
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCation delocalization and photo-isomerization enhance the uncaging quantum yield of a photocleavable protecting group†
Albert Marten
Schulte
 a,
Lianne M.
Smid
a,
Georgios
Alachouzos
*a,
Wiktor
Szymanski
*ab and
Ben L.
Feringa
*a
a,
Lianne M.
Smid
a,
Georgios
Alachouzos
*a,
Wiktor
Szymanski
*ab and
Ben L.
Feringa
*a
aCentre for Systems Chemistry, Stratingh Institute for Chemistry, Faculty for Science and Engineering, University of Groningen, Nijenborgh 4, Groningen 9747 AG, The Netherlands. E-mail: alachouzos.georgios@gmail.com; w.c.szymanski@rug.nl; b.l.feringa@rug.nl
bDepartment of Radiology, Medical Imaging Center, University Medical Center Groningen, University of Groningen, Hanzeplein 1, Groningen 9713 GZ, The Netherlands
First published on 14th December 2023
Abstract
Photocleavable protecting groups (PPGs) enable the light-induced, spatiotemporal control over the release of a payload of interest. Two fundamental challenges in the design of new, effective PPGs are increasing the quantum yield (QY) of photolysis and red-shifting the absorption spectrum. Here we describe the combination of two photochemical strategies for PPG optimization in one molecule, resulting in significant improvements in both these crucial parameters. Furthermore, we for the first time identify the process of photo-isomerization to strongly influence the QY of photolysis of a PPG and identify the cis-isomer as the superior PPG.
Using light as a stimulus to control (bio)chemical processes offers unique advantages, most notably spatial control over the process through direction of the irradiation source, along with the benefit of the non-invasive nature of light itself.1 On the molecular level, a commonly used way to achieve optical control over these processes is through the use of photocleavable protecting groups (PPGs).2–4 PPGs are chemical moieties that feature the unique ability to release a cargo of interest (payload) upon irradiation. Careful synthetic introduction of a PPG to a payload of interest can silence the activity of the payload, which can then be conveniently restored through light irradiation. PPGs have been successfully applied in various research areas including local activation of drugs in the field of photopharmacology,5–10 reagents in synthetic chemistry11 and specific molecules of interest in molecular biology.12,13
Depending on the application of the PPG, the wavelength of light used for activation can be a crucial parameter.14,15 This is most clearly exemplified in the field of photopharmacology, where the wavelength of light is strongly connected to its tissue penetration depth, as lower energy, red-shifted photons exhibit a higher penetration depth.16 The development of efficient PPGs that respond to low energy, long wavelength photons is crucial for the successful further development of photopharmacology.
With this in mind, numerous research efforts have focused on the development of red-shifted PPGs, resulting in the successful development of green- to red-light responsive PPGs, such as those derived from BODIPYs,17–21 cyanines22–24 and coumarins.25,26 A common characteristic shared among these red-shifted PPGs is an extended conjugated system, allowing these compounds to absorb lower energy photons. Unfortunately, this often causes these families of PPGs to suffer from poor aqueous solubility, hindering their application in fields that necessitate their use in water such as photopharmacology.
Perhaps even more strikingly, red-shifting of the activation wavelength often comes at the unfortunate cost of a diminished uncaging efficiency of the PPG. The Quantum Yield (QY) of payload release of these PPGs is usually far lower than one percent.17,18,20,23,24,26,27 Lowering a PPGs QY of payload release is highly undesirable, since it diminishes the capacity of these compounds to carry out the exact function they were designed for, negatively affecting or even preventing their practical application.28 While sparse examples exist of red-shifted PPGs featuring higher QYs, these are often rigid conjugated systems, lowering their aqueous solubility as described previously.19,29,30
One of the most widely used families of PPGs is that of coumarins, a member of the large class of heterolytic PPGs that after photon absorption undergo heterolytic bond cleavage in the first singlet excited state (Fig. 1, k1).31,32 After heterolysis, a Contact Ion Pair (CIP) is formed, featuring a positive charge on the coumarin alpha-carbon and a negative charge on the payload. Depending on the stability of this CIP intermediate, it can either productively diffuse to result in payload release (Fig. 1, k2), or unproductively recombine to re-form the substrate (Fig. 1, k−1), a process that reduces the efficiency, and therefore the QY, of the payload release process.
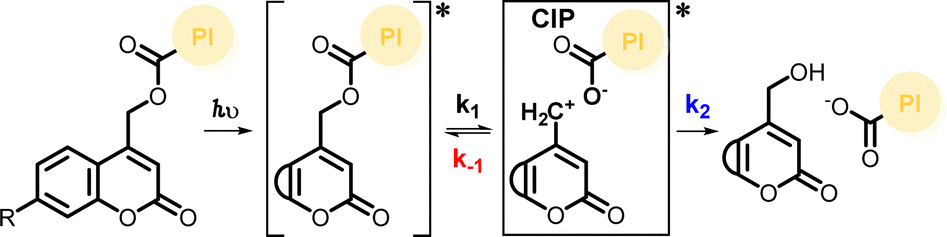 | ||
| Fig. 1 Schematic representation of the heterolytic coumarin photocleavage mechanism. Shown is a model carboxylic acid payload (yellow, Pl). The fused aromatic ring of the coumarin chromophore is abbreviated by a semicircle. | ||
Recently, Bojtár et al. described a new way of simultaneously increasing the activation wavelength of a coumarin PPG as well as improving its aqueous solubility,25 two parameters that are usually negatively correlated as laid out above. They accomplished this through synthetically introducing a vinyl-pyridinium moiety to the coumarin core (Fig. 2a, red, PPG I), thereby simultaneously extending the conjugation of the coumarin core (to achieve a bathochromic shift) and introducing a polar group (to increase the aqueous solubility). Despite the favorable activation wavelength and aqueous solubility, their PPG unfortunately also suffered from a low QY of payload release, around 1%. While there might be several reasons for this low QY – such as an increase in the QY of competing excited state relaxation processes like internal conversion – we hypothesized that the low QY of PPG I could be partially caused by the electron withdrawing effect of the pyridinium moiety. This electron withdrawing effect could destabilize the cation formed in the CIP after heterolysis, resulting in fast CIP recombination and a low QY.
 | ||
Fig. 2 (a) Structures of the studied compounds I, Prenyl-I, and actinometer BODIPY. (b) The normalized absorbance of I and Prenyl-I (both 20 μM, water/MeCN 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) at λ = 500 nm upon irradiation with green light (λmax = 530 nm). (c) Acetic acid formation as determined by 1H-NMR signal integration using DMSO2 as an internal standard (I or Prenyl-I in DMSO-d6/D2O 1:1, 1 mM, irradiated with green light, shown are the average values and standard deviations of triplicate measurements). (d) Uncaging quantum yields (%) of all compounds, determined from the slope of AcOH formation as displayed in (c). :1, v/v) at λ = 500 nm upon irradiation with green light (λmax = 530 nm). (c) Acetic acid formation as determined by 1H-NMR signal integration using DMSO2 as an internal standard (I or Prenyl-I in DMSO-d6/D2O 1:1, 1 mM, irradiated with green light, shown are the average values and standard deviations of triplicate measurements). (d) Uncaging quantum yields (%) of all compounds, determined from the slope of AcOH formation as displayed in (c). | ||
Recently, our group developed a strategy that increases the QY of heterolytic PPGs through engineering the stability of the intermediate CIP by way of cation stabilization.33 Through the introduction of an vinylic substituent, delocalization of the cation formed after excited state heterolysis resulted in a great improvement in QY of a coumarin PPG, up to 35-fold. Out of the cation stabilizing substituents tested, the prenyl-substituent resulted in the largest increase in QY. We envisioned that applying this strategy on red-shifted PPG I would result in re-stabilization of the CIP cation, resulting in an increased QY. Therefore, we set out to install the prenyl substituent at the alfa-carbon of PPG I, resulting in Prenyl-I (Fig. 2a, blue). Since the double bond in the prenyl substituent is not in conjugation with the chromophore, we hypothesized that this substituent would not affect the position of the absorption band of PPG I while simultaneously increase its payload release QY.
The synthetic route towards Prenyl-I was designed and carried out as reported in the ESI† (Section S1). Prenyl-I showed excellent water-solubility (Fig. S33, ESI†), illustrating that this favorable property was retained through prenyl-substitution. With both the prenylated PPG Prenyl-I and reference PPG I in hand, we set out to study and compare their photochemical properties. Comparing the absorption spectra of I and Prenyl-I in the dark (ESI,† Section S2.5), we observed that both compounds showed highly similar absorption spectra, illustrating that the introduction of the prenyl substituent minimally affected the position of the absorption band. Installation of the prenyl substituent slightly reduced the molar absorptivity coefficient of the PPG, by ∼27%, with I and Prenyl-I showing epsilon values of 3.4 × 104 and 2.5 × 104, respectively (ESI,† Section S2.6).
Irradiation of both compounds in water/MeCN (9:1, v/v) with green light (λ = 530 nm) for one hour resulted in changes in the absorption spectra of both compounds (ESI,† Section S2.5). For Prenyl-I, these changes occurred significantly faster than for I (blue and red lines respectively, Fig. 2b). However, the absorption spectra alone do not paint the complete picture of uncaging, since vinyl-pyridinium coumarins are known to isomerize upon irradiation, as was revealed through HPLC-MS analysis in the seminal report from Bojtár et al.25 The isomerization process would also result in the changes in the absorption spectra of I and Prenyl-I, and cannot be easily distinguished from the uncaging process. The fact that multiple processes occur simultaneously upon irradiation can be seen most clearly when following the absorbance of I at a single wavelength (Fig. 2b, red). The absorbance of I drops rapidly during the first two minutes – likely caused by isomerization – after which a constant, slower decrease of the absorbance is observed.
Nonetheless, the substantially faster decrease in the absorption spectrum of Prenyl-I, as compared to I, is a good initial indication of its higher release efficiency, since a higher QY of payload release would always result in more rapid changes in the absorption spectrum. Also, we hypothesized that, since the prenyl moiety of Prenyl-I is not in conjugation with the isomerizing photoswitch, it is more likely to positively affect the QY of uncaging through cation stabilization than the QY of photo-isomerization.
Encouraged by these initial results, we set out to deconvolute the progress of the uncaging process from the other processes happening upon irradiation through 1H-NMR analysis. Using dimethylsulfone (DMSO2) as an internal standard, the formation of acetic acid upon irradiation was quantified (Fig. 2c). Interestingly, for both compounds an initial delay in the rate of AcOH formation was observed, after which a constant rate was reached. A linear fit allowed for the determination of the average rate of AcOH formation of this ‘steady state’ (Fig. 2c). To determine the quantum yield of payload release, an actinometer with a known QY (Fig. 2a, BODIPY, QY = 0.14%21) was irradiated in the same setup. To our delight, installing the prenyl moiety resulted in a significant improvement in QY for the vinyl-pyridinium PPG (Fig. 2d). Whereas the QY of I was determined to be 0.20%, the QY of Prenyl-I was improved 7.4-fold, reaching a final QY of 1.50% (see ESI,† Section S2).
Interestingly, the QY we determined for the reference PPG I lacking the prenyl substituent is lower than the one that Bojtár et al. determined for an identical PPG bearing a slightly different payload.25 This could be caused by the different techniques and therefore solvent systems that were used to determine the QY. Whereas we determined the QY by 1H-NMR and therefore used 1:1 D2O/DMSO-d6, Bojtár et al. determined it by HPLC and used a higher water content (95:5 water/MeCN). A higher water content positively influences the QY of heterolytic PPGs.29
Upon closer inspection of the 1H-NMR spectra obtained in the uncaging experiments of both compounds, we observed another signal appear under irradiation close to the acetate CH3 signal of I and Prenyl-I (ESI,† Section S2.2). Since this signal appeared before a substantial amount of AcOH was released, we concluded it could not be any photoproduct resulting from the PPG after payload release. Furthermore, in the aromatic region of the 1H-NMR spectrum of I, a new set of doublets was formed showing a strong roofing effect and a J-coupling of 12 Hz (Fig. 3b, red spectrum), corresponding to a cis-alkene. For Prenyl-I, at the same chemical shift a singlet was observed, likely due to the strong roofing effect of two doublets with highly similar chemical shift (Fig. 3b, blue spectrum).
 | ||
| Fig. 3 (a) Schematic representation of the photoswitching of PPGs I and Prenyl-I (b) Partial 1H-NMR spectra of I (red) and Prenyl-I (blue) in the dark and after irradiation (λmax: 530 nm, 40 min for I, 6 min for Prenyl-I), 1 mM I or Prenyl-I, 1:1 DMSO-d6/D2O. (c) Acetic acid and cis-isomer formation upon irradiation of I (left) and Prenyl-I (right), experimental details see ESI,† Section S2. Shown are the average values and standard deviations of triplicate measurements. For both graphs, a linear fit of the reduced rate of AcOH formation between t0 and t1 is included. A maximal amount of ∼25% of the cis isomer was obtained during irradiation. | ||
The relative integrals of other signals that appeared upon irradiation corresponded to the expected integrals of the acetate CH3 and protons at the PPG alpha-carbon, suggesting that they also belonged to the cis-isomer (ESI,† Section S2.2). Integration of the acetate CH3 signal of the cis-isomer of I (the intact PPG-AcOH conjugate, before uncaging) revealed a highly informative kinetic profile (Fig. 3c, red line), with an initial fast formation of the isomer, followed by its slow consumption. Interestingly, when comparing the kinetic profile of cis-isomer formation with that of payload release (Fig. 3c, grey line), we observed that during the initial stages in which payload release is delayed, the cis-isomer builds up rapidly. Then, when a substantial amount of cis-isomer has been formed, the rate of payload release increases (Fig. 3c, after 40 min). For Prenyl-I, a highly similar kinetic profile was observed, although both processes happen on a much shorter time scale (Fig. 3c, right). These data suggest that the QY of payload release differs considerably between the two isomers. Given that the rate of payload release increases significantly after formation of the cis-isomer, it must be the cis-isomer that is the more efficient PPG. Whereas the rate increases roughly twofold for I and roughly threefold for Prenyl-I, the actual difference in quantum yield between the two isomers must be significantly larger, since the cis-isomer only constitutes a small fraction of the isomer distribution for both samples (∼25% at maximum value).
To understand the origin of the difference in QY between the two isomers, DFT calculations were performed.24,33–36 The energy barriers of heterolysis (k1) and CIP recombination (k−1) were computed in the first singlet excited state. While DFT-calculations of these energy barriers were unsuccessful for reference PPG I, for Prenyl-I they could be computed. Interestingly, a significant difference in CIP stability was found between the two isomers of Prenyl-I. Most striking was the difference in CIP recombination energy barriers between the two isomers; the computed k−1 energy barrier for the cis-isomer was 13.9 kcal mol−1, roughly two-fold higher than the recombination barrier for the trans-isomer at 6.8 kcal mol−1 (ESI,† Section S3). This indicates that after heterolysis and formation of the CIP, the probability for unproductive recombination of this CIP (Fig. 1, k−1) is much lower for cis-Prenyl-I than for trans-Prenyl-I, resulting in an increased QY for the cis-isomer. While the height of the productive k1 heterolysis barrier for cis-Prenyl-I was slightly higher than the height of the same barrier for the trans-isomer, we have discovered previously that -in contrast to the k−1 CIP recombination barrier- the k1 heterolysis barrier has little influence on the QY of coumarin PPGs.33 Therefore, these calculations satisfyingly rationalize the difference in QY between the two isomers, that is achieved through CIP stabilization resulting in an increase in ΔG‡(k−1).
We hypothesized that the stabilization stems from the partial breakage of conjugation upon switching. This was supported by DFT computations, that predicted a planar geometry of the π-system in case of the trans-isomer, whereas in the cis-isomer the pyridinium moiety is pushed out of plane (Fig. S30, ESI†). A reduced degree of conjugation between the pyridinium moiety and the coumarin core likely also reduces the electron withdrawing effect of this pyridinium moiety on the coumarin core that bears the cation in the CIP. This would reduce the destabilization of the cation at the coumarin alpha carbon, and overall result in CIP stabilization.
In this manuscript, we showcase how combining two photochemical strategies for the improvement of a PPG in one molecule results in a system that displays a combination of the favorable properties resulting from these strategies. Employing the QY-enhancing cation stabilization strategy on a green-light responsive coumarin PPG resulted in a large improvement in the QY of this PPG, with Prenyl-I showing an uncaging QY of 1.50%, 7.4 times higher than the parent molecule. Interestingly, we observed an initial delay in payload release upon irradiation that was attributed to the isomerization of the vinyl-pyridinium moiety. For the first time, we show that photo-isomerization of a PPG influences the QY of payload release. Upon photo-isomerization and formation of the cis-isomer, the rate of payload release increases for both I and Prenyl-I, identifying this cis-isomer as the superior PPG with a higher uncaging QY. This was further rationalized through DFT calculations, that most notably predicted a significant difference between the height of the k−1 CIP recombination barrier of the two isomers, a barrier proven to be crucial in determining heterolytic PPG QY. In the future, this information could be used to design PPGs with electron withdrawing substituents locked in the cis configuration, that could exhibit superior photouncaging QYs. Furthermore, potentially photoswitchable moieties are also prevalent among PPGs that have already been developed.17,23,24 This work highlights the universality of the cation stabilization strategy, and it for the first time identifies photo-isomerization as a process strongly influencing the uncaging QY of a PPG, an effect that needs to be considered in the future design of new PPGs.
A.M.S. conceived the project. L.M.S and A.M.S performed the synthesis and photochemical measurements. G.A. assisted in supervising the project and performed the DFT calculations. W.S. and B.L.F assisted in conceiving the study and supervised the project.
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. Brieke, F. Rohrbach, A. Gottschalk, G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2012, 51, 8446–8476 CrossRef CAS PubMed.
- R. Weinstain, T. Slanina, D. Kand and P. Klán, Chem. Rev., 2020, 120, 13135–13272 CrossRef CAS PubMed.
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef PubMed.
- T. Šolomek, J. Wirz and P. Klán, Acc. Chem. Res., 2015, 48, 3064–3072 CrossRef PubMed.
- G. C. R. Ellis-Davies, Nat. Methods, 2007, 4, 619–628 CrossRef CAS PubMed.
- I. M. Welleman, M. W. H. H. Hoorens, B. L. Feringa, H. H. Boersma and W. Szymański, Chem. Sci., 2020, 11, 11672–11691 RSC.
- W. A. Velema, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 2178–2191 CrossRef CAS PubMed.
- M. M. Lerch, M. J. Hansen, G. M. van Dam, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2016, 55, 10978–10999 CrossRef CAS PubMed.
- K. Hüll, J. Morstein and D. Trauner, Chem. Rev., 2018, 118, 10710–10747 CrossRef PubMed.
- J. Broichhagen, J. A. Frank and D. Trauner, Acc. Chem. Res., 2015, 48, 1947–1960 CrossRef CAS PubMed.
- N. Hoffmann, Chem. Rev., 2008, 108, 1052–1103 CrossRef CAS PubMed.
- C. Bier, D. Binder, D. Drobietz, A. Loeschcke, T. Drepper, K.-E. Jaeger and J. Pietruszka, Synthesis, 2016, 42–52 Search PubMed.
- A. Bardhan and A. Deiters, Curr. Opin. Struct. Biol., 2019, 57, 164–175 CrossRef CAS PubMed.
- P. Štacko and T. Šolomek, Chimia, 2021, 75, 873–881 CrossRef PubMed.
- A. Y. Vorobev and A. E. Moskalensky, Comput. Struct. Biotechnol. J., 2020, 18, 27–34 CrossRef CAS PubMed.
- H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620–2640 CrossRef CAS PubMed.
- J. A. Peterson, C. Wijesooriya, E. J. Gehrmann, K. M. Mahoney, P. P. Goswami, T. R. Albright, A. Syed, A. S. Dutton, E. A. Smith and A. H. Winter, J. Am. Chem. Soc., 2018, 140, 7343–7346 CrossRef CAS PubMed.
- K. Sitkowska, M. F. Hoes, M. M. Lerch, L. N. Lameijer, P. Van Der Meer, W. Szymański and B. L. Feringa, Chem. Commun., 2020, 56, 5480–5483 RSC.
- P. Shrestha, K. C. Dissanayake, E. J. Gehrmann, C. S. Wijesooriya, A. Mukhopadhyay, E. A. Smith and A. H. Winter, J. Am. Chem. Soc., 2020, 142, 15505–15512 CrossRef CAS PubMed.
- P. P. Goswami, A. Syed, C. L. Beck, T. R. Albright, K. M. Mahoney, R. Unash, E. A. Smith and A. H. Winter, J. Am. Chem. Soc., 2015, 137, 3783–3786 CrossRef CAS PubMed.
- T. Slanina, P. Shrestha, E. Palao, D. Kand, J. A. Peterson, A. S. Dutton, N. Rubinstein, R. Weinstain, A. H. Winter and P. Klán, J. Am. Chem. Soc., 2017, 139, 15168–15175 CrossRef CAS PubMed.
- A. P. Gorka, R. R. Nani, J. Zhu, S. Mackem and M. J. Schnermann, J. Am. Chem. Soc., 2014, 136, 14153–14159 CrossRef CAS PubMed.
- H. Janeková, M. Russo, U. Ziegler and P. Štacko, Angew. Chem., Int. Ed., 2022, 61, e202204391 CrossRef PubMed.
- G. Alachouzos, A. M. Schulte, A. Mondal, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2022, 61, e202201308 CrossRef CAS PubMed.
- M. Bojtár, A. Kormos, K. Kis-Petik, M. Kellermayer and P. Kele, Org. Lett., 2019, 21, 9410–9414 CrossRef PubMed.
- A. Gandioso, S. Contreras, I. Melnyk, J. Oliva, S. Nonell, D. Velasco, J. García-Amorós and V. Marchán, J. Org. Chem., 2017, 82, 5398–5408 CrossRef CAS PubMed.
- K. Sitkowska, B. L. Feringa and W. Szymański, J. Org. Chem., 2018, 83, 1819–1827 CrossRef CAS PubMed.
- M. Sharma and S. H. Friedman, ChemPhotoChem, 2021, 5, 611–618 CrossRef CAS PubMed.
- A. Egyed, K. Németh, T. Molnár, M. Kállay, P. Kele and M. Bojtár, J. Am. Chem. Soc., 2022, 145, 4026–4034 CrossRef PubMed.
- Q. Lin, L. Yang, Z. Wang, Y. Hua, D. Zhang, B. Bao, C. Bao, X. Gong and L. Zhu, Angew. Chem., Int. Ed., 2018, 57(14), 3722–3726 CrossRef CAS PubMed.
- R. Schmidt, D. Geissler, V. Hagen and J. Bendig, J. Phys. Chem. A, 2005, 109, 5000–5004 CrossRef CAS PubMed.
- R. Schmidt, D. Geissler, V. Hagen and J. Bendig, J. Phys. Chem. A, 2007, 111, 5768–5774 CrossRef CAS PubMed.
- A. M. Schulte, G. Alachouzos, W. Szymański and B. L. Feringa, J. Am. Chem. Soc., 2022, 144, 12421–12430 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- J. Zheng, X. Xu and D. G. Truhlar, Theor. Chem. Acc., 2011, 128, 295–305 Search PubMed.
- H. S. Yu, X. He, S. L. Li and D. G. Truhlar, Chem. Sci., 2016, 7, 5032–5051 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and spectroscopic data. See DOI: https://doi.org/10.1039/d3cc05055f |
| This journal is © The Royal Society of Chemistry 2024 |