
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
[Xe(OTeF5)(pyF)]+: a strong oxidizing xenonium(II) teflate cation with N-donor bases†
Ahmet N.
Toraman
 a,
Lukas
Fischer
a,
Alberto
Pérez-Bitrián
*b,
Anja
Wiesner
a,
Kurt F.
Hoffmann
a and
Sebastian
Riedel
*a
a,
Lukas
Fischer
a,
Alberto
Pérez-Bitrián
*b,
Anja
Wiesner
a,
Kurt F.
Hoffmann
a and
Sebastian
Riedel
*a
aInstitut für Chemie und Biochemie, Freie Universität Berlin, Fabeckstraße 34/36, Berlin 14195, Germany. E-mail: s.riedel@fu-berlin.de
bInstitut für Chemie, Humboldt-Universität zu Berlin, Brook-Taylor-Straße 2, Berlin 12489, Germany. E-mail: alberto.perez-bitrian@hu-berlin.de
First published on 13th December 2023
Abstract
Herein we report on the formation of the adduct salts [Xe(OTeF5)(pyF)][Al(OTeF5)4] (pyF = C5F5N, C5H3F2N) by abstraction of an –OTeF5 group from Xe(OTeF5)2 with the Lewis superacid Al(OTeF5)3 and subsequent adduct formation of the generated [XeOTeF5]+ cation with fluorinated pyridines. These salts represent the first xenonium cations with the weakly coordinating [Al(OTeF5)4]− anion. The strong oxidizing property of these compounds is further assessed.
The strong oxidizing [XeF]+ cation exhibits a high Lewis acidity and therefore readily forms adducts with e.g. nitrogen bases that are resistant to oxidation.1,2 In fact, the chemistry of Xe–N species has mostly been realized by coordination of nitrogen-containing bases to stabilize the [XeF]+ cation, although they can be produced by the reaction of XeF2 with a suitable protic acid and the release of HF.3
The pentafluoroorthotellurate (teflate, –OTeF5) group resembles the fluoride ligand in its high electron withdrawing properties, but with a higher steric demand.4,5 Therefore, a variety of reactive species, including noble-gas compounds,6e.g. Xe(OTeF5)2 and Kr(OTeF5)2, have been synthesized.7,8 The teflate analogue of the [XeF]+ cation, namely [XeOTeF5]+, was first reported by Sladky in the reaction of FXeOTeF5 in combination with the Lewis acid AsF5, which forms the [XeOTeF5][AsF6] salt.9 Later it was shown, that this salt has a significant cation–anion interaction through a fluoride bridge.10 This interaction between the xenon(II) centre and the [AsF6]− anion is indicative of the high Lewis acidity of the [XeOTeF5]+ cation, similar to its well-known fluoride analogue [XeF]+. Efforts to isolate the free [XeOTeF5]+ cation in the solid state by utilizing weakly coordinating anions (WCAs) such as [Sb(OTeF5)6]− were so far unsuccessful. Instead, the solvent adduct [Xe(OTeF5)(SO2ClF)][Sb(OTeF5)6] was always obtained, indicating the near-linear alignment at the xenon centre.11,12 Furthermore, the [XeOTeF5]+ cation has been proved to be a strong two-electron oxidizer, which is e.g. a useful synthon for the preparation of trihalomethyl carbocations.13
Additionally, due to its chemical robustness, the teflate group has been utilized to prepare well-performing WCAs.14 Our group reported on the [Al(OTeF5)4]− anion in 2017,15 which has subsequently allowed the stabilization of highly reactive cations, such as [P4H]+, [(CH3)2Cl]+, [(C5H5P)CH3]+ and [(C(C6F5)3)]+.16–19 Based on these achievements the teflate-based aluminate is a promising candidate for resisting the strong oxidizing property of a xenonium(II) cation. The lack of N-donor adducts of the [XeOTeF5]+ cation, contrary to the well investigated ones for [XeF]+, prompted us to extend the chemistry of the teflate derivative, while pushing the limits of the [Al(OTeF5)4]− anion to withstand strong oxidizing cations.
Herein we report on the formation and characterization of the cationic pentafluoroorthotellurato xenonium(II) adducts with fluorinated pyridines, namely [Xe(OTeF5)(pyF)]+ (pyF = C5F5N, C5H3F2N), as their salts of the weakly coordinating [Al(OTeF5)4]− anion. Furthermore, the oxidizing properties of the cation [Xe(OTeF5)(C5F5N)]+ have been investigated. Moreover, the molecular structure of [Xe(OTeF5)(NC5F5)][Sb(OTeF5)6] was determined via single-crystal X-ray diffraction.
Based on the literature-known synthesis of the [FXe(NC5F5)]+ cation, starting from [HNC5F5][AsF6] and XeF2,20 Xe(OTeF5)2 was reacted with our recently prepared Brønsted acid [HNC5F5][Al(OTeF5)4].21 However, various attempts to obtain [Xe(OTeF5)(NC5F5)][Al(OTeF5)4] (1) with this method were unsuccessful, resulting in multiple teflate- and pentafluoropyridine-containing species. Unfortunately, no xenon-compound was observed by 129Xe NMR spectroscopy.
Alternatively, we performed the abstraction of a teflate group from Xe(OTeF5)2 with the Lewis superacid Al(OTeF5)3,22 whereby both the [Xe(OTeF5)]+ cation and the corresponding WCA, [Al(OTeF5)4]−, should be formed. The equimolar reaction of Xe(OTeF5)2 with Al(OTeF5)3 in SO2ClF at −50 °C successfully results in a non-isolable intermediate with a distinct yellow colour, presumably [Xe(OTeF5)][Al(OTeF5)4]. Characterization of this intermediate was not possible with the low-temperature spectroscopic measurements. By subsequent addition of one equivalent of a fluorinated pyridine (pyF = C5F5N, C5H3F2N), the cationic adducts [Xe(OTeF5)(NC5F5)]+ (1) and [Xe(OTeF5)(NC5H3F2)]+ (2) as salts of the [Al(OTeF5)4]− anion are formed (Scheme 1).
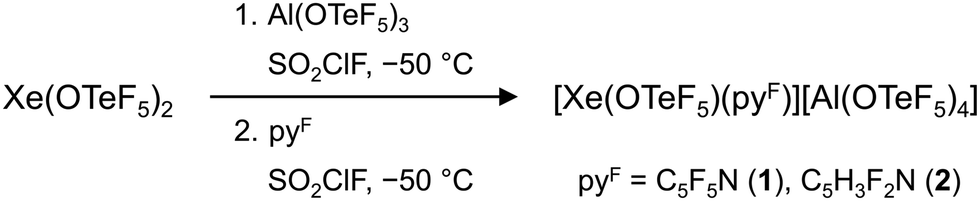 | ||
| Scheme 1 Synthesis of [Xe(OTeF5)(pyF)][Al(OTeF5)4] (pyF = C5F5N, C5H3F2N). | ||
The temperature-sensitive compounds 1 and 2 were characterized by NMR spectroscopy at −50 °C in SO2ClF, confirming the successful formation of the desired species (Table 1). Variable temperature NMR studies showed that compounds 1 and 2 decompose at temperatures higher than −10 °C. In the 19F NMR spectrum of 1, two magnetically inequivalent –OTeF5 groups with overlapping signals are observed. Simulation of the spectrum confirmed the expected two AB4 spin systems assigned to one teflate group in the cation and the four teflate groups located at the aluminium centre forming the WCA (Fig. 1a). The spectroscopic parameters of the latter are in agreement with those previously reported for the [Al(OTeF5)4]− anion.15 The formation of this WCA to stabilize the xenonium(II) cation is additionally proved by 27Al NMR spectra, as both adduct salts 1 and 2 show the characteristic resonance at δ(27Al) ≈ 46 ppm for the [Al(OTeF5)4]− anion.
| Species | Chemical shift (δ)bc [ppm] | Coupling constantb [Hz] | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 19FA | 19FB | 129Xe | 19Fo | 19Fm | 19Fp | 27Al | 1 J(19FA,125Te) | 2 J(19FA,19FB) | 3 J(19Fo,129Xe) | |
| a All NMR spectra were recorded in SO2ClF at −50 °C with [D6]acetone external locking. b The 19F NMR data are reported according to the simulated spectra obtained with the gNMR software (see ESI). c The symbols FA and FB denote equatorial and axial fluorine atoms, respectively, within the AB4 spin system of the –OTeF5 group. The symbols Fo, Fm, Fp represent ortho-, meta- and para-fluorine atoms of the fluorinated pyridine moieties. | ||||||||||
| [Xe(OTeF5)(NC5F5)]+ | −45.4 | −41.6 | −2241 | −86.7 | −152.0 | −108.3 | — | 3598 | 177 | 69 |
| [Al(OTeF5)4]− | −38.4 | −45.7 | — | — | — | — | 45.9 | 3353 | 188 | — |
| [Xe(OTeF5)(NC5H3F2)]+ | −43.1 | −42.5 | −2433 | −67.6 | — | — | — | 3488 | 180 | 57 |
| [Al(OTeF5)4]− | −38.1 | −45.5 | — | — | — | — | 46.2 | 3338 | 186 | — |
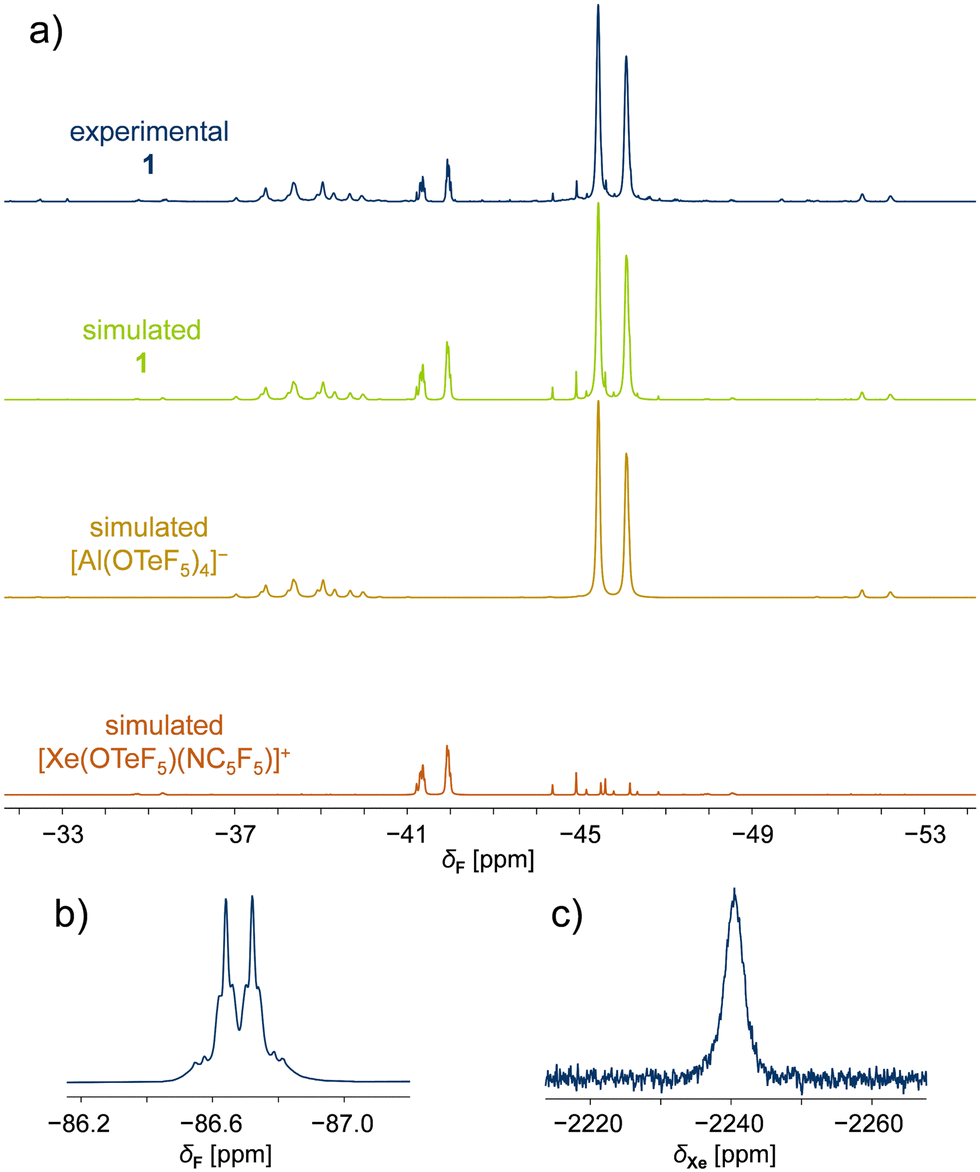 | ||
| Fig. 1 NMR spectra of [Xe(OTeF5)(NC5F5)][Al(OTeF5)4] (1) in SO2ClF ([D6]acetone (external lock), −50 °C). (a) 19F NMR (282 MHz) spectra showing the teflate region of (top to bottom) experimental 1, simulated 1, simulated [Al(OTeF5)4]− anion and simulated [Xe(OTeF5)(NC5F5)]+ cation. (b) 19F NMR (282 MHz) spectrum of 1 depicting the ortho fluorine atoms of the pentafluoropyridine moiety, with the corresponding 129Xe satellites. (c) 129Xe NMR (83 MHz) spectrum of 1 depicting a broad singlet. | ||
The AB4 pattern of the cations is inverted compared to the one of the [Al(OTeF5)4]− anion, but in agreement with the observed 19F NMR spectrum of the previously reported [Xe(OTeF5)(SO2ClF)]+ cation.12 The chemical shifts of the [Xe(OTeF5)(NC5F5)]+ cation in 1 are found to be δ(19FA) = −45.4 ppm and δ(19FB) = −41.6 ppm, with the corresponding 2J(19FA,19FB) coupling constant of 177 Hz (Table 1). The resonances of the teflate group in the [Xe(OTeF5)(NC5H3F2)]+ cation in 2 are less separated, appearing at δ(19FA) = −43.1 ppm and δ(19FB) = −42.5 ppm, with the 2J(19FA,19FB) coupling constant being 180 Hz (Table 1).
Furthermore, the 19F NMR chemical shifts arising from the C5F5N moiety in the [Xe(OTeF5)(NC5F5)]+ cation in 1 (Table 1) are found to be downfield shifted with respect to those of neat C5F5N (Δδ(19Fortho) = 3.0 ppm, Δδ(19Fmeta) = 10.8 ppm, Δδ(19Fpara) = 26.6 ppm). The ortho-fluorine resonance is accompanied by 129Xe (I = ½, 26.4%) satellites, arising from the 3J(19F,129Xe) spin–spin coupling (Fig. 1b). The presence of xenon satellites unambiguously demonstrates the coordination of C5F5N to the xenon(II) centre. In the 19F NMR spectrum of 2, two different C5H3F2N species are observed. The signal corresponding to the cation of product 2 is easily identified, as it exhibits the characteristic 129Xe satellites with a 3J(19F,129Xe) coupling constant of 57 Hz. The secondary signal, resonating at a higher field, δ(19Fortho) = −77.8 ppm, than neat C5H3F2N is found to be [HNC5H3F2]+ cation by a control experiment.
The 129Xe NMR spectra of 1 and 2 consist of one xenon resonance appearing as a broad singlet at δ(129Xe) = −2241 ppm (Fig. 1c) and δ(129Xe) = −2433 ppm, respectively, indicating the presence of a single xenon species in each case. When compared to the previously reported [XeOTeF5]+ cation in SO2ClF at −50 °C, which shows a chemical shift of δ(129Xe) = −1459.5 ppm,12 the 129Xe chemical shifts of 1 and 2 are found to be shifted to lower frequencies upon adduct formation with nitrogen bases.
To further characterize our new xenonium cations, concentrated reaction mixtures of 1 and 2 were analysed by low-temperature Raman spectroscopy. The presence of strong fluorescence interference in the spectrum of 1 rendered its interpretation difficult. In the case of 2 (see Fig. S3.1 in ESI†), characteristic bands can be observed, which are in good agreement with the computed wavenumbers for the [Xe(OTeF5)(NC5H3F2)]+ cation at the B3LYP/def2-TZVPP level of theory. In particular, the prominent bands observed at ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 140, 241 and 581 cm−1 are assigned to the stretching modes of O–Xe–N, Te–O–Xe and Xe–N, respectively. According to our experimental measurements, bands at = 303, 432 and 1221 cm−1 belong to remaining free SO2ClF in the sample. On the other hand, the Te–O and Te–F bands are overlapped with those from the [Al(OTeF5)4]− anion, therefore hampering the assignment of the bands in the teflate region, 600–780 cm−1.
= 140, 241 and 581 cm−1 are assigned to the stretching modes of O–Xe–N, Te–O–Xe and Xe–N, respectively. According to our experimental measurements, bands at = 303, 432 and 1221 cm−1 belong to remaining free SO2ClF in the sample. On the other hand, the Te–O and Te–F bands are overlapped with those from the [Al(OTeF5)4]− anion, therefore hampering the assignment of the bands in the teflate region, 600–780 cm−1.
All our crystallization attempts for 1 and 2 to investigate the molecular structure of the new xenonium(II) cationic adducts in the solid state were so far unsuccessful. Consequently, we changed the anion from [Al(OTeF5)4]− to [Sb(OTeF5)6]−, which resulted useful to crystallize the [Xe(OTeF5)(SO2ClF)]+ cation.11,12 The salt [Xe(OTeF5)(NC5F5)][Sb(OTeF5)6] (3) was obtained by a similar procedure as 1 and 2. Namely, the salt [Xe(OTeF5)(SO2ClF)][Sb(OTeF5)6] was treated with one equivalent of C5F5N, forming a yellow solution. Characterization by multinuclear NMR spectroscopy showed that the 19F and 129Xe NMR data of 3 are in good agreement with those of the cation in the salt 1 (see ESI† for the complete data set). Unfortunately, the Raman spectrum of 3 shows strong fluorescence, which did not allow a detailed analysis, likewise in the case of 1.
Upon slowly cooling down the reaction mixture from −50 °C to −80 °C, single crystals of 3 suitable for X-ray diffraction could be obtained. Compound 3 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The O1, Xe1 and N1 atoms of the cation are linearly aligned with an angle of 179.2(2)° (Fig. 2), which is consistent with the AX2E3 VSEPR molecular structure for a xenon(II) species and closer to 180° than the previously reported O–Xe–O angle in the molecular structure of [Xe(OTeF5)(SO2ClF)][Sb(OTeF5)6] (174.2(2)°).12 The Te1–O1–Xe1 angle in 3 is 121.4(2)°, which is similar to the related SO2ClF adduct salt (120.8(2)°).12 The distance between the xenon and the oxygen atom of the teflate group in 3 is slightly elongated (Xe1–O1 = 202.8(4) pm), and the distance from the xenon to the nitrogen atom is shortened (Xe1–N1 = 233.4(5) pm), when compared with [Xe(OTeF5)(SO2ClF)]+ (Xe–Oteflate = 196.9(4) pm, Xe–Osolvent = 247.9(4) pm), and is in agreement with the higher basicity of C5F5N than of SO2ClF. The measured O1–Te1 distance of 188.3(5) pm is found to be in between the values reported for Xe(OTeF5)2 (184.3(11) pm)10 and the [Xe(OTeF5)(SO2ClF)]+ cation (193.8(5) pm).12
. The O1, Xe1 and N1 atoms of the cation are linearly aligned with an angle of 179.2(2)° (Fig. 2), which is consistent with the AX2E3 VSEPR molecular structure for a xenon(II) species and closer to 180° than the previously reported O–Xe–O angle in the molecular structure of [Xe(OTeF5)(SO2ClF)][Sb(OTeF5)6] (174.2(2)°).12 The Te1–O1–Xe1 angle in 3 is 121.4(2)°, which is similar to the related SO2ClF adduct salt (120.8(2)°).12 The distance between the xenon and the oxygen atom of the teflate group in 3 is slightly elongated (Xe1–O1 = 202.8(4) pm), and the distance from the xenon to the nitrogen atom is shortened (Xe1–N1 = 233.4(5) pm), when compared with [Xe(OTeF5)(SO2ClF)]+ (Xe–Oteflate = 196.9(4) pm, Xe–Osolvent = 247.9(4) pm), and is in agreement with the higher basicity of C5F5N than of SO2ClF. The measured O1–Te1 distance of 188.3(5) pm is found to be in between the values reported for Xe(OTeF5)2 (184.3(11) pm)10 and the [Xe(OTeF5)(SO2ClF)]+ cation (193.8(5) pm).12
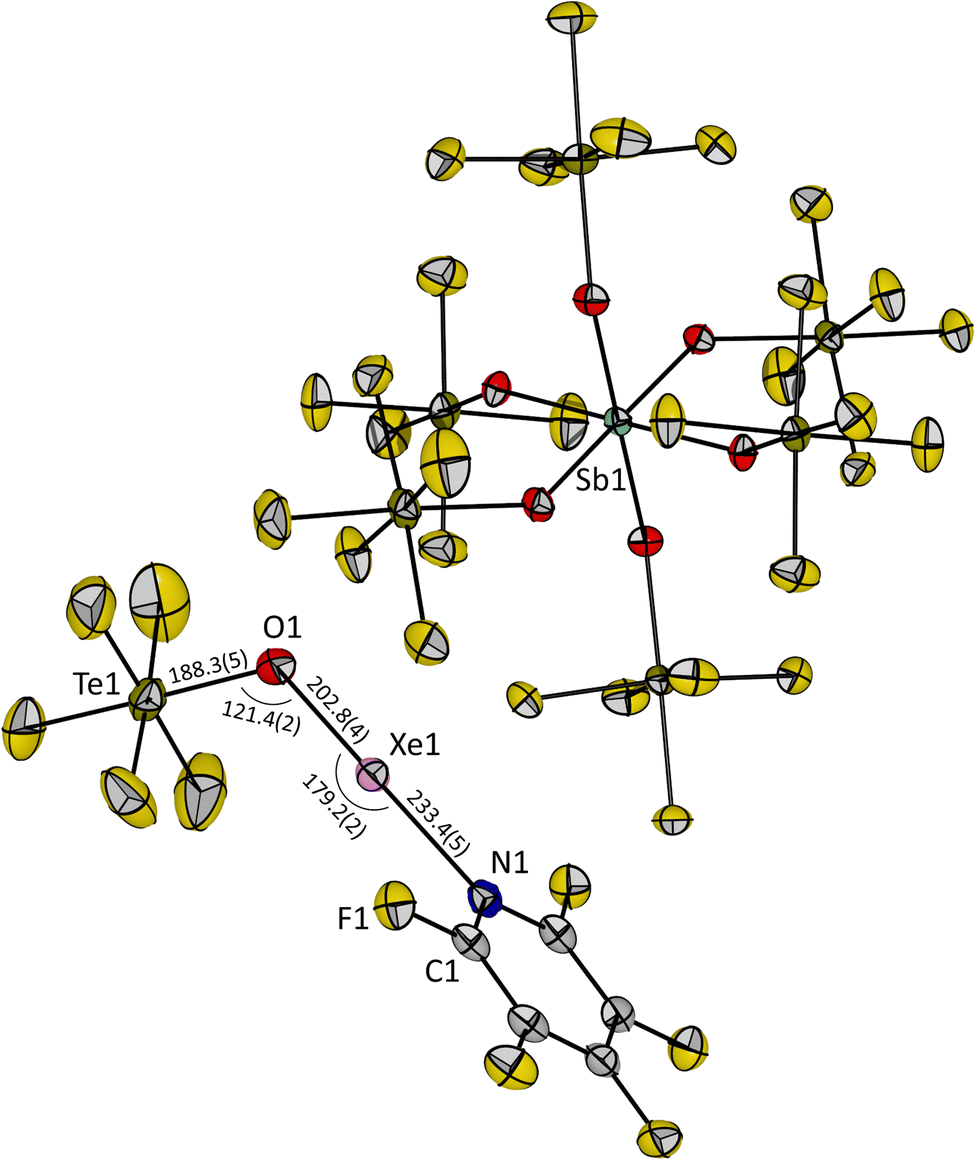 | ||
| Fig. 2 Molecular structure of [Xe(OTeF5)(NC5F5)][Sb(OTeF5)6] (3) in the solid state. Displacement ellipsoids set at 50% probability. Selected bond lengths [pm] and angles [°] are depicted. For crystallographic details see ESI.† | ||
Finally, to demonstrate the oxidation potential of [Xe(OTeF5)(NC5F5)][Al(OTeF5)4] (1), the compound was reacted with an excess of tris(2,4,6-tribromophenyl)amine in order to oxidize it to the corresponding ammoniumyl radical cation, which is a variation of tris(4-tribromophenyl)amine (“magic blue”) with an even higher standard oxidation potential.23 An immediate colour change from brown to deep-purple with gas evolution was observed upon addition of the amine, indicating the formation of the tris(2,4,6-tribromophenyl)ammoniumyl radical cation. This was proved by EPR spectroscopy showing one broad signal with a g value of 2.009, assigned to the radical cation (see Fig. S5, ESI†). Furthermore, the oxidation potential of xenonium cations has been evaluated by adiabatic ionization energy calculations at the B3LYP/def2-TZVPP level of theory and resulted to be XeF/XeF+ 10.7, Xe(OTeF5)/[Xe(OTeF5)]+ 10.6, and Xe(OTeF5)(NC5F5)/[Xe(OTeF5)(NC5F5)]+ 9.2 eV (see also ESI,† Table S4).
In conclusion, we have demonstrated that the Lewis superacid Al(OTeF5)3 is able to abstract a teflate group from Xe(OTeF5)2 to presumably form the [Xe(OTeF5)]+ cation and the weakly coordinating [Al(OTeF5)4]− anion. This intermediate could be subsequently stabilized upon coordination of the oxidation-resistant nitrogen bases C5F5N and C5H3F2N. This way, the [Xe(OTeF5)(pyF)][Al(OTeF5)4] (pyF = C5F5N, C5H3F2N) salts were prepared and characterized by low-temperature NMR and Raman spectroscopy, entailing the first xenonium(II) cations stabilized by the [Al(OTeF5)4]− WCA. Also, the synthesis of the related [Sb(OTeF5)6]− salt of the [Xe(OTeF5)(NC5F5)]+ cation enabled us to structurally characterize this unprecedented cation for the first time. Moreover, we have experimentally shown the high oxidation potential of the adduct salt [Xe(OTeF5)(NC5F5)][Al(OTeF5)4] (1), which oxidizes tris(2,4,6-tribromophenyl)amine to form the corresponding ammoniumyl radical cation.
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project ID: 387284271 (SFB 1349: Fluorine-Specific Interactions) and the European Research Council (ERC) – Project HighPotOx. A. P.-B. thanks the Fonds der Chemischen Industrie for a Liebig Fellowship. Computing resource was provided by the Zentrum für Elektronische Datenbearbeitung (ZEDAT) at Freie Universität Berlin. The authors thank the core facility Biosupramol for analytical measurements and acknowledge MSc Liza Richter (Humboldt-Universität zu Berlin), MSc Deniz Meyer (Freie Universität Berlin) and MSc Johanna Schlögl (Freie Universität Berlin) for performing spectroscopic measurements.
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. A. A. Emara and G. J. Schrobilgen, J. Chem. Soc., Chem. Commun., 1987, 21, 1644 RSC.
- G. L. Smith, H. P. A. Mercier and G. J. Schrobilgen, Inorg. Chem., 2007, 46, 1369 CrossRef CAS PubMed.
- J. F. Sawyer, G. J. Schrobilgen and S. J. Sutherland, J. Chem. Soc., Chem. Commun., 1982, 4, 210 RSC.
- K. Seppelt, Angew. Chem., Int. Ed. Engl., 1982, 21, 877 CrossRef.
- M. Gerken, H. P. A. Mercier and G. J. Schrobilgen, in Advanced Inorganic Fluorides, ed. T. Nakajima, B. Žemva and A. Tressaud, Elsevier, Lausanne, 2000, pp. 117–174 Search PubMed.
- D. S. Brock, G. J. Schrobilgen and B. Žemva, in Comprehensive Inorganic Chemistry II, ed J. Reedijk and K. Poeppelmeier, Elsevier, Oxford, 2nd edn, 2013, ch. 1.25, vol. 1, pp. 755–822 Search PubMed.
- F. Sladky, Angew. Chem., Int. Ed. Engl., 1969, 8, 523 CrossRef CAS.
- J. C. P. Sanders and G. J. Schrobilgen, J. Chem. Soc., Chem. Commun., 1989, 20, 1576 RSC.
- F. Sladky, Angew. Chem., Int. Ed. Engl., 1970, 9, 375 CrossRef CAS.
- B. A. Fir, H. P. A. Mercier, J. C. P. Sanders, D. A. Dixon and G. J. Schrobilgen, J. Fluorine Chem., 2001, 110, 89 CrossRef CAS.
- P. Ulferts and K. Seppelt, Z. Anorg. Allg. Chem., 2004, 630, 1589 CrossRef CAS.
- H. P. A. Mercier, M. D. Moran, J. C. P. Sanders, G. J. Schrobilgen and R. J. Suontamo, Inorg. Chem., 2005, 44, 49 CrossRef CAS PubMed.
- H. P. A. Mercier, M. D. Moran, G. J. Schrobilgen, C. Steinberg and R. J. Suontamo, J. Am. Chem. Soc., 2004, 126, 5533 CrossRef CAS PubMed.
- I. M. Riddlestone, A. Kraft, J. Schaefer and I. Krossing, Angew. Chem., Int. Ed., 2018, 57, 13982 CrossRef CAS PubMed.
- A. Wiesner, T. W. Gries, S. Steinhauer, H. Beckers and S. Riedel, Angew. Chem., Int. Ed., 2017, 56, 8263 CrossRef CAS PubMed.
- A. Wiesner, S. Steinhauer, H. Beckers, C. Müller and S. Riedel, Chem. Sci., 2018, 9, 7169 RSC.
- S. Hämmerling, G. Thiele, S. Steinhauer, H. Beckers, C. Müller and S. Riedel, Angew. Chem., Int. Ed., 2019, 58, 9807 CrossRef PubMed.
- L. Fischer, F. Wossidlo, D. Frost, N. T. Coles, S. Steinhauer and S. Riedel, Chem. Commun., 2021, 57, 9522 RSC.
- K. F. Hoffmann, D. Battke, P. Golz, S. M. Rupf, M. Malischewski and S. Riedel, Angew. Chem., Int. Ed., 2022, 61, e202203777 CrossRef CAS PubMed.
- A. A. A. Emara and G. J. Schrobilgen, J. Chem. Soc., Chem. Commun., 1988, 4, 257 RSC.
- S. Kotsyuda, A. N. Toraman, P. Voßnacker, M. A. Ellwanger, S. Steinhauer, C. Müller and S. Riedel, Chem. – Eur. J., 2023, 29, e202202749 CrossRef CAS PubMed.
- K. F. Hoffmann, A. Wiesner, S. Steinhauer and S. Riedel, Chem. – Eur. J., 2022, 28, e202201958 CrossRef CAS PubMed.
- W. Schmidt and E. Steckhan, Chem. Ber., 1980, 113, 577 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2302485. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc05560d |
| This journal is © The Royal Society of Chemistry 2024 |