
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Co-factor prosthesis facilitates biosynthesis of azido-pseudaminic acid probes for use as glycosyltransferase reporters†
Tessa
Keenan
 a,
Harriet S.
Chidwick
a,
Matthew
Best
a,
Emily K. P.
Flack
ab,
Nicholas D. J.
Yates
a,
Natasha E.
Hatton
a,
Matthew E.
Warnes
a and
Martin A.
Fascione
*a
a,
Harriet S.
Chidwick
a,
Matthew
Best
a,
Emily K. P.
Flack
ab,
Nicholas D. J.
Yates
a,
Natasha E.
Hatton
a,
Matthew E.
Warnes
a and
Martin A.
Fascione
*a
aDepartment of Chemistry, University of York, Heslington, York YO10 5DD, UK. E-mail: martin.fascione@york.ac.uk
bDepartment of Biology, University of York, York, YO10 5DD, UK
First published on 2nd January 2024
Abstract
Truncated thioester N,S-diacetylcysteamine (SNAc) was utilised as a co-factor mimic for PseH, an acetyl-coA dependent aminoglycoside N-acetyltransferase, in the biosynthesis of the bacterial sugar, pseudaminic acid. Additionally, an azido-SNAc analogue was used to smuggle N7-azide functionality into the pseudaminic acid backbone, facilitating its use as a reporter of pseudaminyltransferase activity.
5,7-Diacetyl pseudaminic acid (Pse5Ac7Ac, 1) and its analogues are rare non-mammalian sugars belonging to the nonulosonic acid family, a group of nine-carbon α-keto-acid sugars that are widely distributed in nature and found in diverse cell-surface glycoconjugates.1 Pse glycans are particularly prevalent in pathogenic bacterial species, including Campylobacter jejuni,2Helicobacter pylori,3,4Shigella boydii,5Aeromonas caviae and multidrug resistant ESKAPE pathogens Pseudomonas aeruginosa and Acinetobacter baumannii.6–10 They are also recognised as virulence factors and are essential for correct flagellar assembly and motility in some bacteria,2,4,11 as well as playing a role in immune evasion.12 There is therefore a vital need to understand the biosynthesis of Pse derivatives and how they are incorporated into cell-surface glycoconjugates. Although significant progress has been made in understanding the biosynthesis of CMP-Pse5Ac7Ac 7 (Fig. 1A), the nucleotide activated form of the parent sugar that is the substrate for glycosyltransferases (GTs), the discovery and characterisation of GT enzymes that catalyse the incorporation of the sugar into glycoconjugates,13 termed “pseudaminyltransferases” (PseTs), has lagged behind. This area of research has been hampered by requirement for straightforward access to Pse-based chemical probes, as well as sensitive and straightforward methods to screen for PseT activity. Using metabolic glycan engineering, others have demonstrated the ability to label Pse-containing glycans in bacteria using chemical synthesis to access an ester-protected N5-azidoacetamide-functionalised Pse analogue,14 or a functionalised intermediate in the Pse biosynthetic pathway, 6-deoxy-Alt-NAc4NAz.15,16 To be incorporated into the cell-surface glycans, these analogues would need to enter the Pse biosynthetic pathway in vivo to produce CMP nucleotide activated forms of the sugars, and then be processed by PseTs. In this study we therefore explored whether similarly azido functionalised Pse analogues could serve as bioorthogonal probes that would facilitate preliminary in vitro screening of enzymes for PseT activity, prior to time and resource intensive full characterisation of an enzyme and challenging scale up of the glycoside forming reaction.
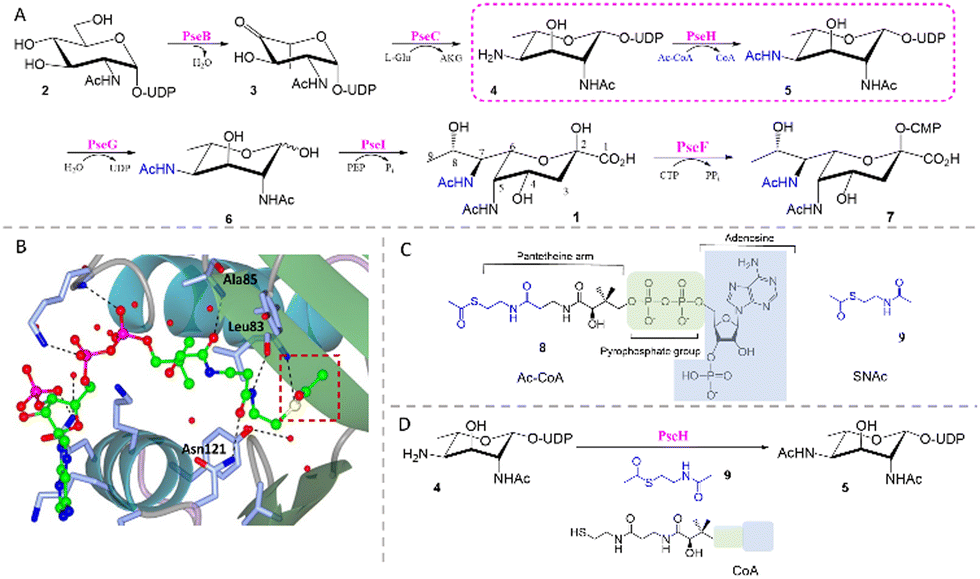 | ||
| Fig. 1 (A) The biosynthetic pathway for CMP-Pse5Ac7Ac 7 reconstituted in vitro. (B) Crystal structure of C. jejuni PseH (PDB 4XPL)17 in complex with its co-factor acetyl-CoA 8 (ball and stick model, carbon-bright green, oxygen-red, nitrogen-blue, sulfur-yellow, phosphorus-magenta) showing hydrogen bonds (black dotted lines) between residues (stick model, carbon-ice blue, oxygen-red, nitrogen-blue) and the pantetheine arm and pyrophosphate group. The acetyl group of 8 is highlighted in a red box. (C) Structural similarity between 8 and SNAc 9 (highlighted in blue). (D) PseH co-factor prosthesis with 9 and CoA. | ||
In previous work we reconstituted a biosynthetic route for the synthesis of CMP-Pse5Ac7Ac 7in vitro, starting from UDP-GlcNAc 2 and using enzymes from C. jejuni (PseBCHGI) and A. caviae (PseF) (Fig. 1A).18 Additionally, we established a method for co-factor regeneration using PseH, the third enzyme in the biosynthetic pathway, demonstrating that the natural co-factor acetyl coenzyme-A (Ac-CoA, 8) could be substituted for S-acetyl thiocholine iodide and coenzyme-A (CoA). PseH is an aminoglycoside N-acetyltransferase that catalyses acetyl transfer to C4 of UDP-4-amino-4,6-dideoxy-β-ι-AltNAc 4, and part of the GNAT enzyme superfamily which utilise 8 as a co-factor.17,19 It has been proposed that the reactivity of the acetyl transfer in these enzymes stems from the pantetheine arm, and is largely unaffected by the distal parts of the Ac-CoA structure.20 This is evident in the crystal structure of C. jejuni PseH in complex with Ac-CoA, where three hydrogen bonds are formed between the pantetheine arm and the surrounding active site residues (Fig. 1B).17 Interestingly, there are examples of CoA thioester utilising acetyltransferases that have shown promiscuity towards truncated thioesters such as N,S-diacetyl cysteamine (SNAc), facilitating the introduction of unnatural functionality, in an approach we denominate “co-factor prosthesis”.21–23 Considering this precedent and the similarity of SNAc 9 to the pantetheine arm of 8 (Fig. 1C) and the positioning of the co-factor in the PseH active site (Fig. 1B), we hypothesised that SNAc 9 might serve as an alternative PseH co-factor (Fig. 1D) and enable us to smuggle unnatural functionality into Pse5Ac7Ac at the N7 position, via our reconstituted biosynthetic pathway.
To investigate the utility of SNAc 9 in a co-factor prosthesis approach for PseH catalysed acetylation, small-scale reactions were assembled containing UDP-GlcNAc 2 (2 mM), PseB (5 μM) and PseC (20 μM), as well as their associated co-factors. Following PseBC product 4 formation, PseH (10 μM), SNAc 9 (5 mM) and Co-A (0.3 mM) were added and the samples were analysed by negative ESI-LC-MS. A peak consistent with the mass of the acetylated PseH product 5 ([M − H]−m/z 631) was observed in the reaction and absent from the no SNAc control (Fig. S1, ESI†), suggesting that 9 could indeed serve as a co-factor for PseH. The utility of 9 was further demonstrated in the one-pot biosynthesis of Pse5Ac7Ac 1 on a semi-preparative scale starting from UDP-GlcNAc 2 (65 mg) with enzymes from C. jejuni (PseBCHGI) and their associated co-factors. The use of SNAc 9 as a PseH co-factor facilitated the isolation of 27 mg of Pse5Ac7Ac 1 in a yield of 76%. Next, functionalised N-acetyl cysteamines SNAc-N310 and SNAc-Alk 11 were investigated in our in vitro biosynthetic pathway, as a route to accessing Pse5Ac7N312 and Pse5Ac7Alk 13 (Fig. 2). In preliminary experiments using SNAc-N310 in a one-pot biosynthesis starting from UDP-GlcNAc 2 with C. jenuni PseBCHGI and the associated co-factors, we observed PseBC product inhibition (data not shown). Therefore, the biosynthesis was split into two parts where PseBC reaction was allowed to proceed first to afford 4 and then PseHGI and the associated co-factors, including the functionalised SNAcs, were added second (Fig. 2A). In reactions assembled with SNAc-N310, a peak consistent with the mass of the desired product, Pse5Ac7N312 ([M − H]−m/z 374) was observed after analysis by negative ESI-LC-MS (Fig. 2B and Fig. S2a, ESI†). In contrast, no Pse5Ac7Alk 13 product was observed in reactions assembled with SNAc-Alk 11 (Fig. S2b, ESI†). Additionally, there was no evidence of the alkyne-functionalised PseH product ([M − H]−m/z 641) suggesting that the biosynthesis had stalled at PseH. This may be attributed to the low solubility of SNAc-Alk 11 or a lack of conformational flexibility. Following the successful demonstration on a reaction scale that SNAc-N310 could be used in co-factor prosthesis in our reconstituted biosynthetic pathway, Pse5Ac7N312 was then synthesised on a semi-preparative scale starting from 21 mg of UDP-GlcNAc 2. As in the small-scale reactions, the PseBC reaction was carried out first. While it was possible to perform the PseGHI reaction without purification of the PseBC product first, we found that it was difficult to achieve pure Pse5Ac7N312 using this strategy. Therefore, PseBC product 4 was first purified by Biogel P2 column chromatography and then converted to Pse5Ac7N312 using SNAc-N310, PseGHI and the associated co-factors. Following a second Biogel P2 column purification, 3.9 mg of Pse5Ac7N312 was isolated in an overall yield of 33%. Next, we sought to demonstrate that Pse5Ac7N312 was a suitable substrate for A. caviae PseF, a CMP-Pse5Ac7Ac synthetase, for the formation of the activated glycosyl donor αCMP-Pse5Ac7N314.24 Pse5Ac7N312 was incubated with PseF in the presence of cytidine triphosphate (CTP) and the formation of the desired crude product 14 was validated by negative ESI-LC-MS (Fig. 2C) and by NMR following treatment with alkaline phosphatase (Fig. S5 and S6a, ESI†). Notably, we observed some evidence of hydrolytic instability of 14, as is a classic feature of many sugar nucleotides.25,26 However, calculation of the difference in chemical shift between the H3ax and H3eq (Δppm H3eq–H3ax = 0.62 ppm) validated the axially orientated O-CMP group (Fig. S5C, ESI†).27 Having unequivocally confirmed turnover of 12 by PseF, we set out to demonstrate the utility of αCMP-Pse5Ac7N314 as a bioorthogonal reporter of PseT activity (Fig. 3). To limit potential CMP-donor hydrolysis, in a proof of concept experiment we opted to use Pse5Ac7N312 in a one-pot reaction with both PseF and a sialyltransferase with PseT activity.
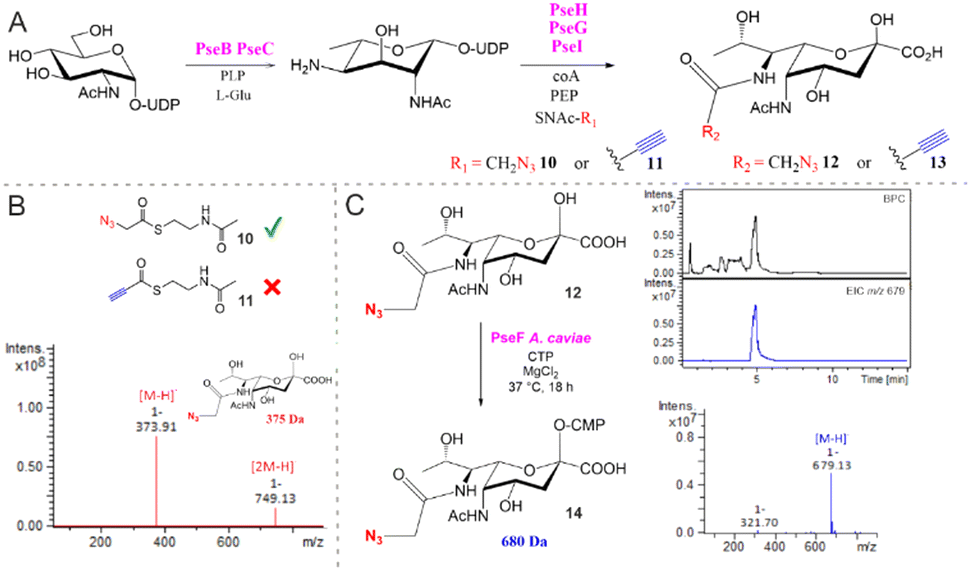 | ||
| Fig. 2 (A) Two-step in vitro biosynthesis of N7-functionalised Pse analogues. (B) Reaction-scale screening of SNAc-N310 and SNAc-Alk 11 shows that only 11 can be used as a co-factor prosthesis with PseH, facilitating the biosynthesis of Pse5Ac7N312 with PseBCHGI in two steps. (C) Pse5Ac7N312 conversion to CMP-Pse5Ac7N314 by A. caviae PseF, as validated by negative ESI-LC-MS. Base peak chromatogram (BPC) shown in black. Extracted ion chromatogram (EIC) for CMP-Pse5Ac7N314 [M − H]−m/z 679 shown in blue. | ||
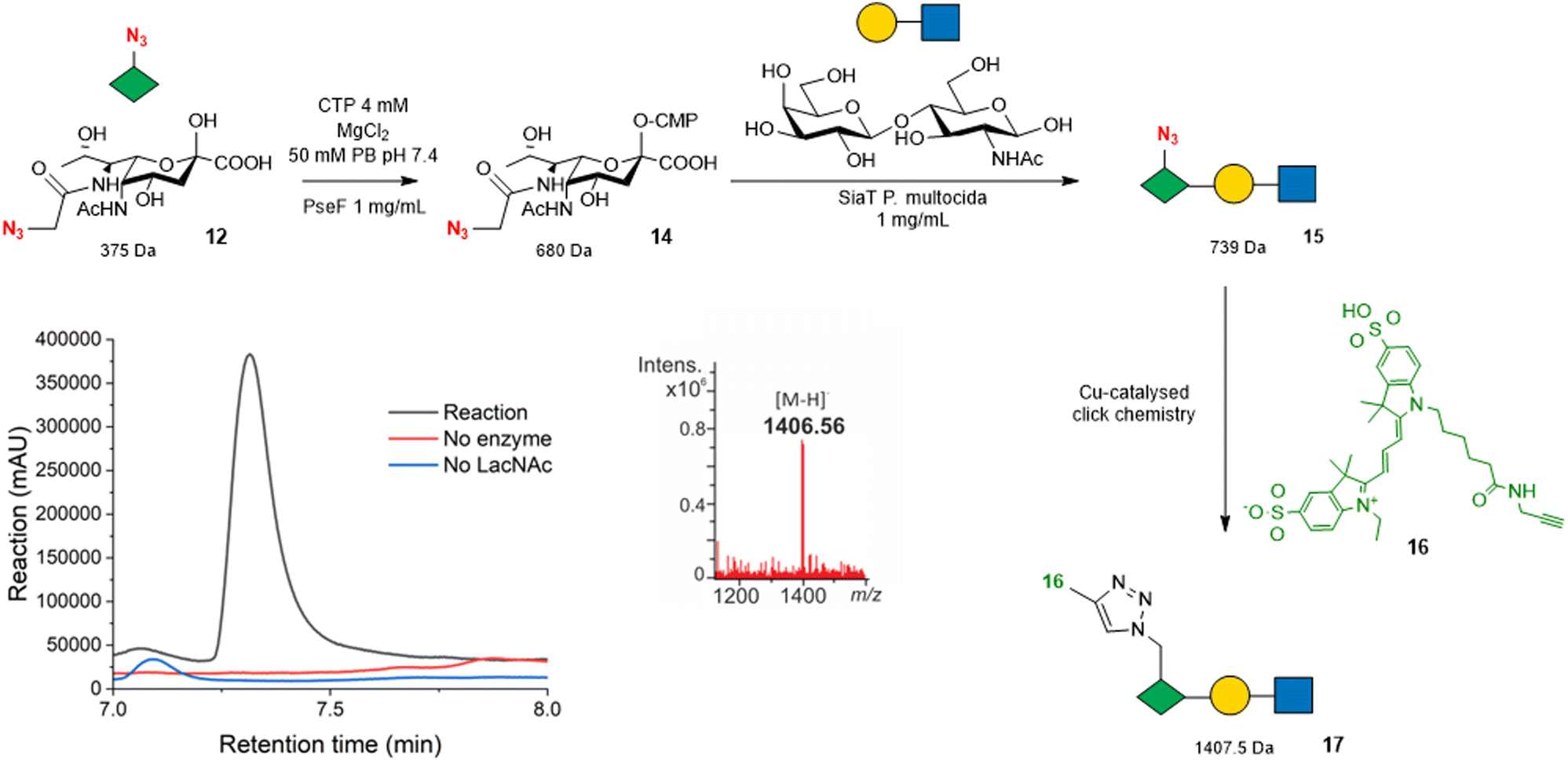 | ||
| Fig. 3 Strategy for using Pse5Ac7N312 as a bioorthogonal reporter for PseT activity. Overlayed 545–565 nm chromatogram shows a peak eluting at 7.25 min in the reaction (black) that is absent from control reactions (red and blue). The mass spectrum under the peak shows an ion at the expected mass of the sulfo-Cy3-conjugated Pse5Ac7N3 -LacNAc 17 [M − H]−m/z 1406.5. Full chromatogram shown in Fig. S10 (ESI†). Note that the regio and stereochemistry of the glycosidic linkage to Pse5Ac7Az in trisaccharide 15 are omitted as they cannot be unambiguously assigned without full NMR characterisation, although sialyltransferase tPm0188Ph was previously demonstrated to afford Pse-β2,3-Gal linkages using αCMP-Pse5Ac7Ac as a donor.24 | ||
We selected the promiscuous inverting sialyltransferase (SiaT) from Pasteurella multocida, tPm0188Ph for this experiment, as it was previously shown to convert αCMP-Pse5Ac7Ac 7 to β2,3-linked Pse5Ac7Ac-terminated glycosides.24 Therefore, Pse5Ac7N312 (1 mM) was first incubated with A. caviae PseF to allow the formation of αCMP-Pse5Ac7N314, as validated by negative ESI-LC-MS (Fig. S7, ESI†), before the addition of SiaT tPm0188Ph and LacNAc to drive the formation of the anticipated product, Pse5Ac7N3-LacNAc 15. However, after 18 h, no evidence of the desired product 15 ([M − H]−m/z 738) was observed (Fig. S8, ESI†), suggesting that either the SiaT tPm0188Ph could not use 14 as a substrate, or that the low-level formation of the Pse5Ac7N3-LacNAc 15 product could not be detected under the conditions of the reaction. To enable detection of low-level PseT activity we therefore used the azide-functionality in the Pse5Ac7N3 moiety to label the reaction products with an alkyne functionalised fluorescent sulfo-cyanine 3 probe 16 using a copper-catalysed alkyne–azide cycloaddition (CuAAC) reaction. We anticipated that bioconjugation to 16 would facilitate the detection of our reaction products by both LC-MS and absorbance at 545–565 nm. Following analysis of the reactions by negative ESI-LC-MS, we observed a clear peak in the UV/Vis chromatogram with a retention time of 7.25 min, that was not present in the no SiaT and no LacNAc control reactions (Fig. 3 and Fig. S9, ESI†). Additionally, the average mass spectrum under the peak showed an ion at m/z 1406.56, which is consistent with the expected [M − H]− for the sulfo-Cy3-conjugated Pse5Ac7N3-LacNAc product 17 at 1407.5 Da. These results demonstrate that sulfo-Cy3 labelling of Pse5Ac7N3-LacNAc 15 also aids the detection of the glycoside by negative ion LC-MS, showcasing the potential of Pse5Ac7N312 as a probe to screen for PseT activity.
In summary, we have shown that the truncated thioester SNAc 9 can serve as a co-factor for PseH in our reconstituted Pse biosynthetic pathway, enabling the facile, one-pot synthesis of Pse5Ac7Ac 1. Additionally, we have demonstrated that SNAc-N39 can be used in an in vitro co-factor prosthesis strategy with PseH, enabling N7-azide functionality to be smuggled into the Pse backbone. In contrast, SNAc-Alk 11 was not a suitable PseH substrate and therefore could not facilitate the introduction of alkyne functionality into Pse. Grimes and colleagues previously demonstrated the introduction of alkyne functionality into N-acetylglucosamine in peptidoglycan using an elongated alkyne-functionalised SNAc.22 It is therefore possible that similar use of a more conformationally flexible SNAc-Alk probe might enable turnover by PseH and subsequently facilitate the introduction of an alkyne functionality into the Pse backbone. Finally, we demonstrated the utility of Pse5Ac7N312, in combination with A. caviae PseF activation, as a bioorthogonal reporter of PseT activity. We therefore anticipate this methodology will facilitate the discovery and validation of new bona fide PseTs.
TK, HC, MB, EF, NY, NH and MW performed the experiments and analysed the data. MAF designed the study and supervised the project. TK, HC and MAF wrote the manuscript.
We thank Dr Ed Bergstrom and The York Centre of Excellence in Mass Spectrometry. The York Centre of Excellence in Mass Spectrometry was created thanks to a major capital investment through Science City York, supported by Yorkshire Forward with funds from the Northern Way Initiative, and subsequent support from EPSRC (EP/K039660/1; EP/M028127/1). This work was supported by The University of York, the EPSRC (PhD award to E. K. F. P.), the Rosetrees Trust (PhD award to H. C.), and a Horizon Europe Guarantee Consolidator award to M. A. F. (selected by the ERC, funded by UKRI; EP/X023680/1).
Conflicts of interest
There are no conflicts to declare.References
- A. L. Lewis, X. Chen, R. L. Schnaar and A. Varki, in Essentials of Glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, P. Stanley, G. W. Hart, M. Aebi, D. Mohnen, T. Kinoshita, N. H. Packer, J. H. Prestegard, R. L. Schnaar and P. H. Seeberger, Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY), 4th edn, 2022 Search PubMed.
- P. Thibault, S. M. Logan, J. F. Kelly, J.-R. Brisson, C. P. Ewing and P. Guerry, J. Biol. Chem., 2001, 276, 34862–34870 CrossRef CAS PubMed.
- A. I. M. Salah Ud-Din and A. Roujeinikova, Cell. Mol. Life Sci., 2018, 75, 1163–1178 CrossRef CAS PubMed.
- M. Schirm, E. Soo, A. Aubry, J. Austin, P. Thibault and S. Logan, Mol. Microbiol., 2003, 48, 1579–1592 CrossRef CAS PubMed.
- Y. A. Knirel, E. V. Vinogradov, V. L. L'vov, N. A. Kocharova, A. S. Shashkov, B. A. Dmitriev and N. K. Kochetkov, Carbohydr. Res., 1984, 133, C5–C8 CrossRef CAS PubMed.
- S. M. B. Tabei, P. G. Hitchen, M. J. Day-Williams, S. Merino, R. Vart, P.-C. Pang, G. J. Horsburgh, S. Viches, M. Wilhelms and J. M. Tomás, J. Bacteriol., 2009, 191, 2851–2863 CrossRef CAS PubMed.
- Y. A. Knirel, E. V. Vinogradov, A. S. Shashkov, B. A. Dmitriev, N. K. Kochetkov, E. S. Stanislavsky and G. M. Mashilova, Eur. J. Biochem., 1987, 163, 627–637 CrossRef CAS PubMed.
- N. Kocharova, A. Shashkov, B. Dmitriev and N. Kochetkov, Bioorg. Khim., 1986, 12, 1384–1390 Search PubMed.
- N. S. Sof'ya, A. V. Popova, A. S. Shashkov, M. M. Shneider, Z. Mei, N. P. Arbatsky, B. Liu, K. A. Miroshnikov, N. V. Volozhantsev and Y. A. Knirel, Carbohydr. Res., 2015, 407, 154–157 CrossRef PubMed.
- J. J. Kenyon, A. M. Marzaioli, R. M. Hall and C. De Castro, Glycobiology, 2014, 24, 554–563 CrossRef CAS PubMed.
- J. L. Parker, M. J. Day-Williams, J. M. Tomas, G. P. Stafford and J. G. Shaw, MicrobiologyOpen, 2012, 1, 149–160 CrossRef CAS PubMed.
- H. N. Stephenson, D. C. Mills, H. Jones, E. Milioris, A. Copland, N. Dorrell, B. W. Wren, P. R. Crocker, D. Escors and M. Bajaj-Elliott, J. Infect. Dis., 2014, 210, 1487–1498 CrossRef CAS PubMed.
- H. S. Chidwick and M. A. Fascione, Org. Biomol. Chem., 2020, 18, 799–809 RSC.
- A. M. Vibhute, H. Tamai, D. Logviniuk, P. G. Jones, M. Fridman and D. B. Werz, Chem. – Eur. J., 2021, 27, 10595–10600 CrossRef CAS PubMed.
- F. Liu, A. J. Aubry, I. C. Schoenhofen, S. M. Logan and M. E. Tanner, ChemBioChem, 2009, 10, 1317–1320 CrossRef CAS PubMed.
- G. Andolina, R. Wei, H. Liu, Q. Zhang, X. Yang, H. Cao, S. Chen, A. Yan, X. D. Li and X. Li, ACS Chem. Biol., 2018, 13, 3030–3037 CrossRef CAS PubMed.
- W. S. Song, M. S. Nam, B. Namgung and S.-I. Yoon, Biochem. Biophys. Res. Commun., 2015, 458, 843–848 CrossRef CAS PubMed.
- H. S. Chidwick, E. K. Flack, T. Keenan, J. Walton, G. H. Thomas and M. A. Fascione, Sci. Rep., 2021, 11, 4756 CrossRef CAS PubMed.
- I. C. Schoenhofen, D. J. McNally, J.-R. Brisson and S. M. Logan, Glycobiology, 2006, 16, 8C–14C CrossRef CAS PubMed.
- Y. Modis and R. Wierenga, Structure, 1998, 6, 1345–1350 CrossRef CAS PubMed.
- R. M. Mizanur, F. A. Jaipuri and N. L. Pohl, J. Am. Chem. Soc., 2005, 127, 836–837 CrossRef CAS PubMed.
- Y. Wang, K. M. Lazor, K. E. DeMeester, H. Liang, T. K. Heiss and C. L. Grimes, J. Am. Chem. Soc., 2017, 139, 13596–13599 CrossRef CAS PubMed.
- X. Xie, K. Watanabe, W. A. Wojcicki, C. C. Wang and Y. Tang, Chem. Biol., 2006, 13, 1161–1169 CrossRef CAS PubMed.
- E. K. Flack, H. S. Chidwick, G. Guchhait, T. Keenan, D. Budhadev, K. Huang, P. Both, J. Mas Pons, H. Ledru and S. Rui, ACS Catal., 2020, 10, 9986–9993 CrossRef CAS.
- C.-H. Lin, B. W. Murray, I. R. Ollmann and C.-H. Wong, Biochemistry, 1997, 36, 780–785 CrossRef CAS PubMed.
- G. K. Wagner, T. Pesnot and R. A. Field, Nat. Prod. Rep., 2009, 26, 1172–1194 RSC.
- Y. A. Knirel, E. T. Rietschel, R. Marre and U. Zahringer, Eur. J. Biochem., 1994, 221, 239–245 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc05924c |
| This journal is © The Royal Society of Chemistry 2024 |