
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Site-selective peptide functionalisation mediated via vinyl-triazine linchpins†
Jack D.
Sydenham
 a,
Hikaru
Seki
a,
Sona
Krajcovicova
ab,
Linwei
Zeng
a,
Tim
Schober
a,
Tomas
Deingruber
a and
David R.
Spring
*a
a,
Hikaru
Seki
a,
Sona
Krajcovicova
ab,
Linwei
Zeng
a,
Tim
Schober
a,
Tomas
Deingruber
a and
David R.
Spring
*a
aYusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, UK. E-mail: spring@ch.cam.ac.uk
bDepartment of Organic Chemistry, Palacky University in Olomouc, Tr. 17. Listopadu 12, Olomouc, Czech Republic
First published on 6th December 2023
Abstract
Herein we introduce 3-vinyl-1,2,4-triazines derivatives as dual-reactive linkers that exhibit selectivity towards cysteine and specific strained alkynes, enabling conjugate addition and inverse electron-demand Diels–Alder (IEDDA) reactions. This approach facilitates site-selective bioconjugation of biologically relevant peptides, followed by rapid and highly selective reactions with bicyclononyne (BCN) reagents.
The reactivity variations among amino acids have been effectively utilised in the realm of selective peptide and protein modifications. Extensive methods have been developed for modification of lysine,1,2 cysteine,3,4 tryptophan,5 methionine,6,7 tyrosine,8 histidine9 and the N- and C-terminal residues.10,11 Among these, cysteine's exceptional nucleophilicity under mild and biocompatible conditions has made it an attractive and dependable candidate for orthogonal manipulation. Numerous reagents have been developed specifically for cysteine modification, encompassing bromomaleimides,12 vinylsulfones,13 and others.14–17 Despite the emergence of these advanced cysteine conjugation technologies, maleimide reagents remain a highly favoured choice due to their rapid reaction kinetics.18 Nevertheless, they exhibit limited chemoselectivity and tend to react with other nucleophilic residues present on the protein surface. Additionally, the thiosuccinimide linkages formed during maleimide conjugation can result in inadequate plasma stability. Premature release of payloads compromises their intended drug delivery properties, leading to off-target effects. To mitigate these detrimental side effects and enhance the therapeutic window of such treatments, the development of more stable linkages becomes crucial. Therefore, there is a need for improved cysteine modification reagents that can address these challenges. In addition to direct modification with payloads, alternative modification strategies introduce functional groups capable of performing bioorthogonal click reactions.19–21
Commonly employed bioorthogonal click reactions include the copper-catalysed azide–alkyne cycloaddition (CuAAC),22,23 strain-promoted azide–alkyne cycloaddition (SPAAC),24 and inverse electron-demand Diels–Alder (IEDDA) reactions between tetrazines and strained alkynes and alkenes.25 While IEDDA reactions have found utility in modifying various biomolecules, the high reactivity of tetrazine groups can pose challenges due to their decomposition in buffer or reactivity with thiol groups.25 Recently, 1,2,4-triazines have emerged as effective IEDDA reagents, exhibiting a favourable balance between rapid reaction kinetics and reagent stability. They have proven successful in the modification of proteins and oligonucleotides.20,21
Strained alkynes such as bicyclo[6.1.0]non-4-yne (BCN) and dibenzocyclooctyne (DBCO) have been demonstrated to react with unprotected cysteines;26–28 however, there are large limitations to the technique stability issues.27 The growing interest in the reactivity of strained alkynes and the stability of the resulting products formed through their click reactions, combined with the appealing nature of cysteine for peptide modifications, has sparked a desire to create selective, dual-reactive linkers capable of connecting the two.29
In our previous study, we introduced the utilisation of vinyl heteroarenes for achieving site-selective protein modification via cysteine.15 This methodology allowed for the selective and stable modification of proteins and antibodies. However, the previous approach required the presence of an external reactive handle for payload attachment, in addition to the use of copper for facilitation of the attachment process (Fig. 1; Previous work).
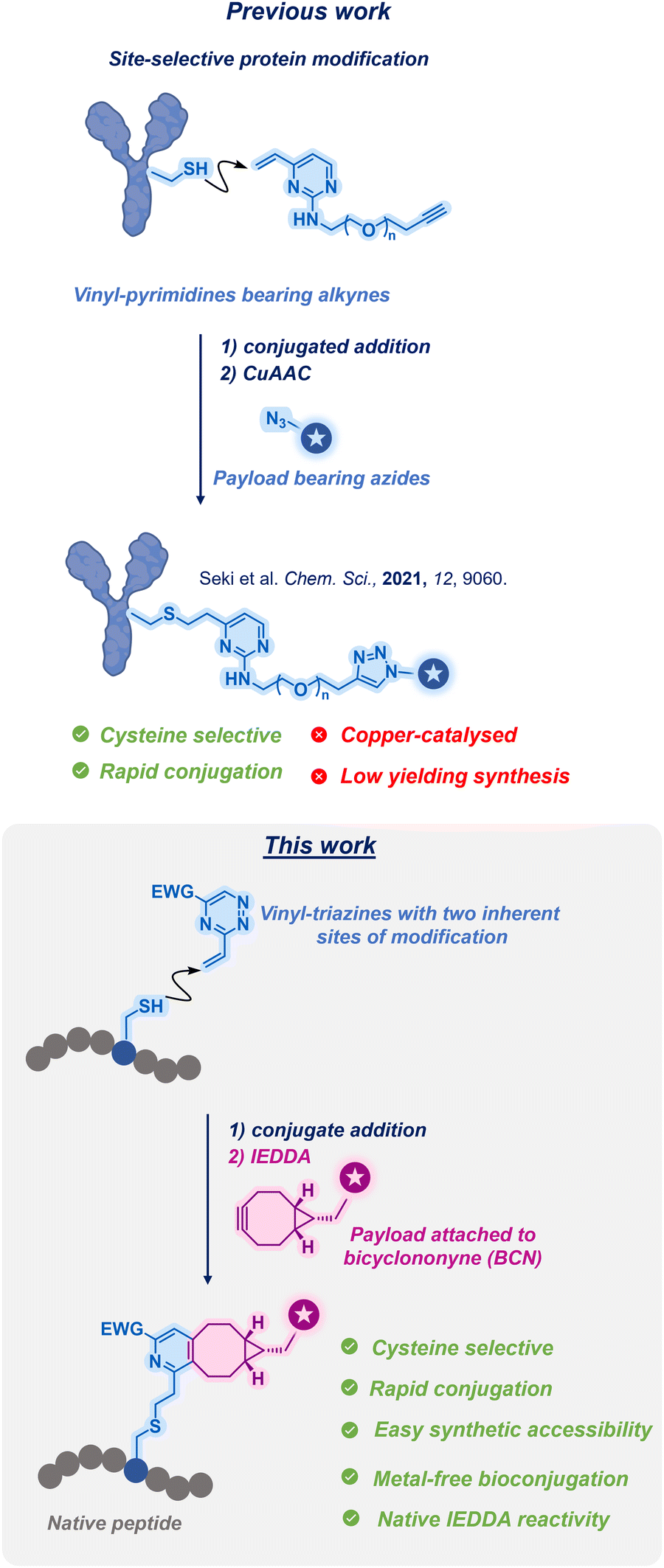 | ||
| Fig. 1 Previous work vs. this work. | ||
Herein, we present a distinct approach by employing 3-vinyl-1,2,4-triazines (vinyl-triazines thereafter) as dual-reactive linkers. This innovative strategy involves the initial vinyl conjugation via cysteine followed by a rapid IEDDA reaction of the triazine. Through this process, a diverse range of functionalised biomolecules can be efficiently generated (Fig. 1; This work).
To initiate the investigations, a collection of vinyl-triazines was synthesised. It was crucial to evaluate the influence of triazine ring electronics, as the introduction of electron-withdrawing groups was expected to enhance the conjugate addition rate of cysteine groups, as well as the rate of IEDDA reaction with strained alkynes, being influenced via previous work looking at IEDDA rates.30 Furthermore, the vinyl group was attached on the electron deficient 3-position to increase the propensity towards conjugate addition. For this reason, highly electronegative 4-fluorophenyl and 4-trifluoromethylphenyl substituents were chosen. The synthesis commenced from methyl hydrazinecarbimidothioate hydroiodide 1 which was in the first step subjected to reaction with glyoxal derivatives to afford 2a–c (Scheme 1). The Liebeskind-Srogl coupling was then used to afford 3a–c. To determine reaction kinetics of the vinyl-triazines towards cysteine addition, compounds 3a–c were incubated with an equimolar quantity of Ac-Cys-OMe in a mixture of sodium phosphate and MeCN-d3 (Scheme 1, Conjugate addition) following by 1H NMR. A clear trend of increasing rate constants was observed with the introduction of electron-withdrawing substituents, with a threefold increase observed from 3a to 3c, and with highest value for the latter, as anticipated (Table 1; entries 1–3) with the expected, almost full conversion (95%) after less than 2 hours under tested conditions. The rate constants obtained are comparable to those commonly observed bioconjugations.31 Importantly, after 24 hours of incubation under the same reaction conditions, minimal reactivity with lysine was observed for the most reactive 3c, which indicates a high selectivity of these scaffolds towards cysteine.
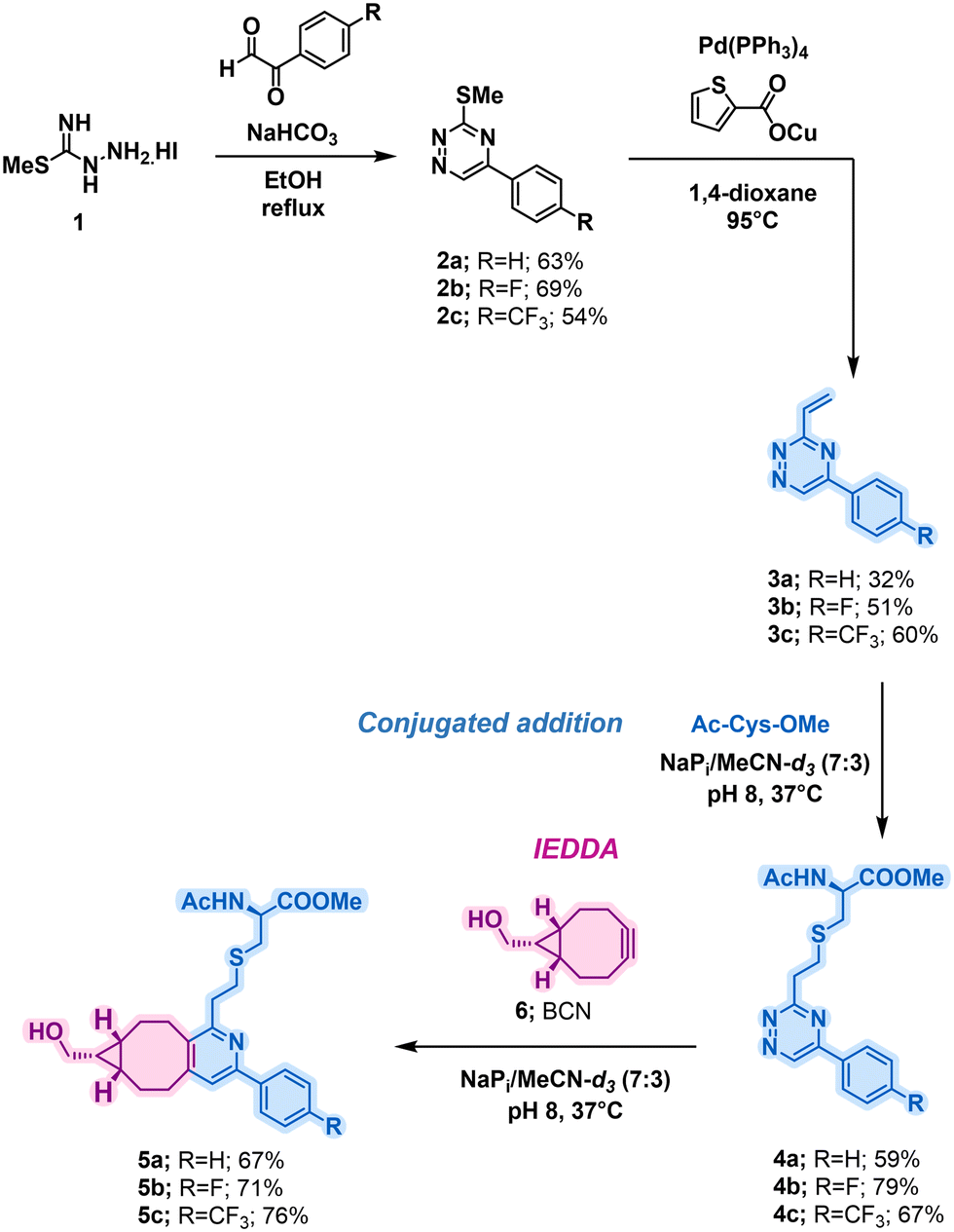 | ||
| Scheme 1 Synthesis of vinyl-triazines linker and their reactivity in conjugate addition and IEDDA reaction, respectively. | ||
| Entry | Starting compound | Reaction | Rate constant [× 10−3 M−1 s−1] |
|---|---|---|---|
| 1 | 3a | CA | 77.4 ± 0.0003 |
| 2 | 3b | CA | 49.0 ± 0.0057 |
| 3 | 3c | CA | 237 ± 0.136 |
| 4 | 4a | IEDDA | 2.43 ± 0.0044 |
| 5 | 4b | IEDDA | 3.75 ± 0.0008 |
| 6 | 4c | IEDDA | 1.64 ± 0.0021 |
After establishing the fast bioconjugation kinetics with cysteine, it was crucial to evaluate the reactivity of the compounds towards IEDDA reaction with BCN reagents. To investigate this, compounds 4a–c were incubated with equimolar amounts of 6 under identical solvent conditions as previously described (Scheme 1, IEDDA).
Consistent with the trends observed in literature for IEDDA reaction rates,32 the rate of reaction was found to increase as expected with greater electron deficiency, with compound 4b exhibiting the highest rate constant (Table 1; entries 4–6).
The potential cross-reactivity of DBCO, another popular strained-alkyne reagent, with the vinyl-triazines was then investigated with no reaction observed for 4c at 37 °C (Fig. S17–S19, ESI†). This selectivity is attributed to the increased steric bulk of DBCO. Such encouraging findings allows for the opportunity to selectively form dual functionalisation of molecules, using functional groups such as azides that would react with DBCO and then introducing BCN to react with the vinyl-triazines.
Having established the viability of triazines for selective cysteine modification and their reactivity with BCN, the stability of the resulting thioether linkages followed. To evaluate their stability, model substrates (thioethers 7a–c; Fig. 2) were synthesised from vinyl-triazines 3a–c. As a benchmark, the maleimide adduct 8 was also synthesised. These model compounds were subjected to incubation at pH 7.4 and 37 °C for 10 days in the presence of excess dithioerythritol. Analysis by 19F NMR revealed remarkable stability for compounds 7a–c, with less than 5% degradation observed over the 10-day period (Fig. 2). In contrast, thiosuccinimide 8 was significantly less stable, with over 60% degradation observed within the same timeframe. The improved stability of the vinyl-triazine conjugates highlights the tremendous potential of this method for biological applications.
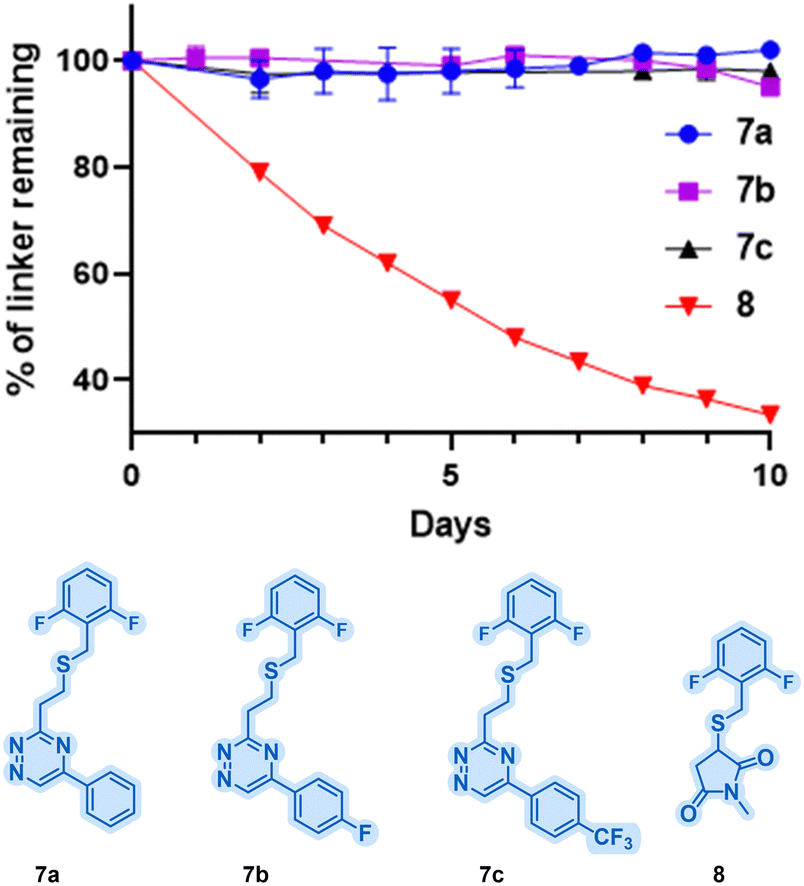 | ||
| Fig. 2 Stability study of vinyl-triazine linkers 7a–cvs. maleimide 8. | ||
To assess the robustness of these dual reactive vinyl-triazine linkers, a bioactive peptide substrate was chosen (Scheme 2 and Scheme S2, ESI†)‡. The design of the peptide 9 was inspired by antimicrobial peptides previously reported in the literature and modified in both a two and one-pot fashion (Scheme 2 and Scheme S2–S4, ESI†). Pleasingly, a single modification event was observed for the reaction between the most reactive vinyl-triazine 3c and peptide 9, demonstrating an exclusive selectivity in presence of various lysine residues, free N-terminal amine as well as C-terminal amide. The modified peptide S4 was then incubated with BCN 6 yielding compound 10 in 5 hours (Scheme 2). This highlights the effectiveness of this technique and its potential applications to append biologically active peptides with payloads through BCN derivatives.
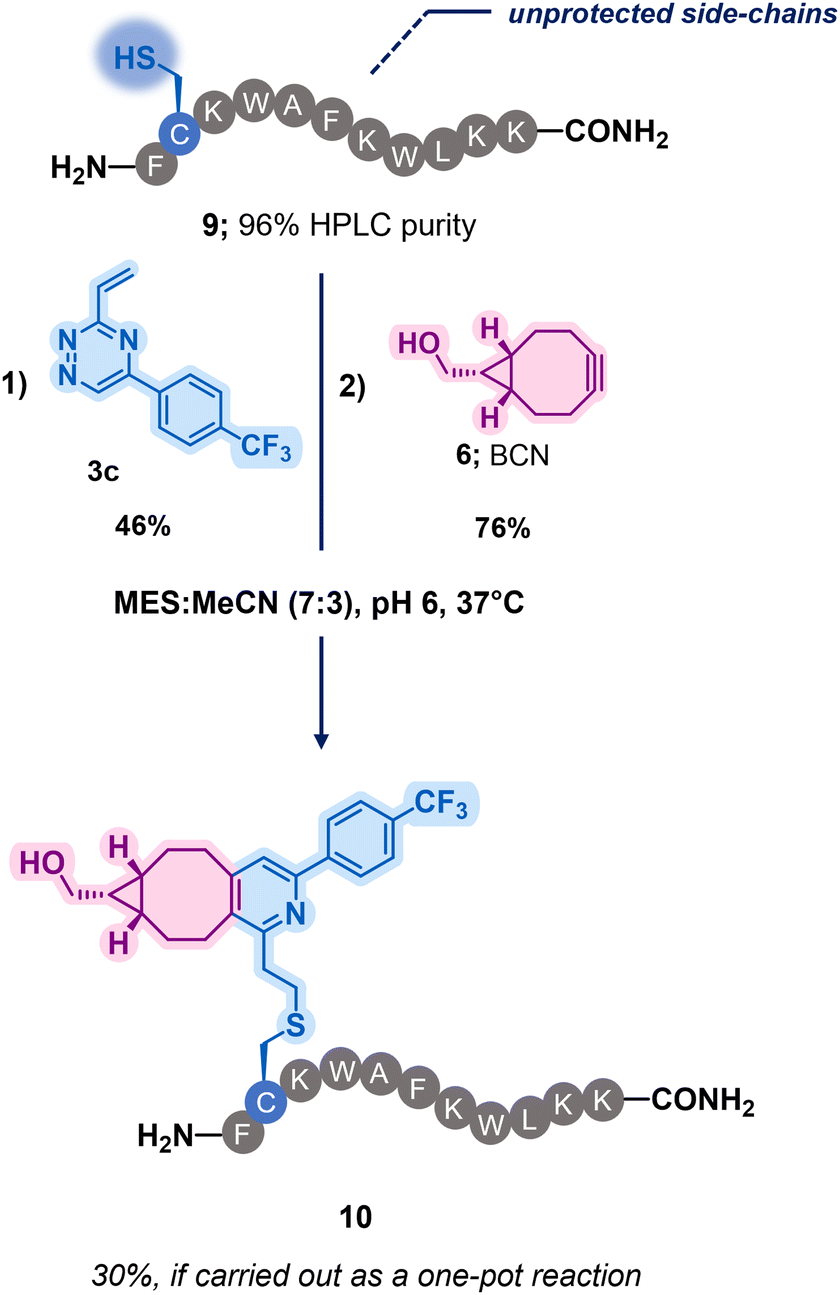 | ||
| Scheme 2 Application of the dual orthogonal conjugation on a model peptide 9. | ||
To summarise, we have successfully established a remarkably orthogonal bioconjugation approach utilising dual-reactive 3-vinyl-1,2,4-triazines that display selectivity towards cysteine residues. This methodology enables the generation of modified peptides incorporating a stable triazine handle, which readily reacts with BCN derivatives in an inverse electron-demand Diels–Alder (IEDDA) reaction. Importantly, the observed rate constants for these reactions are comparable to widely employed techniques for protein modification. This technique was used to selectively modify a cysteine in an antimicrobial peptide in the presence of many lysine residues and a free N-terminus allowing for a controlled reaction with 6 which could enable the introduction of different payloads. Following application of the method in protein and antibody modifications is currently a subject of our interest.
J. D. S. was involved in conceptualization, investigation, data curation, formal analysis, methodology, and writing – original draft. H. S. was involved in conceptualization. S. K. was involved in peptide investigation, visualization and writing – editing and review. L. Z. was involved in investigation. T. D. and T. S. were involved in conceptualisation and supervision. D. R. S was involved in conceptualization, supervision, project administration and funding acquisition.
J. D. Sydenham acknowledges support from the UK Engineering and Physical Sciences Research Council (EPSRC) Centre of Doctoral Training in Automated Chemical Synthesis Enabled by Digital Molecular Technologies (SynTech) [EP/S024220/1], University of Cambridge, UK. T. Schober acknowledges EU funding by the H2020-MSCA-RISE-2020 through the ALISE project (grant 101007256). S. Krajcovicova is grateful to the Experientia Foundation, Czech Science Foundation (GA CR 22-07138O) and Cambridge Isaac Newton Trust (Grant Ref No: 22.39(l)) for their financial support. We are grateful to P. Gierth, A. Mason and D. Howe for their support in NMR analysis. T. Deingruber acknowledges a studentship from AstraZeneca. The authors acknowledge Nathanael Smalley for preliminary work on vinyl heteroarene. The Spring group research was supported by grants from UKRI. For the purpose of Open Access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript (AAM) version arising.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. J. Matos, G. Jiménez-Osés and G. J. L. Bernardes, Methods in Molecular Biology, Humana, New York, NY, 1st edn, 2019, pp. 25–37 Search PubMed.
- C. L. Tung, C. T. T. Wong, E. Y. M. Fung and X. Li, Org. Lett., 2016, 18, 2600–2603 CrossRef.
- Y. Zhang, X. Zhou, Y. Xie, M. M. Greenberg, Z. Xi and C. Zhou, J. Am. Chem. Soc., 2017, 139, 6146–6151 CrossRef.
- J. Yu, X. Yang, Y. Sun and Z. Yin, Angew. Chem., Int. Ed., 2018, 57, 11598–11602 CrossRef PubMed.
- S. J. Tower, W. J. Hetcher, T. E. Myers, N. J. Kuehl and M. T. Taylor, J. Am. Chem. Soc., 2020, 142, 9112–9118 CrossRef PubMed.
- M. T. Taylor, J. E. Nelson, M. G. Suero and M. J. Gaunt, Nature, 2018, 562, 563–568 CrossRef PubMed.
- M. Q. Zhang, P. Y. He, J. J. Hu and Y. M. Li, J. Pept. Sci., 2023, 29, 1075–2617 Search PubMed.
- K. Maruyama, T. Ishiyama, Y. Seki, K. Sakai, T. Togo, K. Oisaki and M. Kanai, J. Am. Chem. Soc., 2021, 143, 19844–19855 CrossRef.
- X. Chen, F. Ye, X. Luo, X. Liu, J. Zhao, S. Wang, Q. Zhou, G. Chen and P. Wang, J. Am. Chem. Soc., 2019, 141, 18230–18237 CrossRef.
- S. Bloom, C. Liu, D. K. Kölmel, J. X. Qiao, Y. Zhang, M. A. Poss, W. R. Ewing and D. W. C. Macmillan, Nat. Chem., 2018, 10, 205–211 CrossRef.
- C. T. T. Wong, J. C. H. Chu, S. Y. Y. Ha, R. C. H. Wong, G. Dai, T. T. Kwong, C. H. Wong and D. K. P. Ng, Org. Lett., 2020, 22, 7098–7102 CrossRef PubMed.
- M. E. B. Smith, F. F. Schumacher, C. P. Ryan, L. M. Tedaldi, D. Papaioannou, G. Waksman, S. Caddick and J. R. Baker, J. Am. Chem. Soc., 2010, 132, 1960–1965 CrossRef.
- J. Morales-Sanfrutos, J. Lopez-Jaramillo, M. Ortega-Muñoz, A. Megia-Fernandez, F. Perez-Balderas, F. Hernandez-Mateo and F. Santoyo-Gonzalez, Org. Biomol. Chem., 2010, 8, 667–675 RSC.
- M. J. Matos, C. D. Navo, T. Hakala, X. Ferhati, A. Guerreiro, D. Hartmann, B. Bernardim, K. L. Saar, I. Compañón, F. Corzana, T. P. J. Knowles, G. Jiménez-Osés and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2019, 58, 6640–6644 CrossRef PubMed.
- H. Seki, S. J. Walsh, J. D. Bargh, J. S. Parker, J. Carroll and D. R. Spring, Chem. Sci., 2021, 12, 9060–9068 RSC.
- M. A. Kasper, M. Glanz, A. Stengl, M. Penkert, S. Klenk, T. Sauer, D. Schumacher, J. Helma, E. Krause, M. C. Cardoso, H. Leonhardt and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2019, 58, 11625–11630 CrossRef.
- V. Chudasama, M. E. B. Smith, F. F. Schumacher, D. Papaioannou, G. Waksman, J. R. Baker and S. Caddick, Chem. Commun., 2011, 47, 8781–8783 RSC.
- J. T. W. Tong, P. W. R. Harris, M. A. Brimble and I. Kavianinia, Molecules, 2021, 26, 5847 CrossRef.
- P. E. Brandish, A. Palmieri, S. Antonenko, M. Beaumont, L. Benso, M. Cancilla, M. Cheng, L. Fayadat-Dilman, G. Feng, I. Figueroa, J. Firdos, R. Garbaccio, L. Garvin-Queen, D. Gately, P. Geda, C. Haines, S. Hseih, D. Hodges, J. Kern, N. Knudsen, K. Kwasnjuk, L. Liang, H. Ma, A. Manibusan, P. L. Miller, L. Y. Moy, Y. Qu, S. Shah, J. S. Shin, P. Stivers, Y. Sun, D. Tomazela, H. C. Woo, D. Zaller, S. Zhang, Y. Zhang and M. Zielstorff, Bioconjug. Chem., 2018, 29, 2357–2369 CrossRef PubMed.
- K. A. Horner, N. M. Valette and M. E. Webb, Chem. – Eur. J., 2015, 21, 14376–14381 CrossRef PubMed.
- D. N. Kamber, Y. Liang, R. J. Blizzard, F. Liu, R. A. Mehl, K. N. Houk and J. A. Prescher, J. Am. Chem. Soc., 2015, 137, 8388–8391 CrossRef.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef PubMed.
- M. R. Karver, R. Weissleder and S. A. Hilderbrand, Bioconjug. Chem., 2011, 22, 2263–2270 CrossRef PubMed.
- H. Tian, T. P. Sakmar and T. Huber, Chem. Commun., 2016, 52, 5451–5454 RSC.
- R. Van Geel, G. J. M. Pruijn, F. L. Van Delft and W. C. Boelens, Bioconjug. Chem., 2012, 23, 392–398 CrossRef.
- C. Zhang, P. Dai, A. A. Vinogradov, Z. P. Gates and B. L. Pentelute, Angew. Chem., Int. Ed., 2018, 57, 6459–6463 CrossRef PubMed.
- A. M. Tallon, Y. Xu, G. M. West, C. W. am Ende and J. M. Fox, J. Am. Chem. Soc., 2023, 145, 16069–16080 CrossRef.
- D. N. Kamber, S. S. Nguyen, F. Liu, J. S. Briggs, H. W. Shih, R. D. Row, Z. G. Long, K. N. Houk, Y. Liang and J. A. Prescher, Chem. Sci., 2019, 10, 9109–9114 RSC.
- K. Lang and J. W. Chin, ACS Chem. Biol., 2014, 9, 16–20 CrossRef PubMed.
- M. L. W. J. Smeenk, J. Agramunt and K. M. Bonger, Curr. Opin. Chem. Biol., 2021, 60, 79–88 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc05213c |
| ‡ pH 6 buffer was used due to the solubility of the peptide dependant on acidic pH. |
| This journal is © The Royal Society of Chemistry 2024 |