening between E and C and double bond isomerization back to Z (deactivation) – we can selectively control switching between all three fulgimide isomers (Scheme 3).
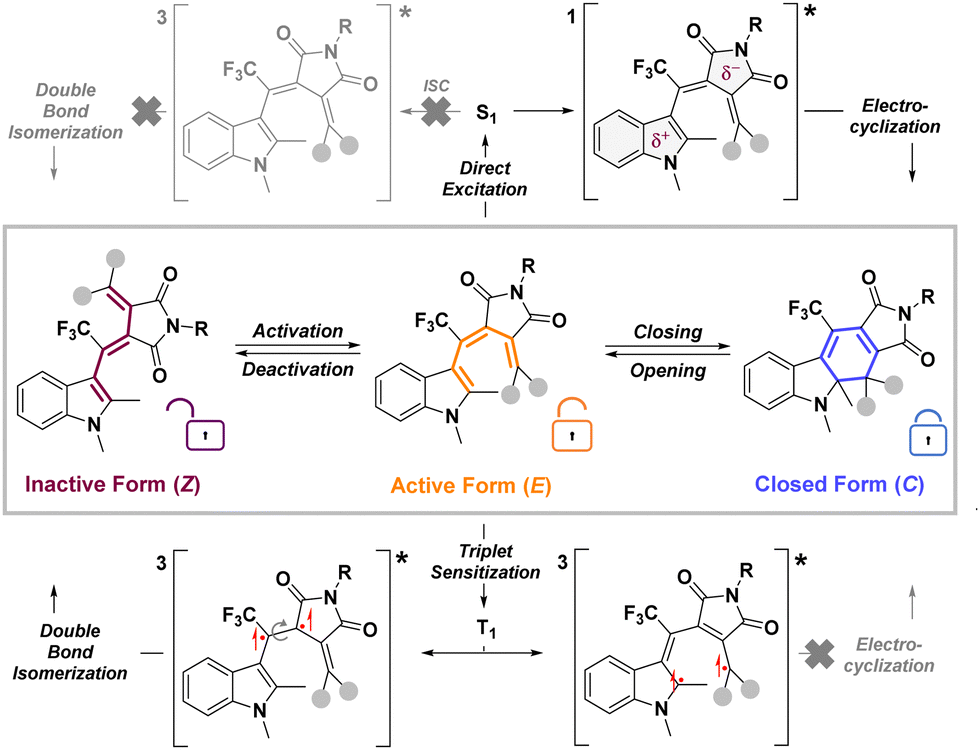 | ||
| Scheme 3 Photoinduced fulgimide isomerization and outcome of E isomerization based on excited-state multiplicity. | ||
Since our one-pot synthetic procedure led only to Z, closing was not triggered by daylight, conveniently enabling further modifications. However, the fulgide core required activation to facilitate closing and opening. Because thermal activation from Z to E took approximately 3 days at 100 °C in DMSO, activation was accelerated by purple light irradiation (0.2–0.6% quantum yields at 400 nm depending on R, Table 1). However, the absorptions of Z and E overlapped, so the incident light also triggered ring closing, resulting in a mixture of active E and closed C forms with less than 5% of Z (Table S2A, ESI†).
| R = | Absorption maxima/nm (ε/M−1 cm−1) | Ratio of isomers (Z![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :E:C) :E:C) |
Quantum yields/%a | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Z | E | C | PSS(420nm) | PSS(626nm) | PSS(deactiv.) | Φ 1 | Φ 2 | Φ 3 | ||
| a Φ 1 = Z–E, 400 nm, activation. Φ2 = E–C, 400 nm, closing. Φ3 = C–E, 626 nm, opening. | ||||||||||
| 1a |

|
397 (1800) | 414 (2700) | 550 (3750) | 3:17:80 |
3:97:0 |
89:11:0 |
0.3 | 9 | 49 |
| 1b |

|
400 (2500) | 416 (3900) | 554 (4600) | <1:13:87 |
<1:100:0 |
89:11:0 |
0.3 | 8 | 44 |
| 1c |

|
398 (2200) | 413 (3450) | 552 (4400) | <1:13:87 |
<1:100:0 |
97:3:0 |
0.4 | 9 | 49 |
| 1d |

|
406 (1700) | 418 (2200) | 561 (2700) | 4:15:81 |
4:96:0 |
90:10:0 |
0.3 | 12 | 46 |
| 1e |

|
402 (2400) | 415 (3600) | 555 (5000) | 5:19:76 |
5:95:0 |
88:12:0 |
0.5 | 9 | 53 |
| 1f |

|
405 (2200) | 418 (3300) | 560 (4300) | 3:14:83 |
3:97:0 |
90:10:0 |
0.6 | 10 | 60 |
| 1g |

|
409 (3350) | 420 (4300) | 561 (5650) | 4:18:78 |
4:96:0 |
81:19:0 |
0.2 | 5 | 31 |
| 1h |

|
403 (2000) | 419 (3400) | 559 (4650) | 4:15:81 |
4:96:0 |
39:61:0 |
0.3 | 8 | 46 |
Nevertheless, the closed form was easily removed from the mixture by irradiation with red light (626 nm). Upon activation with dual wavelength irradiation (purple + red light), the resulting closed C isomer was immediately switched to the active E form (Table S2B, ESI†). In all cases, fulgimide activation bathochromically shifts the maxima and increases the extinction coefficient by approximately 50% (Scheme 2D).
The primary photochromic mode of action of fulgides, ring closing and opening, readily occurs from the singlet state of the activated system.21 As for ring closure, two new absorption bands of C with maximum around 350 and 550 nm (Scheme 2D) emerged after exposing E to purple light (5–12% quantum yields at 400 nm depending on R, Table 1 and Table S2C, ESI†). Under 626 nm irradiation, C opens with high quantum yields ranging from 31 to 60% (vs. 4–7% of fulgimides bearing methyl substitution at R2 and R3).22 This marked increase in quantum yield may be explained by the high strain of the closed form introduced by geminal cyclopropyl groups.13 Throughout the photoswitching cycles, excellent photostationary states were attained at 420 nm, with up to 87% of the closed isomer. Moreover, no thermal opening occurred under ambient conditions without light exposure. Irradiation with red light quantitatively opened the closed form, leaving mainly the active E form (with less than 5% Z, Table S2D, ESI†). Multiple cycles of photoswitching can be performed without photobleaching (Scheme 2D). This demonstrates that introduction of imide does not affect the excellent photostability of fulgimide, as previously demonstrated on other fulgimide derivatives.23
Accessing the second photochromic mode of fulgides requires facilitating double bond isomerization of E back to Z. This isomerization involves a large structural change, markedly altering the fulgide steric demands without significantly affecting the color. The E–Z isomerization together with electrocyclization gives fulgides option to undergo a large structural change or a vast absorption profile shift, depending on the isomerization conditions. This deactivation cannot be induced thermally, because E is thermally more stable than Z, or by irradiation of E, which triggers rapid cyclization to C. Nevertheless, we hypothesized that opting for the triplet excited state could solve this problem. Judging from phosphorescence measurements of a similar fulgide,24 the triplet state energy of Z should be approximately 3 kcal mol−1 lower than that of E. Accordingly, under adiabatic conditions, relaxation from the triplet state of E may yield Z.25 As direct fulgide excitation leads only to the singlet state, which decays on ultrafast scales (τ ∼ 0.4 ps),21 the triplet state is only accessible by sensitization. Anthracene was selected as a sensitizer because its triplet-state energy should be higher than that of fulgimides.26 Considering its biradical character, the triplet state may permit double bond isomerization while precluding electrocyclization as the two unpaired spins cannot recombine to form a new sigma bond (Scheme 3). Anthracene was added to the solution of either E or C, and the mixture was degassed to prevent oxygen from quenching triplet states. The solution was then simultaneously exposed to 385- and 626-nm irradiation. The first wavelength directly excited anthracene, which rapidly underwent intersystem crossing into the triplet state and transferred its energy to fulgimide. Simultaneously, the same wavelength excited the active form of fulgimide (E), yielding the closed form (C). Furthermore, in its singlet excited state, anthracene may transfer its singlet state energy to fulgimide via Förster resonance energy transfer (FRET), again triggering closing (Fig. S1 for anthracene fluorescence quenching, ESI†). Therefore, additional irradiation with 626-nm light was used to quantitatively open the closed form. Under these conditions, a photo-equilibrated state was reached, primarily consisting of Z, except for 1h, where the larger delocalization of π-electrons might have facilitated different relaxation decays of the triplet state (Table 1). Energy transfer from the triplet state of anthracene was confirmed by its quenching by 1b (kq = (1.37 ± 0.18) × 108 M−1 s−1), as shown by transient absorption spectroscopy (for detail, see ESI† – transient measurements). Oxygen played a key role in controlling the outcome of photoisomerization. In its absence, sensitization of E efficiently generated the triplet excited state of fulgimide, resulting in deactivation to Z. Conversely, in the presence of oxygen, any triplet excited state of the sensitizer was rapidly quenched. Hence, aeration completely blocked deactivation, while fulgides could still undergo photochemical activation, closing and opening, even in the presence of sensitizer. Alternatively, anthracene could be easily disposed by filtration through silica.
Our results show that combining simple and dual irradiation with triplet sensitization enables us to precisely control both photochromic modes with large geometrical and spectral changes (E–Z isomerization and electrocyclization) in almost quantitative photoisomerization yields. This monochromophoric system can undergo two photochromic pathways and be prepared in a short, high-yielding, one-pot reaction, making it possible to easily introduce various substituents. The unique three-state photoswitching, monochromophoric system may reignite interest in developing and applying fulgide photoswitches as photoresponsive compounds prompting advances in materials science and programmable photochemistry.
The authors acknowledge the MEYS CR (project No. LTAIN19166) for funding, Lucie Ludvíková for the triplet quenching experiment and Carlos V. Melo for editing the manuscript.
Conflicts of interest
There are no conflicts to declare.References
- M.-M. Russew and S. Hecht, Adv. Mater., 2010, 22, 3348–3360 CrossRef CAS PubMed.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- R. Göstl, A. Senf and S. Hecht, Chem. Soc. Rev., 2014, 43, 1982–1996 RSC.
- M. J. Fuchter, J. Med. Chem., 2020, 63, 11436–11447 CrossRef CAS PubMed.
- B. L. Feringa, W. F. Jager and B. de Lange, Tetrahedron, 1993, 49, 8267–8310 CrossRef CAS.
- H. G. Heller, K. Koh, C. Elliot and J. Whittall, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 1994, 246, 79–86 CrossRef CAS.
- M. A. Wolak, N. B. Gillespie, R. R. Birge and W. J. Lees, Chem. Commun., 2003, 992–993 RSC.
- M. Reinfelds, V. Hermanns, T. Halbritter, J. Wachtveitl, M. Braun, T. Slanina and A. Heckel, ChemPhotoChem, 2019, 3, 441–449 CrossRef CAS.
- Y. Yokoyama and K. Takahashi, Chem. Lett., 1996, 1037–1038 CrossRef CAS.
- Y. Yokoyama, Chem. Rev., 2000, 100, 1717–1740 CrossRef CAS PubMed.
- C. J. Thomas, M. A. Wolak, R. R. Birge and W. J. Lees, J. Org. Chem., 2001, 66, 1914–1918 CrossRef CAS PubMed.
- J. Harada, M. Taira and K. Ogawa, Cryst. Growth Des., 2017, 17, 2682–2687 CrossRef CAS.
- Y. Yokoyama, T. Inoue, M. Yokoyama, T. Goto, T. Iwai, N. Kera, I. Hitomi and Y. Kurita, Bull. Chem. Soc. Jpn., 1994, 67, 3297–3303 CrossRef CAS.
- M. M. Lerch, M. J. Hansen, W. A. Velema, W. Szymanski and B. L. Feringa, Nat. Commun., 2016, 7, 12054 CrossRef CAS PubMed.
- A. Fihey, A. Perrier, W. R. Browne and D. Jacquemin, Chem. Soc. Rev., 2015, 44, 3719–3759 RSC.
- S. Jia, H. Ye, P. He, X. Lin and L. You, Nat. Commun., 2023, 14, 7139 CrossRef CAS PubMed.
- P. Bharmoria, S. Ghasemi, F. Edhborg, R. Losantos, Z. Wang, A. Mårtensson, M. Morikawa, N. Kimizuka, Ü. İşci, F. Dumoulin, B. Albinsson and K. Moth-Poulsen, Chem. Sci., 2022, 13, 11904–11911 RSC.
- M. Reimann, E. Teichmann, S. Hecht and M. Kaupp, J. Phys. Chem. Lett., 2022, 13, 10882–10888 CrossRef CAS PubMed.
- A. Kaneko, A. Tomoda, M. Ishizuka, H. Suzuki and R. Matsushima, Bull. Chem. Soc. Jpn., 1988, 61, 3569–3573 CrossRef CAS.
- N. I. Islamova, X. Chen, S. P. Garcia, G. Guez, Y. Silva and W. J. Lees, J. Photochem. Photobiol. Chem., 2008, 195, 228–234 CrossRef CAS.
- H. Port, P. Gärtner, M. Hennrich, I. Ramsteiner and T. Schöck, Mol. Cryst. Liq. Cryst., 2005, 430, 15–21 CrossRef CAS.
- M. A. Wolak, C. J. Thomas, N. B. Gillespie, R. R. Birge and W. J. Lees, J. Org. Chem., 2003, 68, 319–326 CrossRef CAS PubMed.
- R. Matsushima and H. Sakaguchi, J. Photochem. Photobiol. Chem., 1997, 108, 239–245 CrossRef CAS.
- A. V. Metelitsa, M. I. Knyazhanskii, N. V. Volbushko, I. Y. Grishin, Y. M. Chunaev and N. M. Przhiyalgovskaya, Chem. Heterocycl. Compd., 1991, 27, 1012–1015 CrossRef.
- H.-D. Ilge and R. Paetzold, J. Für Prakt. Chem., 1984, 326, 705–720 CrossRef CAS.
- D. F. Evans, J. Chem. Soc. Resumed, 1957, 1351–1357 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc05975h |
| This journal is © The Royal Society of Chemistry 2024 |