
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure–property–function relationships of stabilized and persistent C- and N-based triaryl radicals
Anna
Vasiļevska
ab and
Tomáš
Slanina
*a
aInstitute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, 166 10 Prague 6, Czech Republic. E-mail: tomas.slanina@uochb.cas.cz
bDepartment of Organic Chemistry, Charles University, 128 00 Prague 2, Czech Republic
First published on 5th December 2023
Abstract
Structurally similar C- and N-based triaryl radicals are among the most commonly used structural motifs in stable, open-shell, organic molecules. The application of such species is associated with their stability, properties and structural design. This study summarizes the basic stabilization and persistence principles of C- and N-based triaryl radicals and highlights recent advances in design strategies of radicals tailored for specific applications.
 Anna Vasiļevska | Anna Vasiļevska is a PhD student at the Department of Organic Chemistry, Faculty of Science, Charles University. She conducts her doctoral research at the Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences (IOCB Prague) under the supervision of Dr Tomáš Slanina. She joined his Redox Photochemistry group after graduating with a MSc in Chemistry at the University of Latvia in 2019. Since then, her research has focused on trityl-based photoredox switches, triarylamine radical cations and their application in reversible, photoinduced electron transfer processes. |
 Tomáš Slanina | Tomáš Slanina is an Adjunct Professor at Charles University and a junior group leader of the Redox Photochemistry group at IOCB Prague. Previously, following his MSc studies at Masaryk University, he joined a joint PhD program between his alma mater and the University of Regensburg, under the supervision of Professors Klán and König, graduating in 2015. Subsequently, he completed two postdoctoral stays, first with Professor Heckel, at Goethe University, and then with Professor Ottosson, at Uppsala University. Currently, his research interests lie at the intersection of photo-, organic and radical chemistry and spectroscopy. |
1. Introduction
Isoelectronic C- and N-centred triaryl radicals have numerous applications in magnetic and multifunctional materials, catalysis, macromolecular chemistry, spectroscopy, spin labelling and trapping, molecular electronics and photovoltaics.1–3 However, organic radicals are typically highly reactive and their reactivity is difficult to control. For this reason, their stabilization is crucial for their applications.In radical chemistry, a radical is deemed stable when it can be isolated, characterised, and stored as a pure compound. Although the word “stability” is used to describe lack of degradation, this term overlooks the origin, characteristics, and mechanism of the processes that lead to decomposition. So, when describing the “stability” of radicals, we typically evaluate the combination of two independent factors: thermodynamic stabilization and kinetic persistence (Fig. 1). Thermodynamic stabilization derives from spin density delocalization over the molecule, while kinetic persistence is introduced by steric shielding of the reactive centers.4
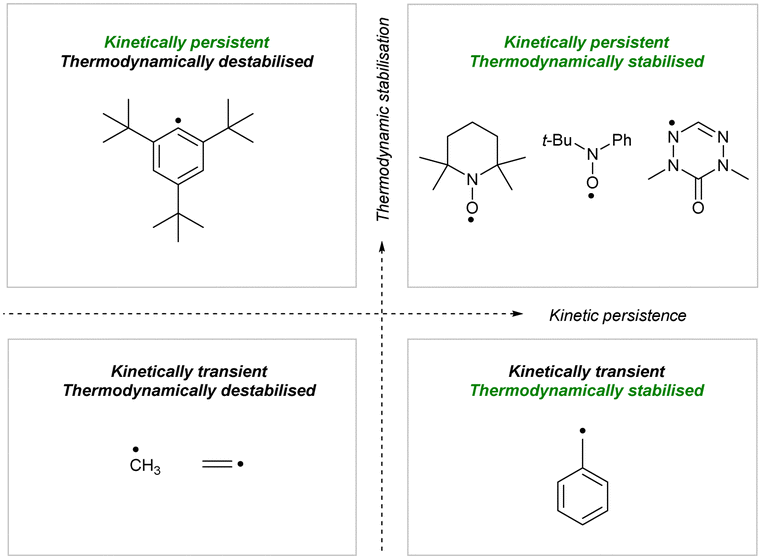 | ||
| Fig. 1 Mapping organic radicals based on thermodynamic stabilization and kinetic persistence.4 | ||
Numerous stable organic radicals have been developed for synthetic and catalytic transformations.5–7 More specifically, several efficient design approaches have been established to ensure the stabilization and persistence of C- and N-based triaryl radicals.6,8–10 But novel applications of the radicals demand further advances in design strategies to yield targeted physical, photophysical and electronic properties, chemical reactivity and response output.
Most studies describing organic radicals only focus on chemical properties and potential applications. Yet, by carefully studying chemical reactivity, including degradation pathways, we may understand the main factors that explain the stability or resistance to degradation of these radicals. In this study, we discuss the main stability trends of structurally similar triphenylmethyl (trityl) radicals and triphenylamine radical cations by analysing their reactivity and degradation pathways and derive essential design principles suitable for novel applications of such radicals.
2. Triarylmethyl radicals
Research on persistent radical chemistry started in 1900 when Gomberg first reported the triphenylmethyl (trityl) radical.11 The trityl radical can be prepared from trityl chloride 1 (Scheme 1) by reduction with zinc. This radical 2 is stable under inert conditions but trapped by atmospheric oxygen, forming bis(triphenylmethyl)peroxide 3.11,12 Molecular oxygen has a triplet (biradical) electronic ground state and reacts in a coupling reaction with other free radicals. Upon heating, peroxide homolytically cleaves the O–O bond and rearranges, forming diether 4.13 Density functional theory (DFT) calculations have revealed that the O-centred triphenylmethoxyl radical 5 isomerises through the oxaspiro intermediate 6 to a more stable C-centred diphenyl(phenoxy)methyl radical 7, which in turn undergoes dimerization.14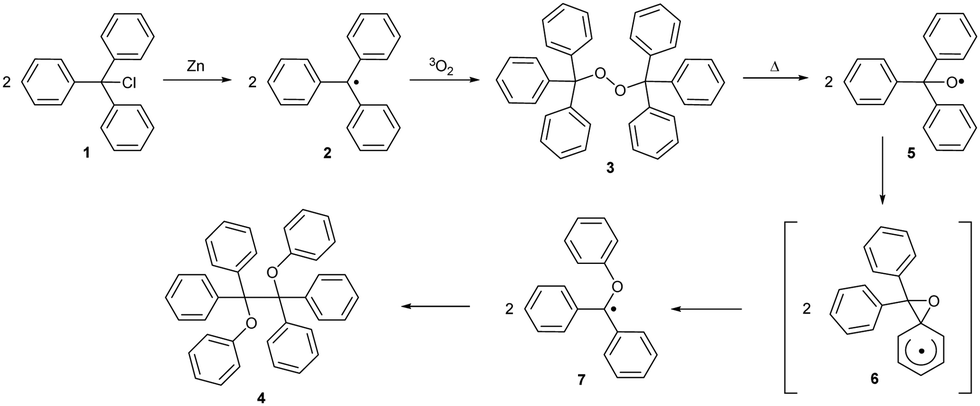 | ||
| Scheme 1 Trityl peroxide 3 formation and rearrangement. | ||
The structure of peroxides formed in a reaction with molecular oxygen can differ depending on the substitution pattern of the initial trityl radical. The tris(2,6-dimethoxyphenyl)methyl radical 8a (R = OMe) has an unusual geometry, resembling the enantiomerisation transition state 2‡ of Ar3X-like propellers; two phenyl rings are considerably twisted out of the plane of central (α) carbon (Scheme 2a, grey rectangle) by 61°, inducing shielding of the central carbon, and the third phenyl ring is almost planar, with a twist angle of only 12°.15 The less substituted analogue, bis(2,6-dimethoxyphenyl)(phenyl) methyl radical 8b (R = H), has a similar geometry to that of the 8a with a higher twist angle (30°) and less hindered rotation of the unsubstituted phenyl ring. This conformational flexibility promotes spin delocalization and increases the reactivity at the para-position of the unsubstituted phenyl ring. Therefore, 8b reacts with oxygen exclusively in para-α positions, forming “head-to-tail” peroxide 9, while the more shielded, fully ortho-substituted trityl radical 8a forms “tail-to-tail” para–para peroxide 10 (Scheme 2b).
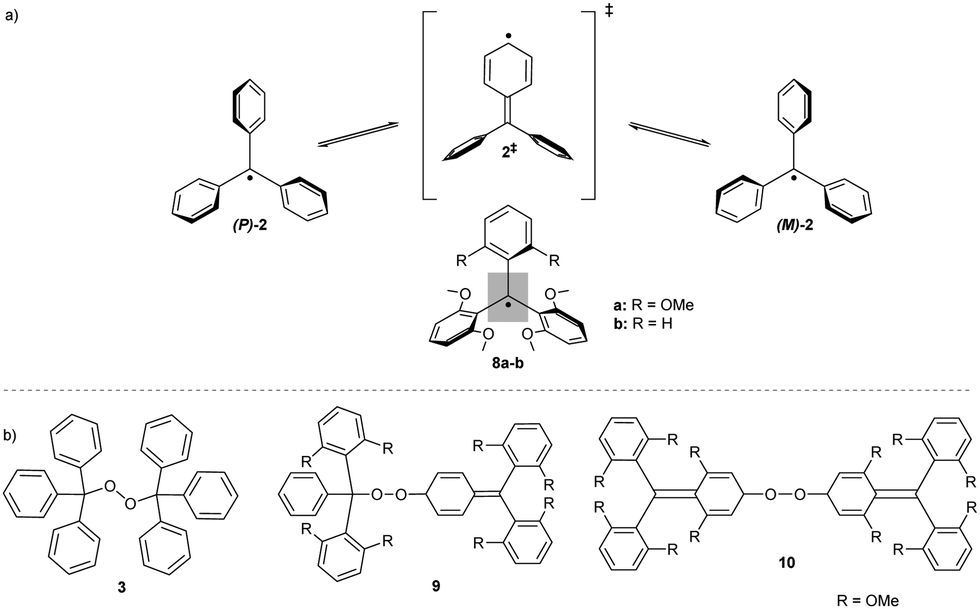 | ||
| Scheme 2 (a) Ring-flip enantiomerisation of CPh3 propellers; the transition state has a similar geometry to o-methoxy trityls 8. The grey rectangle highlights the plane of the central carbon. (b) Chemical structures of unsubstituted and o-methoxy substituted trityl peroxides 3, 9 and 10. | ||
2.1. Halogenated trityls
Full or partial chlorination of phenyl rings increases the kinetic persistency of the radical due to additional steric shielding of the reactive carbon centre by ortho-chlorines. Thus, perchlorinated triphenylmethyl (PTM) radical 11 is air-stable. Bulky chlorine atoms additionally twist the phenyl rings, diminishing the spin density delocalization over the π-system. Because the molecule contains electron withdrawing chlorine atoms, PTM radicals are good electron acceptors and can be easily reduced to the respective anions 11− (Scheme 3a). Oxidation to PTM cations 11+ is also possible, albeit at higher potentials than those required by the parent trityl radical.6,16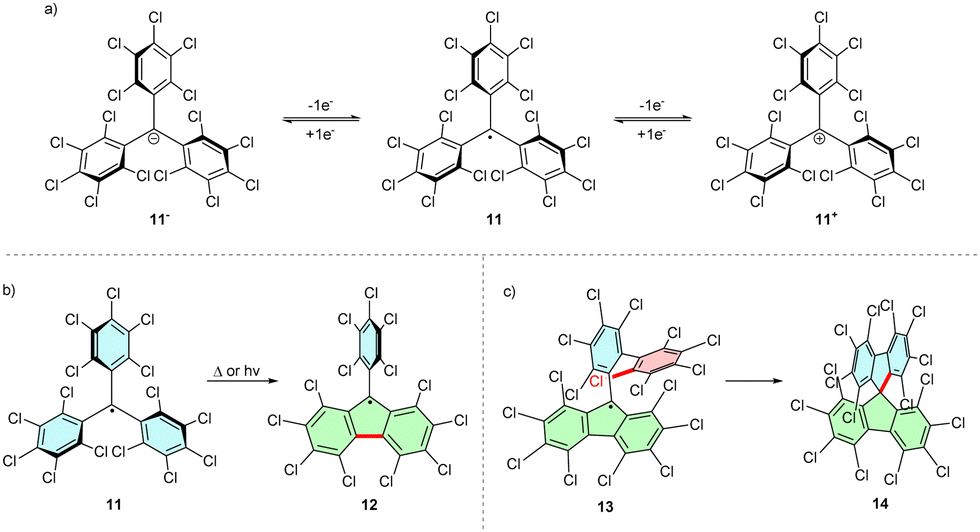 | ||
| Scheme 3 (a) Structure of polychlorinated trityl radical 11 and its respective anion and cation. (b) Formation of perchlorinated fluorenyl radical 12 upon photoirradiation or thermolysis of 11. (c) Cyclization of perchlorinated 9-(2-biphenylyl)fluorenyl radical 13 to form bispiro compound 14. | ||
The fully chlorinated trityl radical shows a high thermal stability and only decomposes upon thermolysis over 300 °C. The main product of thermal and UV-light induced decomposition is a product of cyclization, perchloro-9-phenylfluorenyl (PPF) radical 12 (Scheme 3b). The increased conjugation within the fluorenyl moiety increases the stability of 12, which decomposes only at 350 °C. Conversely, introducing further substituents at 12 can destabilize the system by providing alternative reactive pathways. For example, perchlorinated 9-(2-biphenylyl)fluorenyl radical 13 readily cyclizes, forming the spiro analogue 14, even in the solid state (Scheme 3c). This reactivity can be explained by the close proximity of the biphenyl ortho-C–Cl bond and the trityl unpaired electron.17
Thanks to their exceptional persistence, PTM radicals can be structurally modified using covalent organic chemistry without affecting the radical centre (Scheme 4). The scope of known transformations includes acyl chloride PTM-COCl generation from acid 15 and subsequent synthesis of esters 16 and acid anhydrides 17,18 amide bond formation with ammonia/amino acid (18/19),19 alkylation of amino group20 and both acidic19 and basic21 ester hydrolysis (15). The radical is also preserved during reactions on peripheric heteroaromatic substituents (20), such as halogenation (21)22 and Friedel–Crafts acylation (20).22
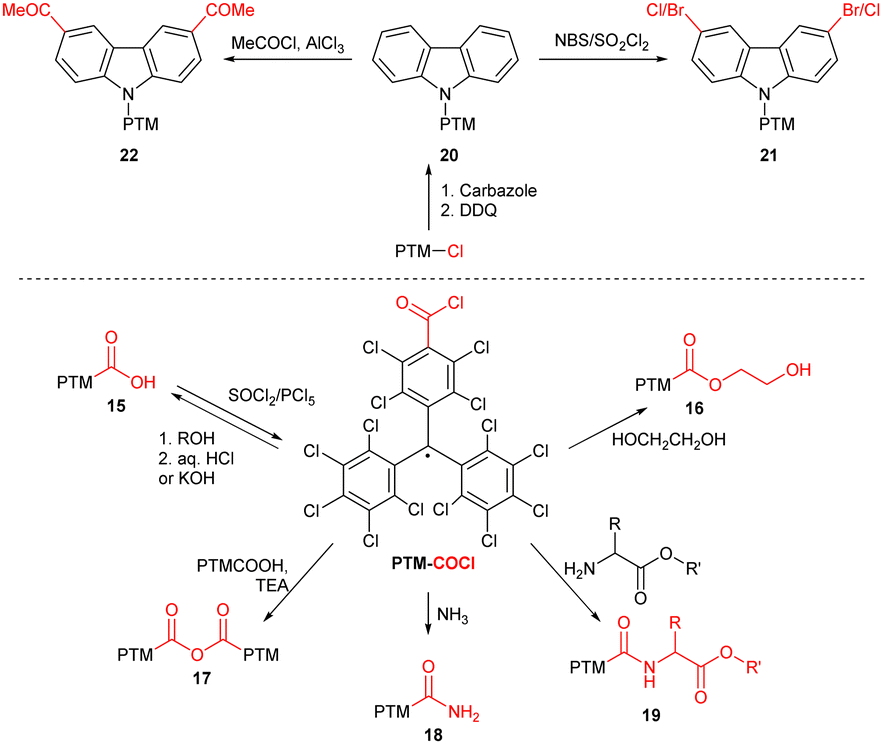 | ||
| Scheme 4 Chemical reactivity of PTM radicals. | ||
Installing larger halogens on trityl aryl rings introduces even greater steric shielding. Partly substituting chlorine atoms for bromines in the tris(2,4,6-trichlorophenyl)-methyl (TTM) radical increases the stability of the radical against photobleaching whilst simultaneously enabling functionalisation by cross-coupling.3 As propeller-shaped trityl radicals are intrinsically chiral, increased steric hindrance and, thus, a higher racemisation barrier, make it possible to isolate both (+)-P and (−)-M atropoisomers of the tris(2,4,6-tribromophenyl)-methyl radical. This radical does not racemise (similar enantiomerisation is shown in Scheme 2a) at high temperatures of up to 60 °C, in contrast to the chlorinated analogue (t1/2 = 18 s at 60 °C).23
2.2. Trityl stabilization by steric hindrance and delocalization
Resistance to degradation can also be achieved via other bulky substituents. Avoiding heteroatoms is beneficial for high field electron paramagnetic resonance (EPR) dipolar distance measurements because the spin density, otherwise delocalized on heteroatoms at positions 2 and 6, concentrates at the triphenylmethyl core, which significantly decreases the g-anisotropy.21 Therefore, polycyclic aryl substituents are used to efficiently shield and localize the spin density at the central carbon atom.An example of this substitution is anthryl-substituted derivative 23 (Scheme 5a),24 with a high dihedral angle of 48.5° between the central carbon and the anthryl groups. Substituting one of the mesityl group in 23 for a TIPS-ethynyl group results in 24, which maintains the high persistence of radical and is susceptible to further modification via [4+2] Diels–Alder reaction.25 In 24, steric hindrance generated by introducing the capping TIPS group is crucial for the resistance to degradation of the molecule. Substituting the TIPS group for a smaller TMS group yields less stable 25, which forms the ‘tail-to-tail’ dimer 26 (Scheme 5b).
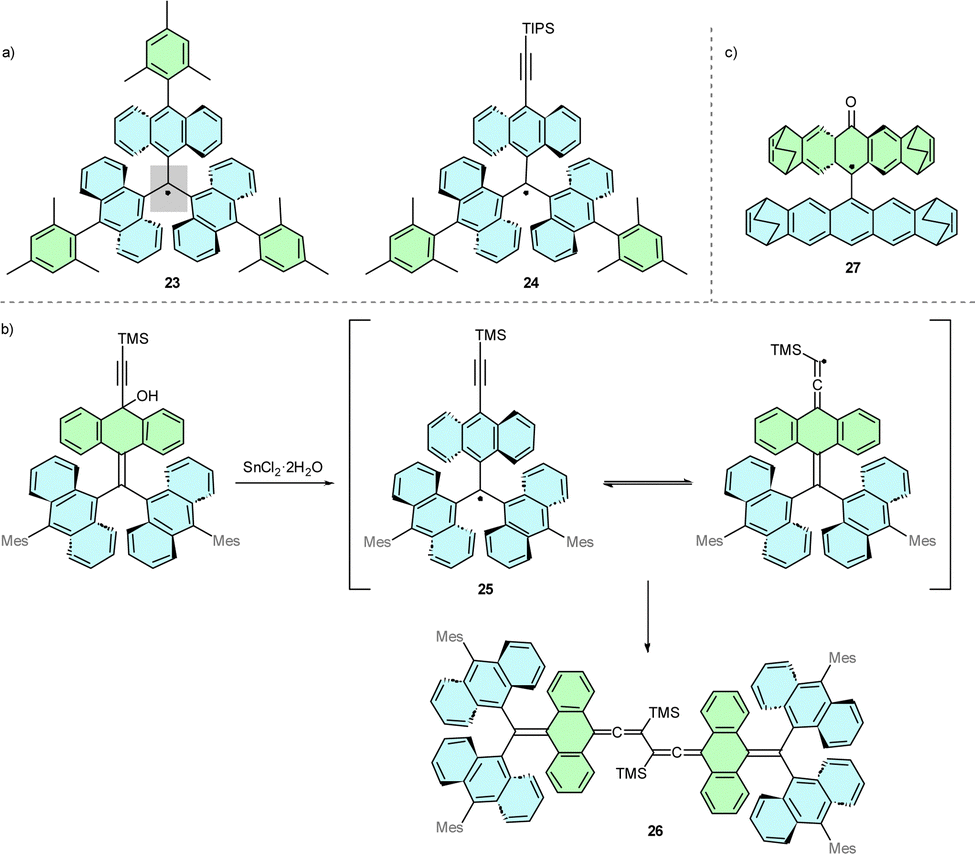 | ||
| Scheme 5 (a) Chemical structure of anthryl-based trityl radicals 23 and 24. Grey rectangle highlights the plane of the central carbon. (b) Decomposition of TMS-ethynyl-capped trityl radical 25 to form dimer 26. (c) Chemical strucure of anthryl-based trityl radical 27. | ||
9,9′-Anthryl-anthroxyl radical 27 exemplifies a geometric structure that affects the localisation of unpaired spin (Scheme 5c).26 While 27 may contain a phenoxyl radical structural motif, X-ray crystal structure and EPR measurements have confirmed that the highest spin density is localised on the α carbon, not on the oxygen atom. This localisation derives from orthogonal anthryl moieties with bulky peripheral bicyclo[2.2.2]octadiene (BCOD) groups that generate significant steric hindrance of the carbon-based radical centre.
Neutral radicals are usually electron deficient and, therefore, stabilized by electron-donating substituents. However, delocalization of the spin density over the vacant p-orbital of the trivalent boron atom also leads to stabilization caused by increased delocalization of the spin density.27 Combining neutral triphenylborane and the trityl radical (28) with subsequent planarization of the core yields derivative 29, which is stable under ambient conditions (Scheme 6).28 Spin delocalization on boron is confirmed by the values of hyperfine coupling constants assessed by EPR.
 | ||
| Scheme 6 Stabilization of the trityl radical by introducing electron deficient boron atom. Numbers in bold indicate the value of hyperfine coupling constants (EPR) for each related atom; the value for boron atom indicates significant spin delocalization on the atom. | ||
2.3. Triarylmethane dyes
Triphenylmethane-based cationic dyes (including xanthene- and acridine-based compounds, Chart 1) are widely used as staining agents in bioimaging29,30 and catalysts in photochemistry.31 The respective radicals are mainly observed as transient species formed by one-electron photoreduction.31 In turn, chemical reduction leads to the leuco (reduced, hydrogenated, formal adduct of a hydride anion to the central carbon) form of the dyes, which does not absorb visible light.32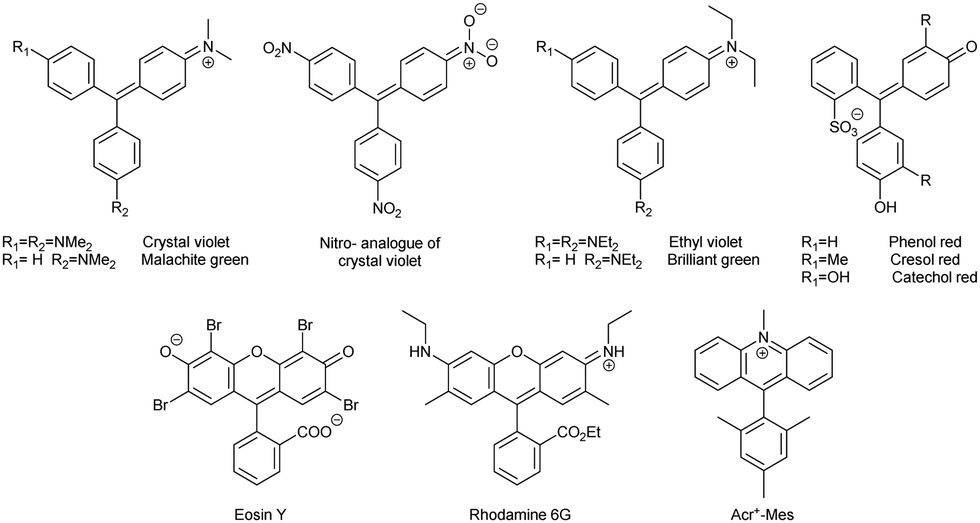 | ||
| Chart 1 Chemical structures of triphenylmethane dyes. | ||
Radicals of triarylmethane dyes have been used as persistent radicals in materials chemistry. For example, the radical of the electron rich crystal violet dye and its electron poor nitro-substituted analogue co-crystallise to form an organic semiconducting material. In this material, both components can be independently reversibly oxidized and reduced with a large potential difference of 0.52 V between the two species. In addition, magnetic susceptibility measurements in the solid state have revealed that the magnetic moment of nitro-substituted compound is close to that of the free electron, indicating minimal spin–spin interactions. By contrast, the magnetic moment of amino-substituted radical is 0 μB, suggesting loss of magnetic character and formation of π-dimers.33
2.4. Finland trityls
Advances in in vivo EPR imaging (EPRI) and the need for compatible spin labels have prompted studies of the trityl radicals with tetraoxy- and tetrathiaacetonide (30, 31, Finland trityl, Chart 2) modifications. In addition to their excellent persistence and redox stability, provided by the lone pairs on heteroatoms in ortho- and meta-positions, these radicals lack hydrogen atoms adjacent to the radical centre. Given the absence of signal-broadening coupling of an unpaired electron with adjacent hydrogen nuclear spins, the EPR signal of Finland trityls is sharper than that of parent trityls. Therefore, Finland trityls can be used as spin labels in in vivo systems.34 | ||
| Chart 2 Chemical structures of electron-rich Finland trityls. | ||
Adding dendritic substituents to the Finland trityl core in 32 increases the steric shielding and improves the stability of the radical in the presence of oxidants, such as ROS (hydroxyl, peroxyl radical, superoxide), Fe3+ and horseradish peroxidase, and reductants, such as Fe2+, glutathione and ascorbic acid. Such a high stability towards oxidants enables us to use 32 as extremely sensitive sensors for O2 concentration in tissues,35 monitoring the broadening of the EPR signal in the presence of oxygen.
2.5. Trityl dimerization
Initially, trityl radicals were predicted to reversibly dimerize, forming hexaphenylethane (Scheme 7a). Only later, with the development of 13C NMR analysis methods, was the trityl dimer identified as a “head-to-tail” (α,p) dimer, 33, also known as Gomberg's dimer. The steric hindrance of the ortho-hydrogen atoms plays a key role in the dimerization regioselectivity of trityls. Despite its partial dearomatization, trityl dimer 33 is more thermodynamically stable than the highly sterically hindered “head-to-head” (α,α) dimer 34.36 Reducing steric hindrance by partly or fully bridging the ortho-positions of phenyl rings (fluorene 3536 or triangulenes 36,37 respectively) facilitates α,α-dimerization (to 37 and 38, respectively) (Scheme 7b).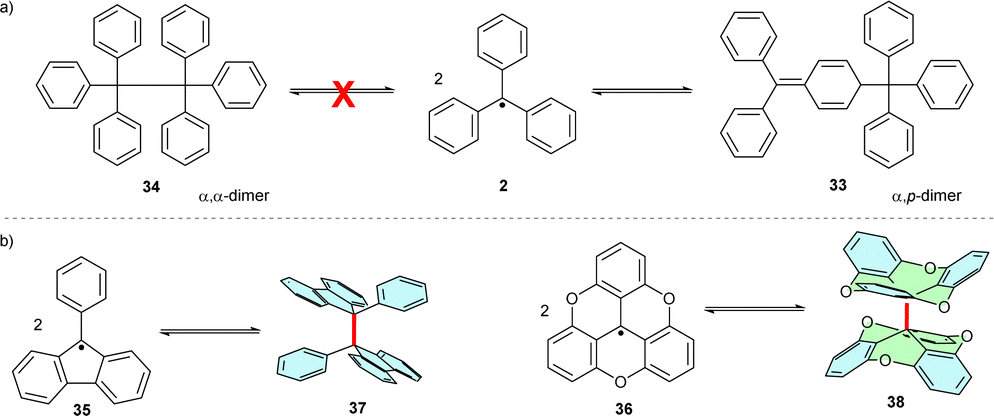 | ||
| Scheme 7 (a) Unsubstituted trityl radical dimerizes in “head-to-tail” (α,p) fashion. (b) Several examples of α,α-dimerization of partially or fully planarized trityl radicals. | ||
Yet, despite being much more sterically hindered, the fully meta-tert-butyl substituted trityl radical counterintuitively dimerizes in an α,α fashion and forms 39,38 while fully para-tert-butyl substituted trityl radical 40 remains monomeric (Fig. 2).39 This contradictory effect was explained by further studies, which showed that radical 2 dimerization is promoted by strong London dispersion (LD) interactions. The calculated energies, as well as the experimentally observed stability trends of variously meta-substituted hexaphenylethanes, directly correlated with the substituent-induced polarizability and, therefore, with the strength of LD interactions and the substituent size (Fig. 2, closed circles). When disregarding LD effects, all energy values were negative, and the trend reversed (Fig. 2, open circles).40 Therefore, London dispersion is essential for the α,α-dimerization mode, especially for bulky (spherical) substituents, such as tert-butyl and adamantyl.41
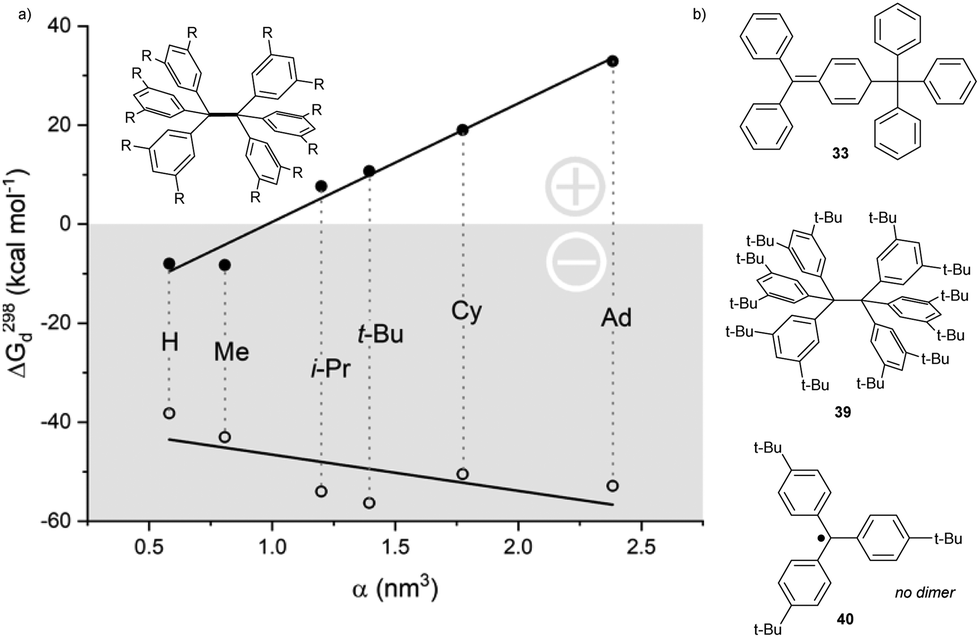 | ||
| Fig. 2 (a) Plot of computed free dissociation energies with (●) and without (○) dispersion corrections vs. alkyl group polarizability (i.e., its size and London dispersion). Experimental data support a significant contribution of LD forces to hexaphenylethane -type dimer stability. DFT calculation method used: B3LYP, Becke Johnson damped dispersion correction D3, hybrid GGA functional M06-2X, cc-pVDZ basis set. (b) Structures of dimerization products highlighting structural differences: classical Gomberg's dimer, LD-induced hexaphenylethane dimer and trityl monomer (which is not prone to dimerization). | ||
2.6. Biradicals
After the initial Gomberg's studies of the trityl radical, Thiele and Tschitschibabin reported trityl biradicals 41 linked through para-positions of phenyl rings (Fig. 3a).42,43 These series were further expanded by Müller hydrocarbons, bearing a longer conjugated phenylene linker. Because both radical centres are conjugated, these hydrocarbons can be drawn either as open-shell (OS) biradical or as closed-shell (CS) quinoid isomers. However, experimental and computational studies indicate that these molecules exist in a semi-quinoid form in the ground state. In this form, the molecules simultaneously display both quinoid and biradical properties.44 Nevertheless, the radical character is maintained, so Thiele's and Tschitschibabin's hydrocarbons are relatively unstable upon isolation and react with other radicals, e.g., triplet oxygen. Substituting the central biphenyl unit of Tschitschibabin's hydrocarbon for more π-delocalized bianthracene results in quinoidal structure 42, which can be synthesized as a pure closed-shell isomer 42-CS (Fig. 3b).45 | ||
| Fig. 3 (a) Chemical structures of Thiele's, Tschitschibabin's and Müller's hydrocarbons. (b) Synthesis of anthracene-based Tschitschibabin's hydrocarbon 42; OS= open shell; CS = closed shell. | ||
A different synthetic procedure (Fig. 3b, bottom right)45 yields 42 as the metastable biradical isomer 42-OS, which slowly decays to the more thermodynamically stable, EPR-silent 42-CS. The closed-shell structure is more stable because it contains two more Clar's sextets.45
2.7. Non-Kekulé compounds
Schlenk and Brauns connected two trityls through the meta-position of the aryl substituents, preventing full conjugation of the unpaired spins. The resulting bis(triphenylmethyl) biradicals 43 and 44 (Scheme 8a)46,47 are the first examples of non-Kekulé molecules that cannot be drawn as closed-shell structures without inducing severe steric strain. Therefore, 43 and 44 exist as diradicals in the ground state.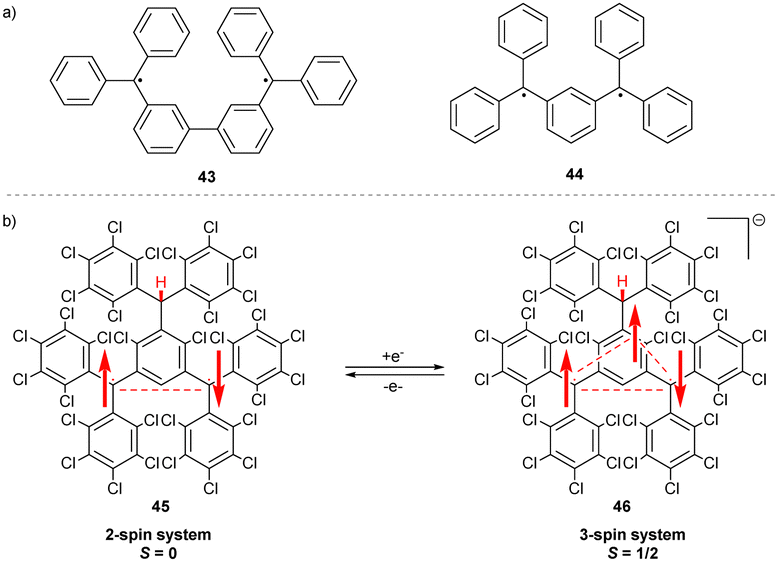 | ||
| Scheme 8 (a) Chemical structures of Schenk and Brauns hydrocarbons. (b) Structure and spin density distribution upon one-electron reduction of the neutral diradical 45. | ||
The trityl-based diradical 45 has a similar non-Kekulé structure (Scheme 8b) and shows a unique behaviour upon one-electron reduction to 46. Instead of forming a spin pair, the electron system of 46 is unsymmetrical, containing 3 unpaired electrons, with an overall multiplicity of S = 1/2.48
2.8. Hexaphenylethane switches
Linking two trityl radicals via the ortho-positions of their respective phenyl rings brings their α carbons close to each other, thereby promoting a spin–spin coupling reaction, forming a C–C bond. The product of such formal radical coupling is 9,9,10,10-tetraphenyl-9,10-dihydrophenanthrene, 47 (Fig. 4a), with an elongated C–C single bond between the sp3-hybridized carbon atoms. Connecting all 3 pairs of phenyl rings results in hexabenzo[4.4.4]propellane 48.49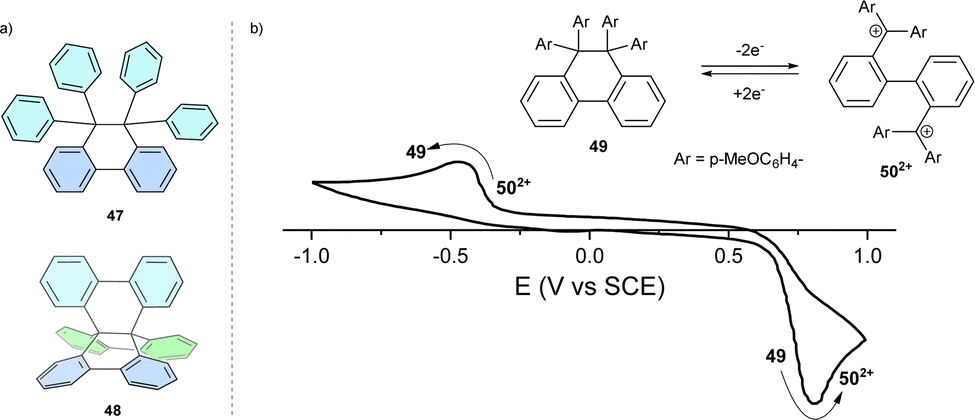 | ||
| Fig. 4 (a) Chemical structures of dihydrophenanthrene 47 and propellane 48. (b) Cyclic voltammogram of hexaphenylethane switch 49 in DCM. | ||
One-electron oxidation of 47 analogues lowers the activation energy of the elongated C–C bond cleavage. Consequently, the respective radical cations tend to undergo mesolytic (forming a radical and the respective cation) bond cleavage with higher rates than that of the homolytic bond cleavage of the neutral compound.50,51 Electron donating methoxy- and dimethylamino-substituted analogues of 47 have lower oxidation potentials, which further promote the oxidative cleavage of the elongated C–C bond whilst maintaining their stability to thermal homolysis.52 In symmetrically substituted hexaphenylethanes, both radical centres have the same chemical surroundings and, thus, the same oxidation potential, showing a two-electron oxidation wave in cyclic voltammetry. The rigidified isomer 48 is inert to both oxidative and reductive C–C bond cleavage.
Reversible two-electron oxidation and reduction of hexaphenylethanes, which leads to C–C bond cleavage and formation, respectively, is sometimes referred to as redox switching. Redox switching is accompanied by a marked change in geometry, which accounts for the hysteric redox behaviour (compounds 49 and 502+, Fig. 4b) manifested as the difference between the potential of the anodic and cathodic half-reaction monitored by cyclic voltammetry. The difference between these two potentials is termed the potential window and indicates a potential range wherein both neutral and dicationic forms can coexist. Thanks to the large structural reorganization between 49 and 502+, the all-para-methoxy-substituted analogue can reach a potential window as high as 1.5 V.
2.9. Alternative radical coupling modes of diradicals
In addition to α,α-coupling, observed in hexaphenylethane switches, two trityl radicals can also couple the central α-carbon with the ortho- and para-position of the second aryl ring. Calculations suggest that both α,p- and α,o-dimers are similarly sterically hindered, but only α,p-dimer is usually detected (Chart 3).53 α,o-dimerization can be observed only when both α,p- and α,α-dimerization is hindered. For example, substituting the hexaphenylethane 2,2′-biphenyl linking scaffold for naphthalene-1,8-diyl makes the resulting diradical more rigid and prevents α,p-coupling. Moreover, ring annulation at peri-positions of the naphthalene core in 51 induces angle strain and causes the ‘scissor effect’, elongating the distance between the radical centres and preventing the formation of the α,α-coupling product. The only possible coupling mode of 51 is thus α,o-coupling (Scheme 9, right pathway), but installing substituents next to ortho-positions suppresses even α,o-coupling. As a result, the reactivity of ortho-substituted analogue of 51 is restricted to intermolecular radical interactions, including H-abstraction and peroxide formation (Scheme 9, left and middle pathway).53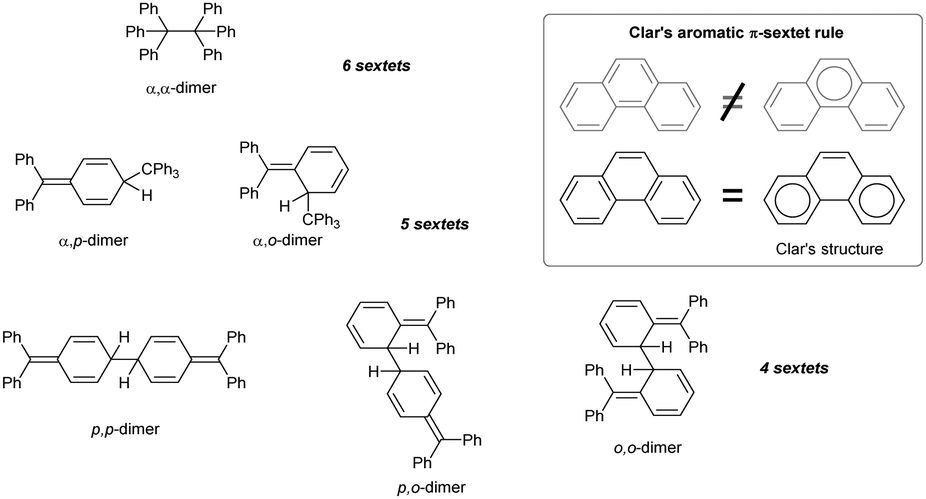 | ||
| Chart 3 Chemical structures of possible trityl dimers sorted by number of Clar's sextets. | ||
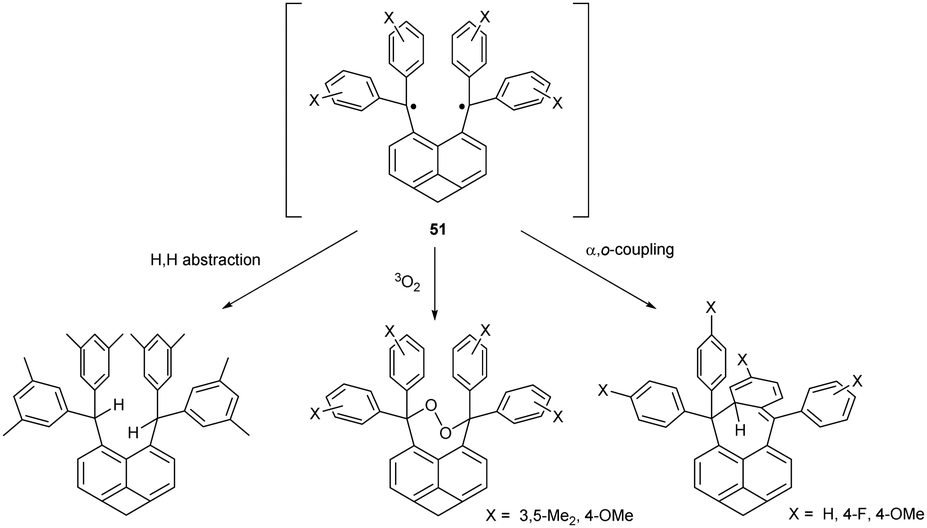 | ||
| Scheme 9 Chemical reactivity of the rigid diradical 51. | ||
Other modes of coupling, omitting the α-carbon position and proceeding exclusively through aryl substituents (p,p-, p,o- and o,o-dimerization, Chart 3) are much less energetically favourable because the resulting dimers have fewer Clar's sextets and are hence less aromatic than the corresponding α-dimers.54
3. Triarylamine radical cations
Triarylamine radical cations, derived from triphenylamine (TPA), are isoelectronic analogues of trityls. The main difference in their electronic structure is the positive charge at the nitrogen atom. Therefore, not only the spin density but also the positive charge (i.e., electron hole) must be delocalized and stabilized by the π-system.The stability of TPA-based radical cations can be conveniently studied by cyclic voltammetry (CV) of their reduced precursors – neutral amines – by monitoring the reversibility of the oxidation/reduction cycle and the appearance of new signals corresponding to side products. A fully reversible CV wave means that the radical cation generated by anodic oxidation at the working electrode does not decompose before being cathodically reduced back to the neutral amine. In fact, CV signal irreversibility is a sign of decomposition pathways and side reactions.
The parent TPA forms an unstable radical cation TPA˙+ upon anodic oxidation (Scheme 10). TPA instability mainly derives from increased spin density in ortho- and para-positions of the phenyl rings. The ortho-position is sterically hindered and thus less accessible, while the para-position is much more reactive. In addition, unlike in trityls where α,α-dimerization is the main radical coupling pathway, triarylamine radical cations do not α,α-dimerize, likely due to the positive charge at the nitrogen atoms, inducing Coulombic repulsion. Consequently, the main reaction pathway of triarylamine radical cations is intermolecular p–p coupling (σ-dimerization) and subsequent two-proton loss, forming benzidine-like species, such as N,N,N′,N′-tetraphenylbenzidine.55
 | ||
| Scheme 10 Dimerization mechanism of TPA˙+. | ||
3.1. Substituent effect
σ-Dimerization can be mitigated or prevented by blocking the para-position of the phenyl ring with a functional group. In addition to sterically blocking the reactive sites, the functional group strongly electronically affects the stability and reactivity of the radical cation. The first studies of mono para-substituted TPAs showed that the 4-methoxytriphenylamine radical cation, generated by electrolysis, is more stable than the parent TPA and that 4-nitrotriphenylamine is much less stable upon anodic oxidation.55 The variation of benzidine coupling rate constants of 4-substituted triarylamines as a function of Hammett substituent constants (σ+ or σ) shows that the corresponding radical cations, as expected, are stabilized by a strong EDG (Fig. 5, black dataset).56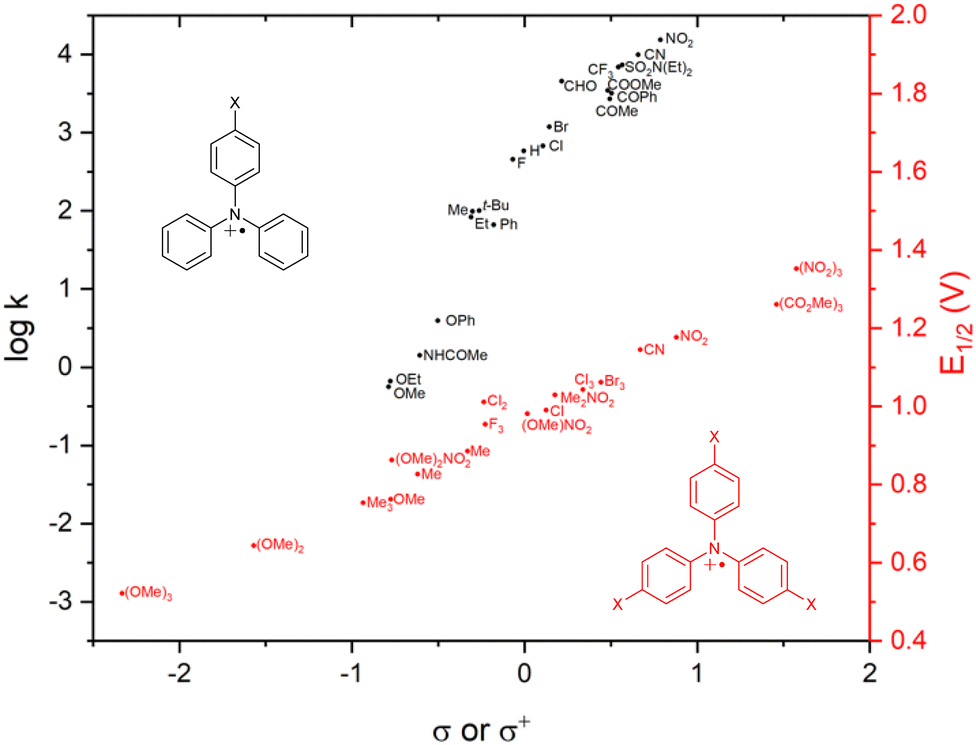 | ||
| Fig. 5 Plot of coupling rate constants (black) and first oxidation potentials (red) of para-substituted TPA˙+vs. Hammett substituent constants. | ||
Dimerization of TPA derivatives to benzidines has also been observed upon chemical oxidation with Cu(ClO4)2.57 Oxidative benzidine coupling of mono- and bis-para-substituted triarylamines proceeds with high chemical yields for easily oxidizable, electron-rich substrates but is mitigated or prevented when electron-withdrawing groups (EWGs) such as –NO2 and –CHO are present because the CuII salt cannot oxidise electron-poor substrates to form the corresponding radical cations.57,58
In classical radical chemistry, a radical can be stabilized by combining an EWG (‘’captive”) with an EDG (“dative”). This stabilization accounts for the wide range of resonance structures, delocalizing the spin density over multiple atoms and is known as the captodative effect. However, σ-dimerization studies of R1R2PhN˙+ with different EDG (para-anisyl-) and EWG (para-nitrophenyl-) combinations have shown that the benzidine coupling rate constant of the ‘captodative-substituted’ analogue (p-MeOC6H4)(p-NO2C6H4)PhN˙+ is significantly higher than that of both mono- and bis- anisyl-substituted analogues. The higher coupling rate and hence lower stability of the ‘captodative-substituted’ analogue demonstrates the lack of a strong captodative effect on TPA derivatives.59
Fully para-substituted triphenylamines form stable radical cations, as shown by their fully reversible oxidation/reduction CV signals.59 Installing different EDG and EWG combinations as para-aryl substituents finetunes the electrochemical60 (Fig. 5, red dataset) and spectral61 (Fig. 6) properties of TPA radicals. For this reason, triphenylamine-based radical cations are amongst the most commonly used organic oxidants.
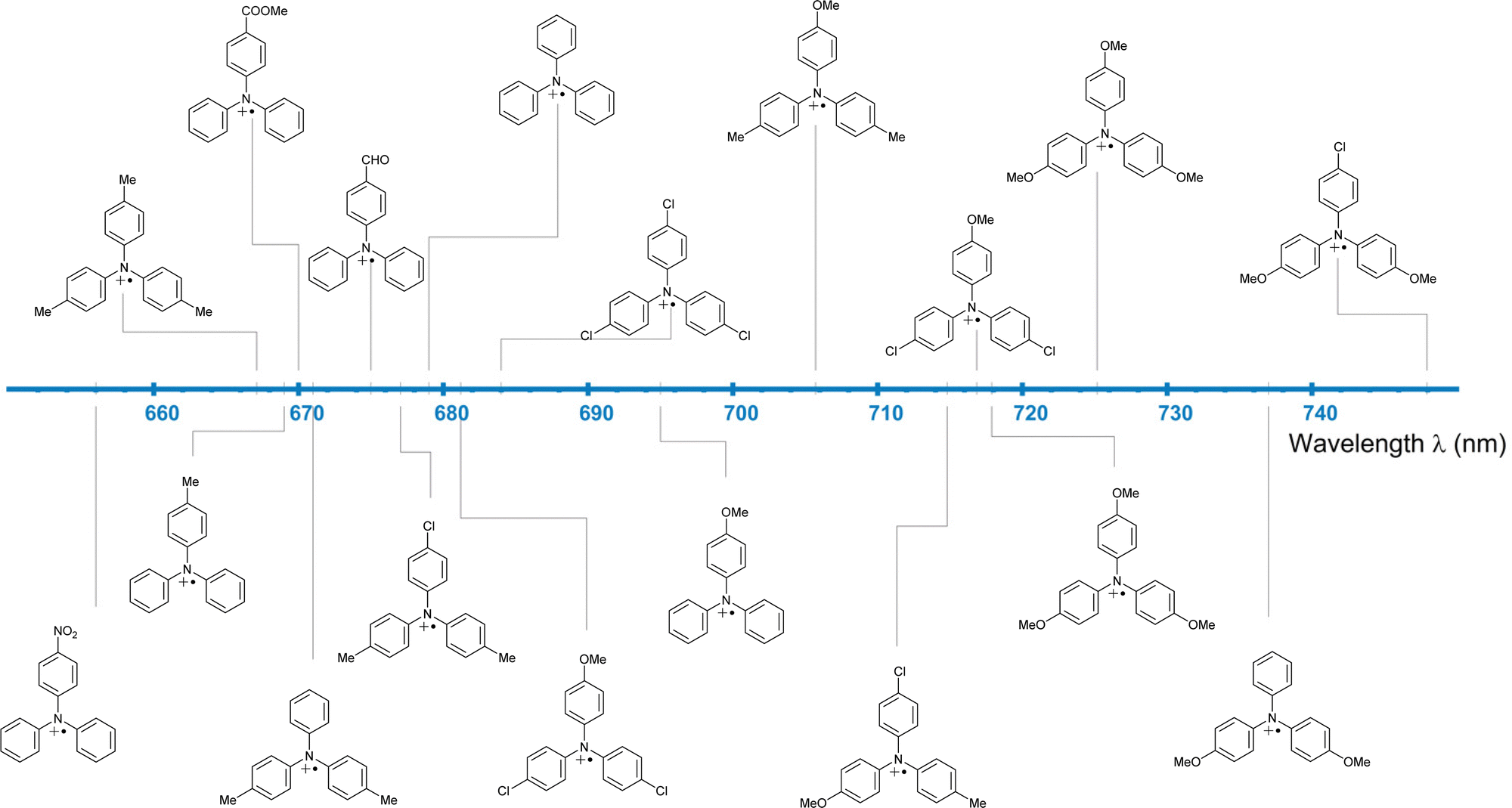 | ||
| Fig. 6 The absorption maxima of p-substituted TPA˙+ in MeCN. | ||
Tris(4-bromophenyl)ammoniumyl hexachloroantimonate, commonly known as “magic blue” (MB; Chart 4), is a widely used and commercially available single-electron oxidant. MB is reasonably stable but known to slowly decompose upon storage in solid state and to abstract hydrogen atoms from organic solvents, decomposing to the corresponding triarylamine.62 In a study of aged commercial MB samples, the tetrakis-(4-bromophenyl)benzidine (TPB) radical cation and its dication were the main decomposition products and thus termed “blues brothers”.63 The instability of MB was explained by the weak C–Br bond in the para-position that cleaves and enables benzidine coupling. As an alternative, the authors proposed “blues cousin”(BC) with bulky, non-labile tert-butyl groups in para-positions and Br atoms in ortho-positions. These structural changes aimed at preventing intermolecular coupling while maintaining the similar oxidation potential (0.70 V for MB and 0.78 V vs. Fc/Fc+ for BC).
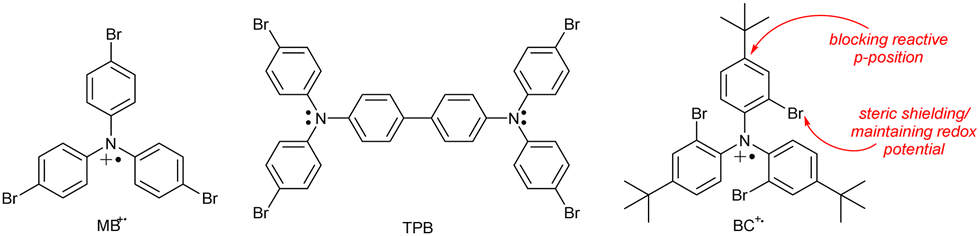 | ||
| Chart 4 Chemical structures of “magic blue”, its dimer TPB (one of the “blues brothers”) and a proposed, more stable alternative “blues cousin”. | ||
3.2. N-Aryl heterocycles
The TPA structural motif is also found in some N-containing heterocycles. Compounds such as N-phenyl phenoxazines, phenothiazines, acridones can be regarded as TPAs connected at ortho-positions of two phenyl rings via O, S atoms or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O linking group, respectively, or linked directly by a σ-bond in carbazoles (Fig. 7a).
O linking group, respectively, or linked directly by a σ-bond in carbazoles (Fig. 7a).
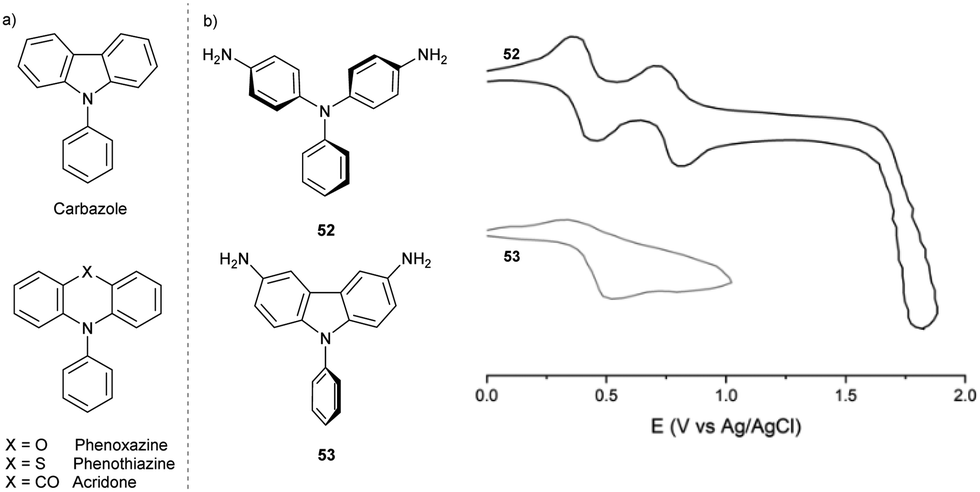 | ||
| Fig. 7 (a) Chemical structures of N-heterocycles with a TPA core. (b) Cyclic voltammograms of 3,6-diaminosubstituted N-phenyl carbazole 53 (bottom) and the respective TPA 52 (top, measured in MeCN containing 0.1 M TBAP, scan rate = 0.1 V s−1). | ||
In contrast to propeller-shaped TPA, which has moderately twisted phenyl rings (47° twist angle in TPA˙+),64 carbazole core planarization forces the N-aryl ring out of the carbazole π-system plane, further twisting the ring (78.4° twist angle in neutral 9-phenylcarbazole;65 no data are available for the respective radical cation) and decreasing the conjugation of the N-aryl substituent with the rest of the molecule (Fig. 7b). This structural change explains the weaker effect of N-phenyl substituents on the properties of the radical cation, such as the absorption maximum or the half-wave potential.66 Furthermore, due to the limited delocalization of the spin density, the carbazole radical cation does not undergo dimerization at the para-positions of the N-phenyl ring.
The difference in stability between TPA and the carbazole radical cation is evident when comparing their CV spectra (Fig. 7b).66 Carbazoles are harder to oxidise than the respective TPAs; their first oxidation potential is more positive by ∼0.3 V.67 In compounds containing more nitrogen atoms, several oxidation waves are expected; the diamino-substituted TPA 52 shows two reversible oxidation waves (leading to the radical cation and the dication), while the analogous carbazole 53 undergoes irreversible oxidation. These outcomes suggest decomposition associated with the anodic oxidation of carbazoles.
In N-heterocycles, such as phenoxazine, phenothiazine and dihydrophenazine (Fig. 7a), conjugation between the N-aryl substituent and the heterocyclic ring is blocked, as in carbazoles. The twist angle between these aromatic moieties is close to 90° which implies that no electron delocalization occurs on the N-aryl. However, electron-rich heteroatoms (O, S, N) used as a linker further stabilize the molecule because their lone pairs stabilize the radical cation. Generally, delocalization on an electronegative heteroatom is more efficient than over a π-system, so the loss of one conjugated phenyl ring is fully compensated for by O or S atoms, which is why these N-heterocyclic radical cations are typically very stable. For their additional stability, N-heterocyclic structural motifs are often installed in OLED materials68,69 and used in photoredox catalysis.31,70,71
3.3. Heterohelicenes
Unlike planar N-containing heterocycles, analogues with two bridges are helical and, thus, may be regarded as electroactive heterohelicenes 54–56 (Chart 5). Carbonyl-bridged amines were studied by cyclic voltammetry, showing quasi-reversible oxidation peaks indicating the high stability of the corresponding radical cations.72 The lack of their dimerization and other side reactions was explained by spin delocalization over the π-system, including the carbonyl groups, and by the bulky tert-butyl substituent, which blocks the central para-position of the phenyl ring. Even higher stabilization was shown in sulphur bridges of fused bis-phenothiazines 57 and 58, which are stable enough to be isolated as a SbF6− salt and characterised by X-ray analysis.73 In addition to stabilizing the radical cation, these structures have a high isomerization barrier and are rare examples of configurationally stable [4]helicenes.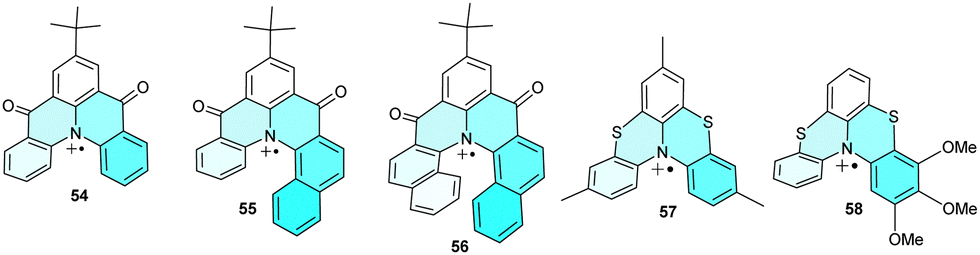 | ||
| Chart 5 Chemical structures of helical-shaped TPAs. | ||
3.4. Planarization/N-heterotriangulenes
The TPA radical cation can be stabilized by introducing three bridges linking all phenyl ortho-positions, thus planarizing the molecule and ensuring spin and charge delocalization over the π-system. Due to spin delocalization, the oxidation potential is lower than in similarly substituted propeller-shaped analogues.74,75 A planar (gem-dimethyl)methylene-bridged radical cation 59 (Scheme 11a) was isolated as a salt of aluminium-based anion [Al(OC(CF3)3)4] and characterised by EPR, UV-Vis and X-ray crystallography. However, methylene bridges alone do not fully stabilize the radical cation. Therefore, the radical cation dimerizes through para positions to the σ-dimer, dication 60, with a strong diradical character.76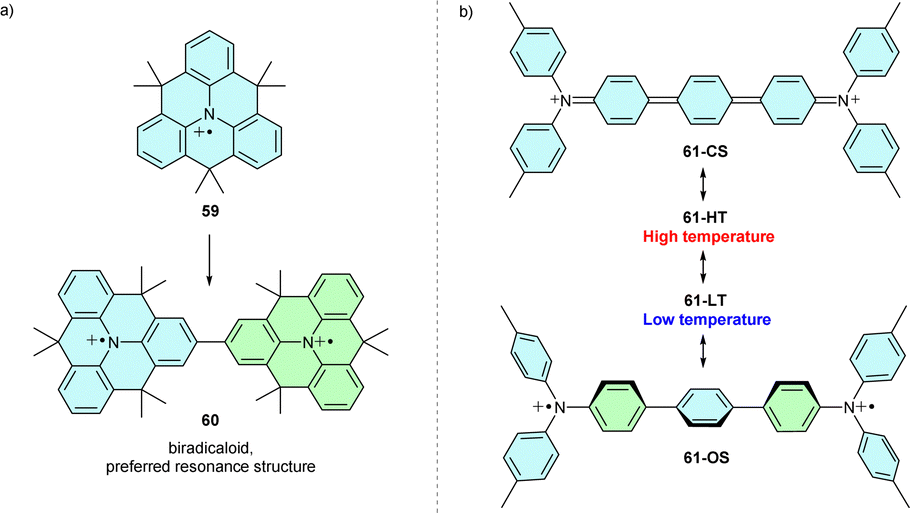 | ||
| Scheme 11 (a) Dimerization of methylene-bridged triangulene 59. (b) Temperature-dependent equilibrium between two singlet states of N-centred Müller hydrocarbon 61. | ||
Planarization of triarylamine derivatives can also be achieved using a different strategy. Connecting two triarylamine to N-centred Müller hydrocarbon analogue 61 can lead to a planar closed-shell structure such as 61-CS (Scheme 11b). However, due to steric hindrance and conformational flexibility, 61 exists in two different singlet states, 61-HT and 61-LT, with a greater open-shell (OS) character at lower temperatures (Scheme 11b).77
Substituting carbon-based bridges for heteroatoms has a dual impact, enabling conjugation with both the π-system and the heteroatom lone pair. Partial or full substitution of methylene bridges for oxygen or sulphur, that is, installing the phenoheteroazine motif (62a–b), prevents radical cation decomposition. In particular, the ether-bridged analogue 62a is a highly stable compound, showing no signs of dimerization or oxygenation (Chart 6).78
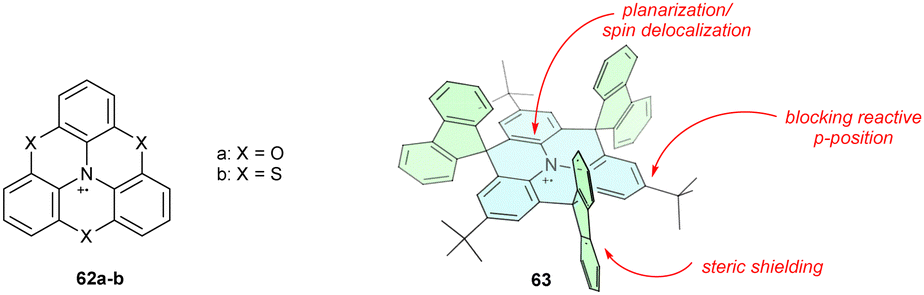 | ||
| Chart 6 Chemical structures of planar triarylamine radical cations. | ||
Planarization can be combined with steric shielding of the heterotriangulene plane and its para-positions to design exceptionally stable triarylamine radical cations. Protecting para-positions sensitive to σ-dimerization with a tert-butyl group and heterotriangulene with sterically demanding fluorenylidene bridges efficiently shields the triarylamine core, yielding 63.75 Because 63 lacks other heteroatoms, the oxidation potential remains relatively high (0.38 V vs. Fc/Fc+).
3.5. Photoreactivity
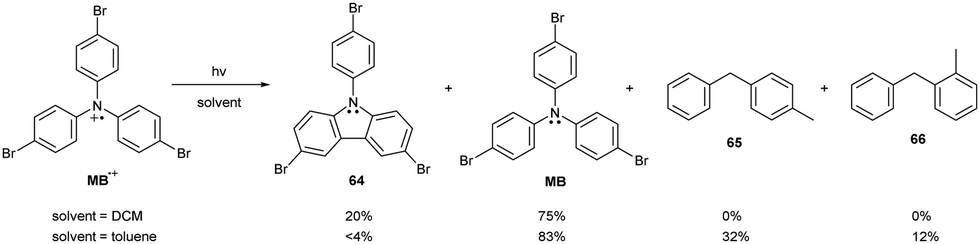
TPA-based radical cations mainly undergo thermal decomposition, but they can also undergo photochemical reactions. The most notable side reaction of TPA is photocyclization of its dications formed by disproportionation, yielding N-phenyl carbazoles and neutral amines.79 In the absence of oxygen, photolysis of bromo- and methoxy-trisubstituted triphenylamine radical cations in DCM generates the corresponding carbazoles and neutral amines in an approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio (Scheme 12). This deviation from the expected 1:1 product ratio suggests that the radical cation undergoes an additional, direct reduction by the solvent. Direct reduction of the radical cation most likely proceeds via hydrogen abstraction. This hypothesis was tested by replacing the rather inert DCM by toluene, which is a better hydrogen atom donor (Scheme 12). Adding toluene suppressed the carbazole 64 formation pathway, yielding not only the neutral TPA derivative MB but also toluene dimerization products 65 and 66. These findings confirm that excited TPA-based radical cations have excellent hydrogen-abstracting properties.
:3 ratio (Scheme 12). This deviation from the expected 1:1 product ratio suggests that the radical cation undergoes an additional, direct reduction by the solvent. Direct reduction of the radical cation most likely proceeds via hydrogen abstraction. This hypothesis was tested by replacing the rather inert DCM by toluene, which is a better hydrogen atom donor (Scheme 12). Adding toluene suppressed the carbazole 64 formation pathway, yielding not only the neutral TPA derivative MB but also toluene dimerization products 65 and 66. These findings confirm that excited TPA-based radical cations have excellent hydrogen-abstracting properties.
 | ||
| Scheme 12 Photoirradiation example of para-substituted TPA radical cationic compounds and conversion of the products. | ||
3.6. π-Dimerization
Introducing the TPA motif into organic polycyclic compounds leads to structurally complex radical cations with unusual geometry and properties. On the one hand, nitrogen-embedded π-extended corannulene 67 is structurally analogous to both TPA and pyrrole and has a bowl-shaped corannulene-like structure (Fig. 8a). At room temperature, the corresponding radical cation exists as a monomer in solution.80 At lower temperatures (−80 °C), reversible σ-dimerization occurs through the carbon adjacent to the nitrogen. On the other hand, the less strained, planar analogue 68 forms a π-dimer 69. The reason for this diverse behaviour of 67 and 68 was explained based on DFT calculations of the electronic and geometric structure of both compounds, finding that σ-dimer 70 formed through strain relief as the connecting carbon atoms of the curved corannulene 67 adopt tetrahedral sp3 hybridisation instead of the initial trigonal sp2. Conversely, σ-dimerization would increase the strain in planar radical cations 68.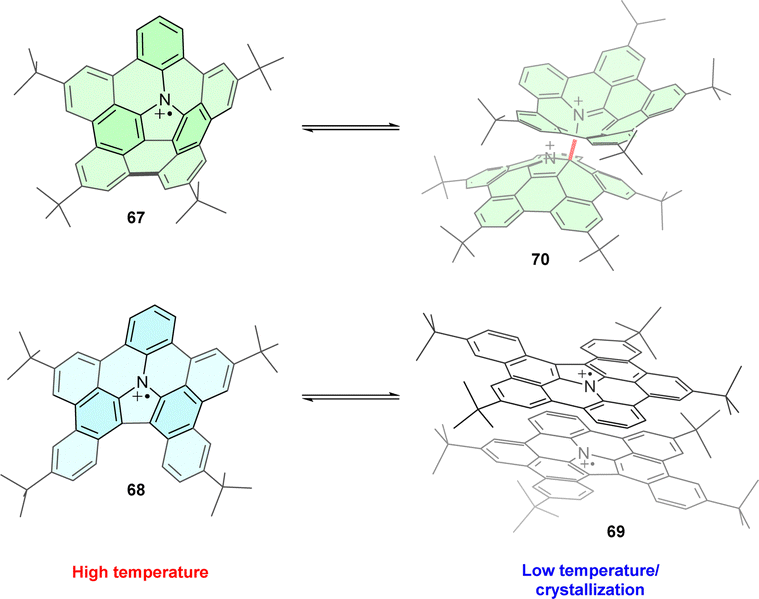 | ||
| Fig. 8 (a) Different dimerization modes of triphenylamine-based corannulene 67 and its planar analogue 68. | ||
3.7. Hypervalent compounds
An alternative approach to stabilization consists of using Lewis basic functional groups near the radical cation centre. This strategy is demonstrated in the design of hypervalent pentacoordinate nitrogen radical cations 71 (Chart 7). X-ray structure analysis and DFT calculations have revealed that the nitrogen centre adopts a trigonal bipyramidal geometry, with weak O–N–O 3-center 5-electron bonding to oxidised nitrogen and electron-rich carbonyl oxygen atoms.81 These compounds are air-stable but decompose upon irradiation with light.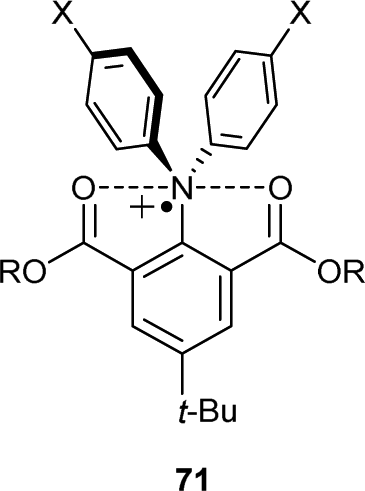 | ||
| Chart 7 Chemical structure of pentacoordinate nitrogen complex 71. | ||
4. Trityl-TPA dyads
Combining trityl and triarylamine centres within one π-system forms molecules benefiting from the properties of both trityl radicals and TPA radical cations. The charged trityl-carbazole dyads have been designed as novel push–pull organic semiconductors with ambipolar properties, e.g., with the ability to transport both electrons and holes (Chart 8).82,83 Trityl-TPA dyad 72 was synthesized by coupling the electron-poor TTM radical with the electron-rich carbazole molecule.22,84 In cyclohexane and chloroform solutions, dyad 72 is thermally stable and has a bathochromically shifted absorption maximum in comparison to the parent trityl radical. This bathochromic shift suggests increased delocalization of an unpaired electron over the molecule.84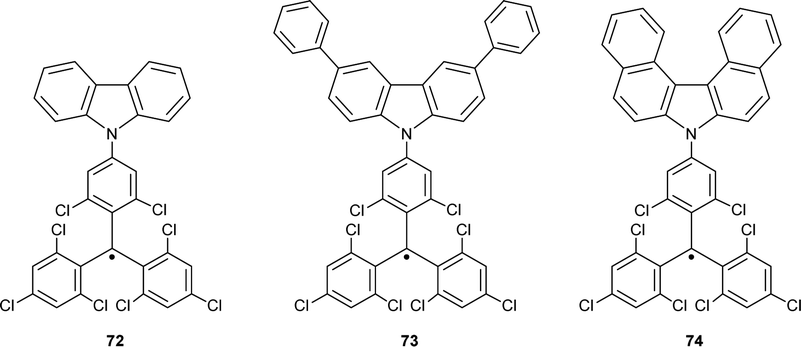 | ||
| Chart 8 Chemical structures of TTM-carbazole dyads. | ||
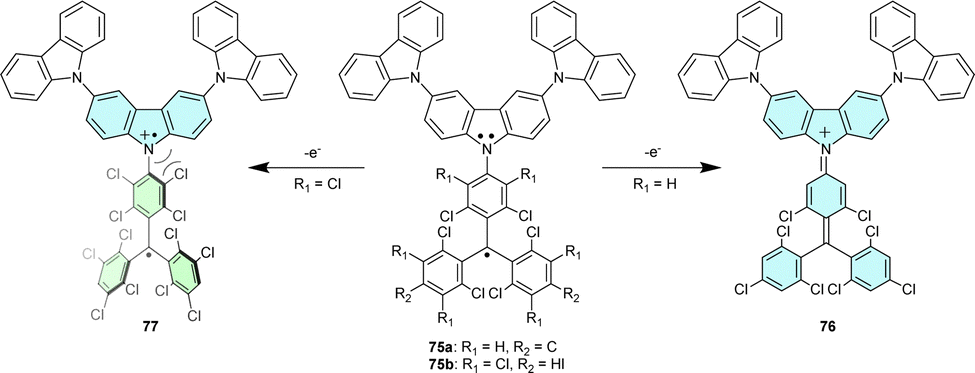
Analogous dyads 73 and 74 with substituents in 3- and 6-positions of the carbazole are highly thermally stable in the solid state and only decompose upon melting (>260 °C).22 Additionally, blocking 3- and 6-positions of the carbazolyl moiety by installing of phenyl groups in 73 also increases its photostability (Scheme 13).85 Unlike 72, with large electron density delocalization between the trityl and TPA moieties, dyads 73 and 74 have EPR parameters (such as g-factor and hyperfine coupling constants) similar to those of the parent TTM radical, thus indicating that the carbazole moiety does not affect the SOMO distribution.
 | ||
| Scheme 13 Oxidation of trityl-carbazole dyads. | ||
In addition to their ambipolar properties, trityl-TPA dyads are also excellent models for studying electron transfer processes because they are stable, neutral and, thus, highly soluble, even in non-polar solvents.86 Two different neutral 3,6-bis(N-carbazolyl)carbazole-substituted chlorinated trityl radicals 75 (Scheme 13) show the same charge distribution and behaviour upon reduction to the corresponding trityl anion. But the oxidation of these dyads is more complex, yielding different products. In 75a, the radical character is completely lost when planar cation 76 is generated. In turn, steric hindrance caused by two chlorine atoms ortho- to carbazolic nitrogen in 75b prevents planarization, and both radical centres remain in cation 77,83 exemplifying the structure-controlled electron configuration of organic molecules. Even a minor change in substitution can have a large impact on the overall multiplicity of the system, leading to different electron configuration isomers.
In these systems, the isomerism of the electron configuration can be even higher. Carbocation 78, a one-electron oxidation product of 72 (Scheme 14), undergoes photoinduced electron transfer from the carbazole nitrogen donor to the trivalent carbon acceptor, yielding transient diradical cation species 79. This intervalence charge transfer (IVCT) involves two redox-active centres in different oxidation states, namely neutral nitrogen and an oxidised carbon.87 As such, these molecules are one of the rare examples of intervalence isomerism of fully organic molecules.
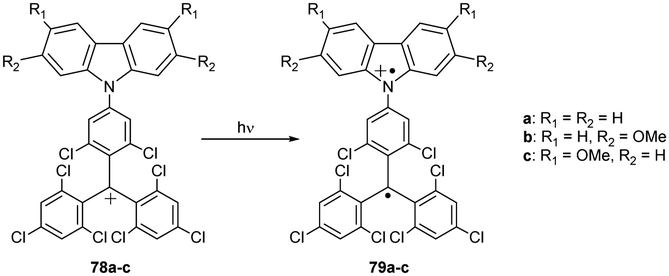 | ||
| Scheme 14 Photoexcitation of the carbazolyl-substituted PTM cation 78. | ||
5. Conclusions
Despite being isoelectronic, triarylamine radical cations and trityl radicals considerably differ in their properties, stability, and reactivity. Table 1 summarises key differences in their structure, absorption and redox properties, and stability.| Trityl˙ | TPA˙+ | |
|---|---|---|
| a In benzene. b In MeCN. c Ph3C˙, electron diffraction. d PTM˙, X-ray. e TPA˙+, B3LYP/6-31++G(d,p). f -CMe2- bridged TPA˙+, X-ray. | ||
| Charge | 0 | 1+ |
| λ max (nm) | 515a,88 | 679b,61 |
| E 0/1+, V vs. SCE | 0.27b,89 | 0.92b,55 |
| Length of X˙–C bond (XC˙, N˙+), Å |
1.48c,90 | 1.415e,92 |
| 1.483–1.486d,91 | 1.419–1.440f,76 | |
| Main destabilizing factor | Unpaired spin | Electron hole (cation) |
| Origin of stability | Kinetic persistence | Thermodynamic stabilization |
| Structural changes | Steric shielding | Planarization |
| Spin density distribution | Mainly localised on central carbon | Delocalized over the molecule |
| Geometry of XAr3 core | Propeller | Planar |
| Substituents | EWG, EDG; bulky halides | EDG; electron-rich heteroatoms |
| Dimerization site | α,p if p-blocked: no dimer bulky meta-Alk, preorganised: α,α | p,p if p-blocked: no dimer |
| Carbazole/fluorene cyclisation | hν/Δ | hν |
| Main application focus | Open-shell structure | Single electron oxidant |
Triphenyl amine and trityl radicals have an unpaired spin, which can be stabilized using two completely orthogonal strategies. The main difference between trityl radicals and the triphenylamine radical cations lies in the positive charge at the nitrogen in TPAs. This positive charge requires additional stabilization. In trityls, persistent barriers are used to capture the spin inaccessible for side-reactivity. By contrast, in triarylamine radical cations, both the spin and charge density tend to delocalize over the whole π-system decorated with EDGs.
The difference in charge is one of the key properties that determine the applications of TPA and trityl radicals. Over the past twenty years, the scope of applications of triphenylamine- and trityl-based radicals have expanded considerably, particularly thanks to chlorinated trityl radicals. These persistent, open-shell species are suitable for further derivatisation. Yet, the structural diversity of these compounds remains limited6 because they experience partial dehalogenation, which, nevertheless, can be solved by introducing the steric bulk.6,26 Other emerging research areas focus on kinetically-protected sulphur-embedded triphenylmethyl radicals with sharp EPR output signals suitable for spin imaging93 and on hexaphenylethane-based molecular redox switches, which leverage trityl dimerization to selectively form and mesolytically cleave a C–C bond.53,94
Triphenylamine radical cations, in turn, are typically used as single-electron oxidants and OLED materials95 and in combination with trityls in dyads with promising luminescent and semiconducting properties.96 While promising, the scope of these dyads is structurally limited to substituted carbazoles and leaves room for screening other TPA-based ED moieties, such as N-containing heterotriangulenes.
While efficient strategies have been developed to enhance the stability and persistence of C- and N-based triaryl radicals and some biradicals, research on polyradicals, dyads and structurally complex open-shell molecules with trityl/triphenylamine fragments is still incipient. Applying design principles summarized in this highlight, future studies must be conducted to develop stabilized and persistent polyradicals with singlet, triplet, or higher spin states and controlled valence tautomerism specifically designed for applications in materials science.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Czech Science Foundation (Project No. 19-20467Y). The authors thank Carlos V. Melo for editing the manuscript.References
- T. Quintes, M. Mayländer and S. Richert, Nat. Rev. Chem., 2023, 7, 75–90 CrossRef PubMed.
- M. M. Haugland, J. E. Lovett and E. A. Anderson, Chem. Soc. Rev., 2018, 47, 668–680 RSC.
- L. Chen, M. Arnold, R. Blinder, F. Jelezko and A. J. C. Kuehne, RSC Adv., 2021, 11, 27653–27658 RSC.
- S. S. V. Sowndarya, P. C. St John and R. S. Paton, Chem. Sci., 2021, 12, 13158–13166 RSC.
- K. Kato and A. Osuka, Angew. Chem., 2019, 131, 9074–9082 CrossRef.
- I. Ratera, J. Vidal-Gancedo, D. Maspoch, S. T. Bromley, N. Crivillers and M. Mas-Torrent, J. Mater. Chem. C, 2021, 9, 10610–10623 RSC.
- J. Hioe and H. Zipse, Org. Biomol. Chem., 2010, 8, 3609–3617 RSC.
- Z. X. Chen, Y. Li and F. Huang, Chem, 2021, 7, 288–332 CAS.
- S. Kataoka, S. Suzuki, Y. Shiota, K. Yoshizawa, T. Matsumoto, M. S. Asano, T. Yoshihara, C. Kitamura and S. Kato, J. Org. Chem., 2021, 86, 12559–12568 CrossRef CAS.
- A. M. Heuer, S. C. Coste, G. Singh, B. Q. Mercado and J. M. Mayer, J. Org. Chem., 2023, 88, 9893–9901 CrossRef CAS.
- M. Gomberg, J. Am. Chem. Soc., 1900, 22, 757–771 CrossRef.
- C. Glidewell, D. C. Liles, D. J. Walton and G. M. Sheldrick, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1979, 35, 500–502 CrossRef.
- H. Wieland, Ber. Dtsch. Chem. Ges., 1911, 44, 2550–2556 CrossRef CAS.
- G. A. DiLabio, K. U. Ingold, S. Lin, G. Litwinienko, O. Mozenson, P. Mulder and T. T. Tidwell, Angew. Chem., Int. Ed., 2010, 49, 5982–5985 CrossRef CAS.
- S.-H. Jang, P. Gopalan, J. E. Jackson and B. Kahr, Angew. Chem., Int. Ed. Engl., 1994, 33, 775–777 CrossRef.
- I. Ratera and J. Veciana, Chem. Soc. Rev., 2012, 41, 303–349 RSC.
- M. Ballester, J. Castaner, J. Riera, J. Pujadas, O. Armet, C. Onrubia and J. A. Rio, J. Org. Chem., 1984, 49, 770–778 CrossRef CAS.
- M. Ballester, I. Pascual, C. Carreras and J. Vidal-Gancedo, J. Am. Chem. Soc., 1994, 116, 4205–4210 CrossRef CAS.
- M. Ballester, J. Riera, J. Castaner, C. Rovira, J. Veciana and C. Onrubia, J. Org. Chem., 1983, 48, 3716–3720 CrossRef CAS.
- N. Roques, D. Maspoch, N. Domingo, D. Ruiz-Molina, K. Wurst, J. Tejada, C. Rovira and J. Veciana, Chem. Commun., 2005, 4801–4803 RSC.
- P. Demay-Drouhard, H. Y. V. Ching, C. Decroos, R. Guillot, Y. Li, L. C. Tabares, C. Policar, H. C. Bertrand and S. Un, Phys. Chem. Chem. Phys., 2020, 22, 20792–20800 RSC.
- D. Velasco, S. Castellanos, M. López, F. López-Calahorra, E. Brillas and L. Juliá, J. Org. Chem., 2007, 72, 7523–7532 CrossRef CAS PubMed.
- P. Mayorga-Burrezo, V. G. Jiménez, D. Blasi, T. Parella, I. Ratera, A. G. Campaña and J. Veciana, Chem. – Eur. J., 2020, 26, 3776–3781 CrossRef CAS.
- T. Nishiuchi, S. Aibara and T. Kubo, Angew. Chem., Int. Ed., 2018, 57, 16516–16519 CrossRef CAS.
- T. Nishiuchi, D. Ishii, S. Aibara, H. Sato and T. Kubo, Chem. Commun., 2022, 58, 3306–3309 RSC.
- T. Aotake, M. Suzuki, N. Aratani, J. Yuasa, D. Kuzuhara, H. Hayashi, H. Nakano, T. Kawai, J. Wu and H. Yamada, Chem. Commun., 2015, 51, 6734–6737 RSC.
- C.-W. Chiu and F. P. Gabbaï, Angew. Chem., Int. Ed., 2007, 46, 1723–1725 CrossRef CAS PubMed.
- T. Kushida, S. Shirai, N. Ando, T. Okamoto, H. Ishii, H. Matsui, M. Yamagishi, T. Uemura, J. Tsurumi, S. Watanabe, J. Takeya and S. Yamaguchi, J. Am. Chem. Soc., 2017, 139, 14336–14339 CrossRef CAS.
- R. W. Sabnis, Handbook of Biological Dyes and Stains: Synthesis and Industrial Applications, John Wiley & Sons, 2010 Search PubMed.
- A. N. Butkevich, M. L. Bossi, G. Lukinavičius and S. W. Hell, J. Am. Chem. Soc., 2019, 141, 981–989 CrossRef CAS.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- R. Muthyala, Chemistry and Applications of Leuco Dyes, Springer Science & Business Media, 2006 Search PubMed.
- W. W. Porter and T. P. Vaid, J. Mater. Chem., 2007, 17, 469–475 RSC.
- T. J. Reddy, T. Iwama, H. J. Halpern and V. H. Rawal, J. Org. Chem., 2002, 67, 4635–4639 CrossRef CAS PubMed.
- Y. Song, Y. Liu, C. Hemann, F. A. Villamena and J. L. Zweier, J. Org. Chem., 2013, 78, 1371–1376 CrossRef CAS PubMed.
- H. A. Staab, H. Brettschneider and H. Brunner, Chem. Ber., 1970, 103, 1101–1106 CrossRef CAS.
- E. Müller, A. Moosmayer, A. Rieker and K. Scheffler, Tetrahedron Lett., 1967, 8, 3877–3880 CrossRef.
- B. Kahr, D. Van Engen and K. Mislow, J. Am. Chem. Soc., 1986, 108, 8305–8307 CrossRef CAS.
- S. Grimme and P. R. Schreiner, Angew. Chem., Int. Ed., 2011, 50, 12639–12642 CrossRef CAS PubMed.
- S. Rösel, C. Balestrieri and P. R. Schreiner, Chem. Sci., 2016, 8, 405–410 RSC.
- S. Rösel, J. Becker, W. D. Allen and P. R. Schreiner, J. Am. Chem. Soc., 2018, 140, 14421–14432 CrossRef PubMed.
- J. Thiele and H. Balhorn, Ber. Dtsch. Chem. Ges., 1904, 37, 1463–1470 CrossRef.
- A. E. Tschitschibabin, Ber. Dtsch. Chem. Ges., 1907, 40, 1810–1819 CrossRef CAS.
- P. Ravat and M. Baumgarten, Phys. Chem. Chem. Phys., 2014, 17, 983–991 RSC.
- Z. Zeng, Y. M. Sung, N. Bao, D. Tan, R. Lee, J. L. Zafra, B. S. Lee, M. Ishida, J. Ding, J. T. López Navarrete, Y. Li, W. Zeng, D. Kim, K.-W. Huang, R. D. Webster, J. Casado and J. Wu, J. Am. Chem. Soc., 2012, 134, 14513–14525 CrossRef CAS PubMed.
- W. Schlenk and M. Brauns, Ber. Dtsch. Chem. Ges., 1915, 48, 661–669 CrossRef CAS.
- W. Schlenk and M. Brauns, Ber. Dtsch. Chem. Ges., 1915, 48, 716–728 CrossRef CAS.
- R. Gaudenzi, J. de Bruijckere, D. Reta, I. de, P. R. Moreira, C. Rovira, J. Veciana, H. S. J. van der Zant and E. Burzurí, ACS Nano, 2017, 11, 5879–5883 CrossRef CAS PubMed.
- P. Debroy, S. V. Lindeman and R. Rathore, Org. Lett., 2007, 9, 4091–4094 CrossRef CAS PubMed.
- P. Maslak, J. N. Narvaez and T. M. Vallombroso, J. Am. Chem. Soc., 1995, 117, 12373–12379 CrossRef CAS.
- P. Maslak, W. H. Chapman, T. M. Vallombroso and B. A. Watson, J. Am. Chem. Soc., 1995, 117, 12380–12389 CrossRef CAS.
- T. Suzuki, J. Nishida and T. Tsuji, Angew. Chem., Int. Ed. Engl., 1997, 36, 1329–1331 CrossRef CAS.
- Y. Uchimura, T. Takeda, R. Katoono, K. Fujiwara and T. Suzuki, Angew. Chem., 2015, 127, 4082–4085 CrossRef.
- E. Clar, The Aromatic Sextet, Wiley, New York, 1972 Search PubMed.
- E. T. Seo, R. F. Nelson, J. M. Fritsch, L. S. Marcoux, D. W. Leedy and R. N. Adams, J. Am. Chem. Soc., 1966, 88, 3498–3503 CrossRef CAS.
- S. C. Creason, J. Wheeler and R. F. Nelson, J. Org. Chem., 1972, 37, 4440–4446 CrossRef CAS.
- K. Sreenath, C. V. Suneesh, V. K. Ratheesh Kumar and K. R. Gopidas, J. Org. Chem., 2008, 73, 3245–3251 CrossRef CAS PubMed.
- H. C. Mruthyunjaya and A. R. Vasudeva Murthy, J. Electroanal. Chem. Interfacial Electrochem., 1968, 18, 200–204 CrossRef CAS.
- R. R. Nelson and R. N. Adams, J. Am. Chem. Soc., 1968, 90, 3925–3930 CrossRef CAS.
- E. M. Arnett, R. A. Flowers, R. T. Ludwig, A. E. Meekhof and S. A. Walek, J. Phys. Org. Chem., 1997, 10, 499–513 CrossRef CAS.
- S. Amthor, B. Noller and C. Lambert, Chem. Phys., 2005, 316, 141–152 CrossRef CAS.
- D. Hidasová and T. Slanina, J. Org. Chem., 2023, 88, 6932–6938 CrossRef PubMed.
- M. R. Talipov, M. M. Hossain, A. Boddeda, K. Thakur and R. Rathore, Org. Biomol. Chem., 2016, 14, 2961–2968 RSC.
- C. B. Duke, J. W.-P. Lin, A. Paton, W. R. Salaneck and K. L. Yip, Chem. Phys. Lett., 1979, 61, 402–406 CrossRef CAS.
- C. Avendaño, M. Espada, B. Ocaña, S. García-Granda, M. d R. Díaz, B. Tejerina, F. Gómez-Beltrán, A. Martínez and J. Elguero, J. Chem. Soc., Perkin Trans. 2, 1993, 1547–1555 RSC.
- S.-K. Chiu, Y.-C. Chung, G.-S. Liou and Y. O. Su, J. Chin. Chem. Soc., 2012, 59, 331–337 CrossRef CAS.
- J. F. Ambrose, L. L. Carpenter and R. F. Nelson, J. Electrochem. Soc., 1975, 122, 876–894 CrossRef CAS.
- P. Ledwon, Org. Electron., 2019, 75, 105422 CrossRef CAS.
- G. Hong, X. Gan, C. Leonhardt, Z. Zhang, J. Seibert, J. M. Busch and S. Bräse, Adv. Mater., 2021, 33, 2005630 CrossRef CAS PubMed.
- S. Wu, J. Žurauskas, M. Domański, P. S. Hitzfeld, V. Butera, D. J. Scott, J. Rehbein, A. Kumar, E. Thyrhaug, J. Hauer and J. P. Barham, Org. Chem. Front., 2021, 8, 1132–1142 RSC.
- J. Žurauskas, S. Boháčová, S. Wu, V. Butera, S. Schmid, M. Domański, T. Slanina and J. P. Barham, Angew. Chem., Int. Ed., 2023, 62, e202307550 CrossRef PubMed.
- J. E. Field, T. J. Hill and D. Venkataraman, J. Org. Chem., 2003, 68, 6071–6078 CrossRef CAS PubMed.
- S. Menichetti, S. Cecchi, P. Procacci, M. Innocenti, L. Becucci, L. Franco and C. Viglianisi, Chem. Commun., 2015, 51, 11452–11454 RSC.
- N. Hammer, T. A. Schaub, U. Meinhardt and M. Kivala, Chem. Rec., 2015, 15, 1119–1131 CrossRef CAS PubMed.
- T. A. Schaub, T. Mekelburg, P. O. Dral, M. Miehlich, F. Hampel, K. Meyer and M. Kivala, Chem. – Eur. J., 2020, 26, 3264–3269 CrossRef CAS PubMed.
- X. Zheng, X. Wang, Y. Qiu, Y. Li, C. Zhou, Y. Sui, Y. Li, J. Ma and X. Wang, J. Am. Chem. Soc., 2013, 135, 14912–14915 CrossRef CAS PubMed.
- Y. Su, X. Wang, L. Wang, Z. Zhang, X. Wang, Y. Song and P. P. Power, Chem. Sci., 2016, 7, 6514–6518 RSC.
- M. Kuratsu, M. Kozaki and K. Okada, Angew. Chem., Int. Ed., 2005, 44, 4056–4058 CrossRef CAS PubMed.
- D. T. Breslin and M. A. Fox, J. Org. Chem., 1994, 59, 7557–7561 CrossRef CAS.
- H. Yokoi, S. Hiroto and H. Shinokubo, J. Am. Chem. Soc., 2018, 140, 4649–4655 CrossRef CAS PubMed.
- C. Yan, M. Takeshita, J. Nakatsuji, A. Kurosaki, K. Sato, R. Shang, M. Nakamoto, Y. Yamamoto, Y. Adachi, K. Furukawa, R. Kishi and M. Nakano, Chem. Sci., 2020, 11, 5082–5088 RSC.
- M. Reig, G. Bagdziunas, D. Volyniuk, J. V. Grazulevicius and D. Velasco, Phys. Chem. Chem. Phys., 2017, 19, 6721–6730 RSC.
- A. Bobet, A. Cuadrado, L. Fajarí, I. Sirés, E. Brillas, M. P. Almajano, V. Jankauskas, D. Velasco and L. Juliá, J. Phys. Org. Chem., 2019, 32, 3974 CrossRef.
- V. Gamero, D. Velasco, S. Latorre, F. López-Calahorra, E. Brillas and L. Juliá, Tetrahedron Lett., 2006, 47, 2305–2309 CrossRef CAS.
- K. Matsuda, R. Xiaotian, K. Nakamura, M. Furukori, T. Hosokai, K. Anraku, K. Nakao and K. Albrecht, Chem. Commun., 2022, 58, 13443–13446 RSC.
- A. Heckmann and C. Lambert, J. Am. Chem. Soc., 2007, 129, 5515–5527 CrossRef CAS PubMed.
- L. Fajarí, R. Papoular, M. Reig, E. Brillas, J. L. Jorda, O. Vallcorba, J. Rius, D. Velasco and L. Juliá, J. Org. Chem., 2014, 79, 1771–1777 CrossRef PubMed.
- W. P. Neumann, Wolfram Uzick and A. K. Zarkadis, J. Am. Chem. Soc., 1986, 108, 3762–3770 CrossRef CAS.
- H. Volz and W. Lotsch, Tetrahedron Lett., 1969, 10, 2275–2278 CrossRef.
- P. Andersen, Acta Chem. Scand., 1965, 19, 629–637 CrossRef CAS.
- J. Guasch, X. Fontrodona, I. Ratera, C. Rovira and J. Veciana, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2013, 69, 255–257 CrossRef CAS PubMed.
- M. U. Munshi, G. Berden, J. Martens and J. Oomens, Phys. Chem. Chem. Phys., 2017, 19, 19881–19889 RSC.
- V. V. Khramtsov and J. L. Zweier, in Stable Radicals, ed. R. G. Hicks, John Wiley & Sons, Ltd, Chichester, UK, 2010, pp. 537–566 Search PubMed.
- T. Suzuki, H. Tamaoki, J. Nishida, H. Higuchi, T. Iwai, Y. Ishigaki, K. Hanada, R. Katoono, H. Kawai, K. Fujiwara and T. Fukushima, in Organic Redox Systems, ed. T. Nishinaga, John Wiley & Sons, Inc, Hoboken, NJ, 2015, pp. 13–37 Search PubMed.
- L. Mao, M. Zhou, X. Shi and H.-B. Yang, Chin. Chem. Lett., 2021, 32, 3331–3341 CrossRef CAS.
- P. Murto and H. Bronstein, J. Mater. Chem. C, 2022, 10, 7368–7403 RSC.
| This journal is © The Royal Society of Chemistry 2024 |