
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePd/Brønsted acid catalysed intramolecular N-allylation of indoles and pyrroles with alkynes for the synthesis of N-fused heterocycles†
Saswat Ranjan
Bhoi‡
,
Chhanda
Debnath‡
and
Shikha
Gandhi
 *
*
Department of Chemical Sciences, Indian Institute of Science Education and Research, Berhampur 760010, India. E-mail: sgandhi@iiserbpr.ac.in
First published on 6th December 2023
Abstract
We, herein, report a Pd(0) and Brønsted acid-catalyzed redox-neutral intramolecular N-allylation of indoles and pyrroles with alkynes for the synthesis of biologically important imidazolidinone-fused N-heterocycles. The allylation is completely atom-economical and is applicable to a wide range of substrates. The methodology eliminates the use of a leaving group or an oxidizing agent, often employed for the allylation of nucleophiles. To the best of our knowledge, N-allylation of indoles and pyrroles with alkynes has not been reported to date.
Allylation of nucleophiles is one of the most widely employed reactions in organic synthesis. The installation of an allyl group is generally accomplished via the allyl-metal intermediate, often generated by the use of a leaving group on an allyl moiety, commonly known as the Tsuji–Trost reaction.1 However, the approach is not atom-economical and often requires an additional step for installing the leaving group. An alternative approach proceeds via allylic C–H oxidation, thus requiring a stoichiometric amount of oxidant.2 Nucleophilic addition to the allyl-metal complex generated from allenes is another alternative, which, however, requires preformed allenes.3 Undoubtedly, one of the best approaches available for these allylations is the atom-economical, redox-neutral addition of nucleophiles to alkynes, with the allyl-metal intermediate being generated via propargylic C–H activation and an in situ generation of allenes.4 This reaction has, since its discovery, been applied to the allylation of several carbon5 and hetero-nucleophiles,6–8 employing either Pd or Rh complexes. The allylation of amines with alkynes has also been explored, mostly focused around intermolecular versions (Scheme 1a).8a–f The intramolecular versions, in comparison, have hardly gained any attention (Scheme 1b).8g–i Despite some progress in the area, the N-allylation of indoles and pyrroles using alkynes as allyl-metal precursors has not found any entries in the literature. The wide prevalence of N-heterocycles and their derivatives in biologically active compounds9 intrigued us to explore the above-mentioned allylation of indoles and pyrroles with alkynes.
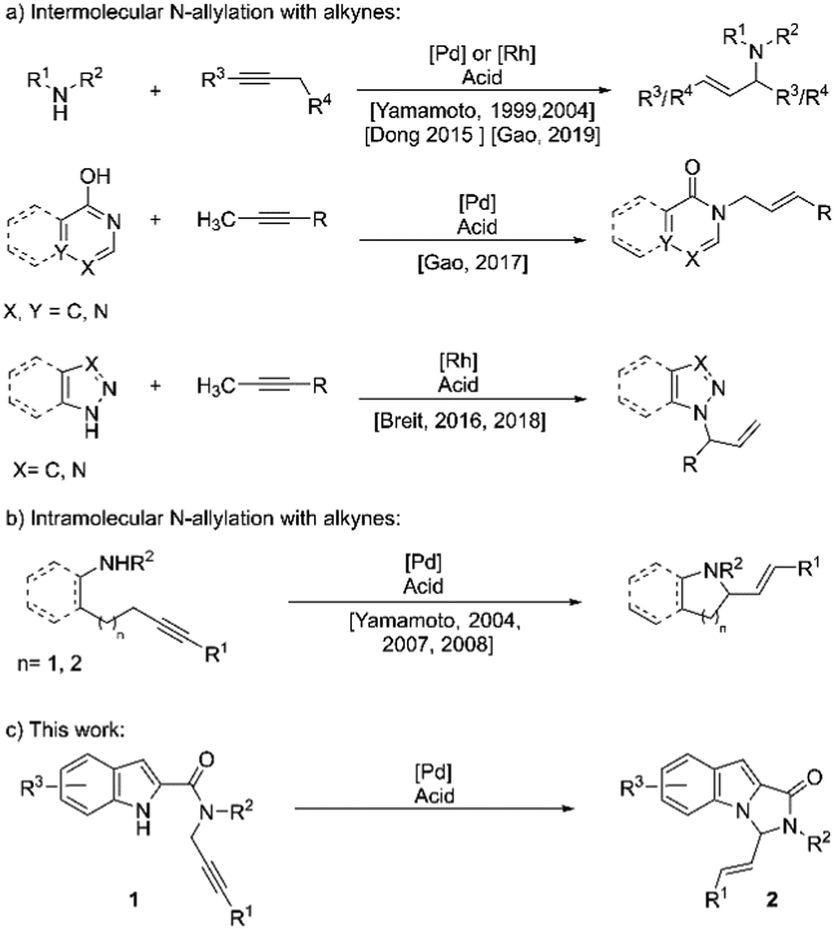 | ||
| Scheme 1 Transition metal catalysed N-allylation with alkynes. | ||
Fused polycyclic N-heterocycles form the structural core of a large number of pharmaceutically relevant compounds.10 N-fused indoles and pyrroles, particularly imidazo-[1,5-a]indoles and pyrrolo-[1,2-c]imidazoles, and their derivatives have been found to exhibit a range of biological activities such as CNS depressants,11 light-dependent TNF-∝ antagonists,12 NPS antagonists,13 and aldose reductase inhibitors,14 among others. These motifs have also found applications in optoelectronics.15
We envisioned that substrates of the type 1 (Scheme 1c), with suitably N-tethered alkyne, would provide direct access to imidazo-[1,5-a]indole and pyrrolo[1,2-c]imidazole skeletons via an intramolecular allylation with the allyl metal complex generated from the alkyne. Considering the regioselectivity desired, a combination of Pd(0) and a Brønsted acid was anticipated to be a suitable catalyst system for the reaction.5–8 We herein report the results of our studies on Pd and acid-catalyzed synthesis of fused N-heterocycles via a direct intramolecular N-allylation with alkynes (Scheme 1c). To the best of our knowledge, this constitutes the first example of an N-allylation of indoles with alkynes.
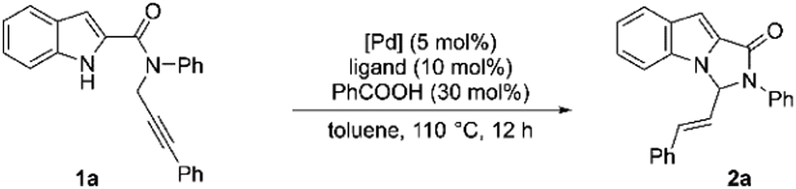
At the outset, our model substrate 1a was heated at 110 °C with 5 mol% of Pd(PPh3)4 and 10 mol% of benzoic acid in toluene under an Argon atmosphere. Gratifyingly, after 12 h, the intended product 2a was isolated as a single E-isomer in 45% yield (Table 1, entry 1). The structure and stereochemistry of 2a were confirmed by NMR and X-ray diffraction analysis. An increase in yield was observed by increasing the amount of acid catalyst to 30 mol% (Table 1, entry 2). Adding 10 mol% of triphenylphosphine additionally to the reaction did not lead to much improvement in the yield (Table 1, entry 3). Test reactions showed that both Pd complex and acid were crucial for the reaction to proceed (Table 1, entries 4 and 5). The use of Pd2(dba)3 as the Pd source led to a slight decrease in the yield (Table 1, entry 6). No reaction was observed when Pd(OAc)2 was used as the catalyst (Table 1, entry 7). We then screened the effect of different phosphine ligands on the reaction using 5 mol% of Pd(PPh3)4 and 30 mol% benzoic acid as the catalysts. The use of 10 mol% of tri(o-tolylphosphine) improved the yield to 70% (Table 1, entry 8). Other phosphines with electron-rich phenyl substituents were found to be inferior ligands than tri(o-tolylphosphine) (Table 1, entries 9–11). To our delight, tris(2-furyl)phosphine was found to be a better ligand and improved the yield to 82% (Table 1, entry 12). The use of dioxane as a solvent with same reaction led to a major drop in the yield (Table 1, entry 13). Tri(p-fluorophenyl)phosphine as ligand with electron-deficient phenyl substituents led to a substantial decrease in yield (Table 1, entry 14). We also tested an aliphatic phosphine, which, however, was found to be an inferior ligand to aromatic phosphines (Table 1, entry 15). A bidentate phosphine was also tested, but the desired product was isolated in only 18% yield after 12 h (Table 1, entry 16).
|
|
|||
|---|---|---|---|
| Entry | [Pd] | Ligand | Yieldb (%) |
| a Unless noted otherwise, reaction conditions: 1a (0.1 mmol), Pd catalyst (5 mol%), ligand (10 mol%), benzoic acid (30 mol%) in dry toluene (0.7 mL) were heated at 110 °C under argon in a Schlenk tube for 12 h. b Isolated yields. c Benzoic acid (0.01 mmol, 10 mol%). d In absence of Pd catalyst. e In absence of benzoic acid. f Dry dioxane as solvent (0.7 mL). | |||
| 1c | Pd(PPh3)4 | — | 45 |
| 2 | Pd(PPh3)4 | — | 56 |
| 3 | Pd(PPh3)4 | PPh3 | 58 |
| 4d | — | — | — |
| 5e | Pd(PPh3)4 | — | — |
| 6 | Pd2(dba)3 | PPh3 | 52 |
| 7 | Pd(OAc)2 | — | — |
| 8 | Pd(PPh3)4 | P(o-tolyl)3 | 70 |
| 9 | Pd(PPh3)4 | P(p-tolyl)3 | 53 |
| 10 | Pd(PPh3)4 | P(2,4,6-trimethylphenyl)3 | 57 |
| 11 | Pd(PPh3)4 | P(o-methoxyphenyl)3 | 62 |
| 12 | Pd(PPh3)4 | P(2-furyl)3 | 82 |
| 13f | Pd(PPh3)4 | P(2-furyl)3 | 46 |
| 14 | Pd(PPh3)4 | P(p-fluorophenyl)3 | 24 |
| 15 | Pd(PPh3)4 | PCy3 | 34 |
| 16 | Pd(PPh3)4 | DPEPhos | 18 |
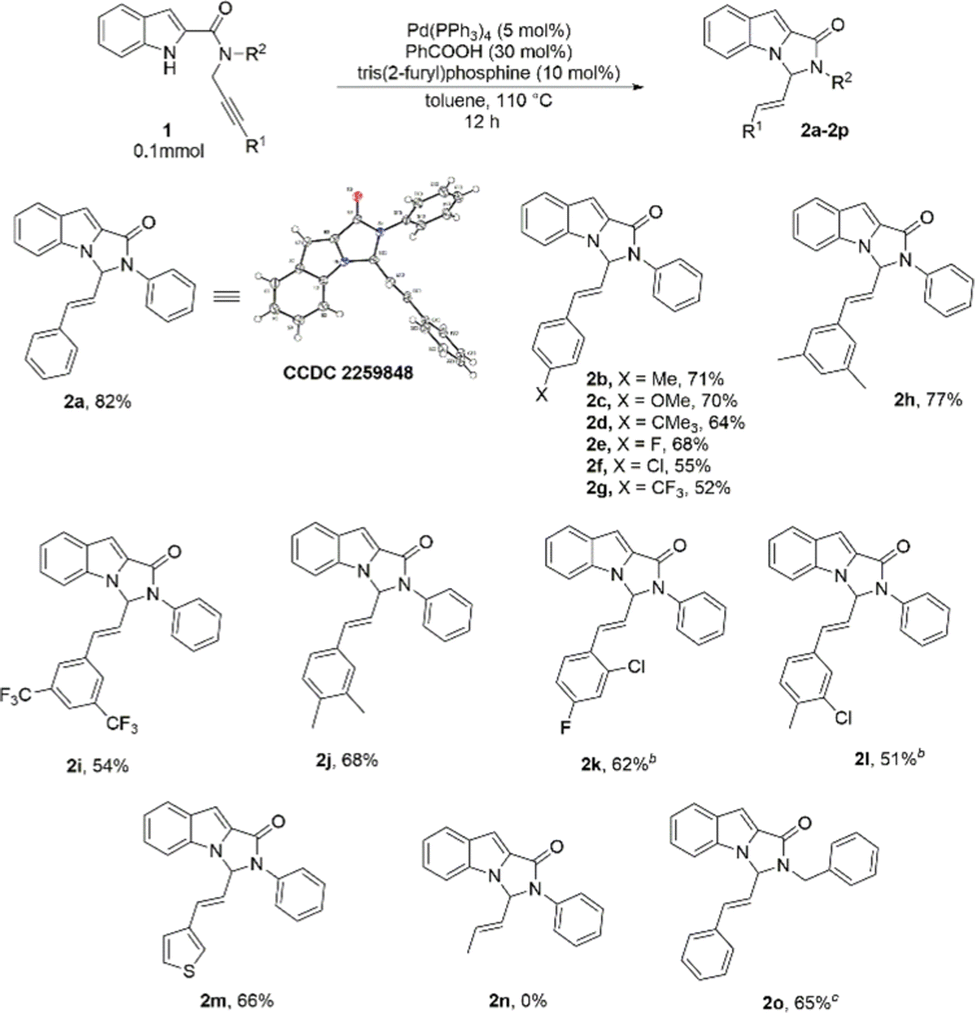
With the optimized reaction conditions, we then set out to explore the substrate scope of the reaction (Table 2). The scope with respect to changing R1 on the alkyne part was first studied. Phenyl rings with electron-donating as well as electron-withdrawing substituents at the 4-position were well tolerated, and the corresponding substrates formed the cyclized products in good yields (Table 2, 2b–2g). 3,5-disubstituted phenyl rings were also tested, and the substrates 1h and 1i reacted well under the standard reaction conditions (Table 2, 2h, 2i). In most cases, phenyl rings with electron-donating groups proved to be slightly better substituents than phenyl rings with electron withdrawing groups. Several other disubstituted phenyl rings, with substituents at different positions of the ring were screened and in all the cases, the products could be isolated in reasonably good yields (Table 2, 2j–2l). A heterocyclic substituent, 3-thienyl, was also explored, and gratifyingly, the product 2m was formed in 66% yield (Table 2, 2m). The aliphatic substituent, methyl, however, was not found to be suitable and the desired product was not formed under the standard reaction conditions when 1n was used as the substrate (Table 2, 2n).4c,8f,16 We also attempted changing R2 substituent on the amide Nitrogen from phenyl to benzyl and the corresponding product was obtained in 65% yield (Table 2, 2o).
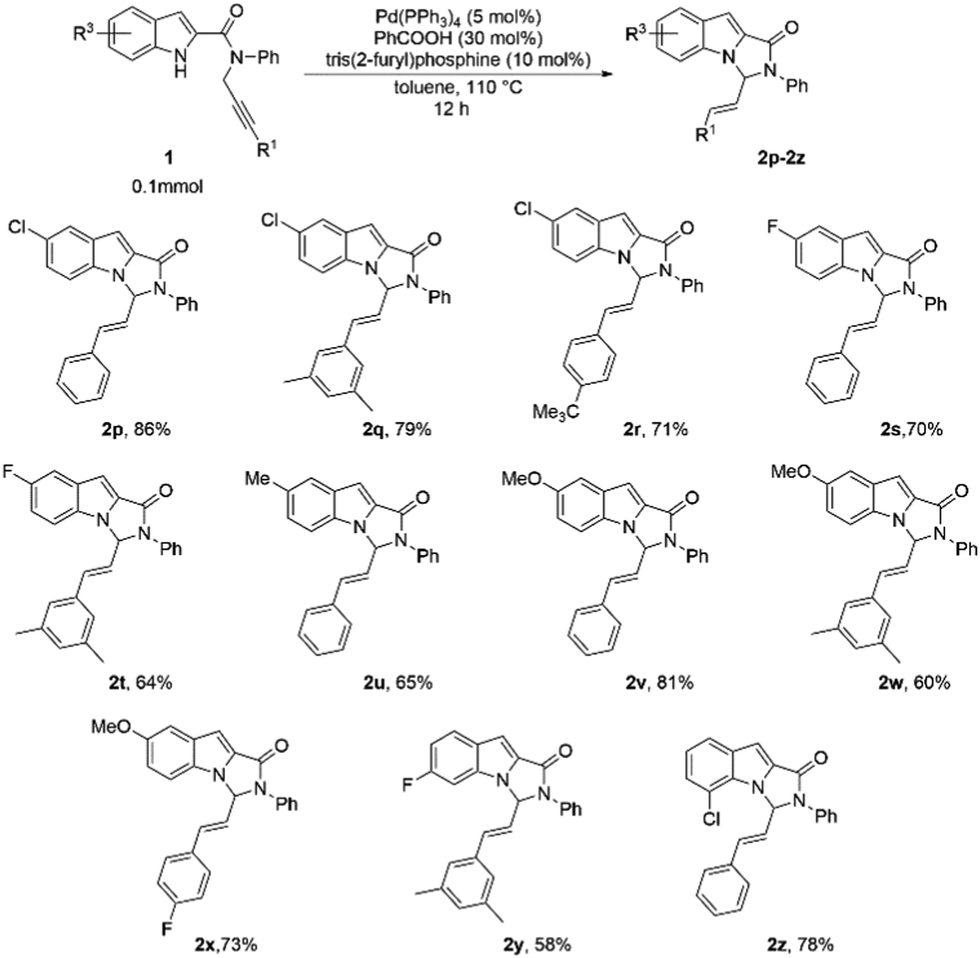
The reaction showed versatility with respect to changing substituents on the indole ring (Table 3). Introduction of electron-withdrawing groups at position 5 of the indole afforded the products in high yields (Table 3, 2p–2t). Notably, a combination of these substituents with different R1 groups was also found to be suitable, and the corresponding substrates formed the products in consistently good yields (64–86%; Table 3, 2q, 2r, 2t). The substrates with electron-releasing substituents at position 5 were also tested and these too reacted remarkably under the standard reaction conditions, even with several different R1 substituents (Table 3, 2u–2x). We also explored the effect of substitutions at positions 6 and 7 of the indole ring, and to our delight, the reaction was well tolerant of these substitutions (Table 3, 2y, 2z).
| a Isolated yields. |
|---|
|
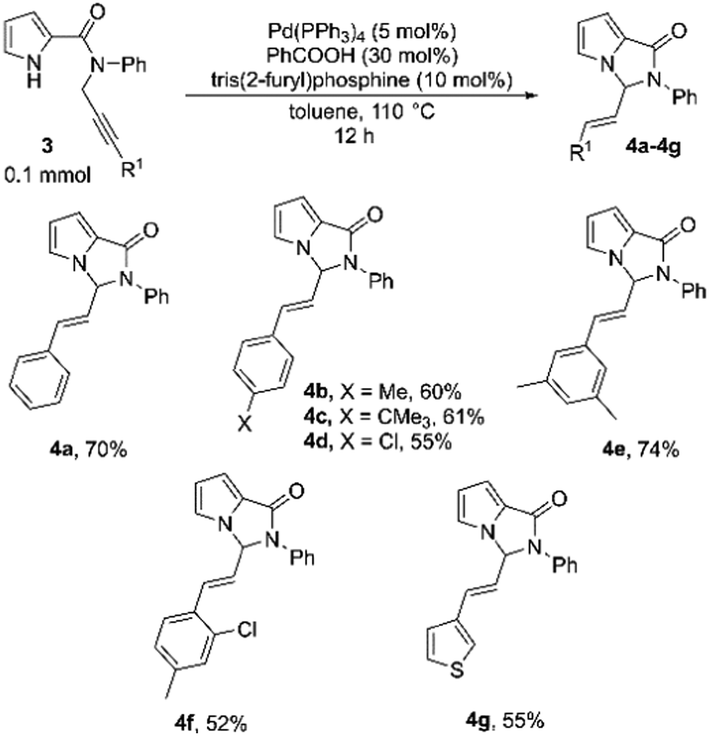
Encouraged by the results on the indole-based N-substituted propargyl amides, we then proceeded to explore the cyclization of pyrrole-based amides 3a–3g (Table 4). The scope of this system with respect to different substituents on the alkyne was screened. The substrate 3a with R1 as phenyl, under the optimized reaction conditions, reacted smoothly, and the product was formed in 70% yield (Table 4, 4a). Satisfyingly, substituted phenyl rings with both electron-releasing as well as electron-withdrawing substituents at different positions could be employed, and the corresponding products were isolated in moderate to good yields (52–74%; Table 4, 4b–4f). The 3-thienyl substituent was also tolerated, and the substrate 3g formed the cyclized product in 55% yield (Tables 4, 4g).
| a Isolated yields. |
|---|
|
To illustrate the synthetic utility of the product, 2a was first subjected to hydrogenation using 10% Pd/C. The reduction of allylic double bond proceeded satisfactorily, and the product 5 was formed in 94% yield (Scheme 2). The indole moiety could also be derivatized further using NBS leading to the product 6, which upon Suzuki coupling with phenylboronic acid led to the product 7 (Scheme 2).
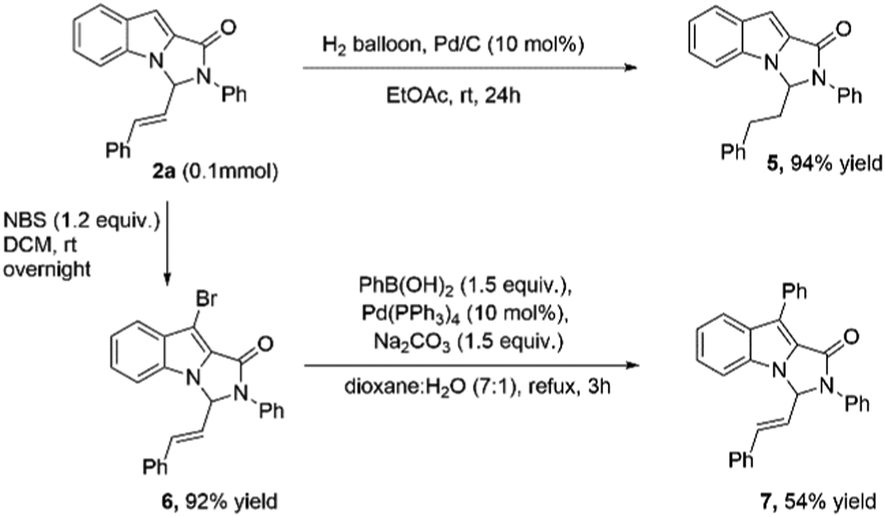 | ||
| Scheme 2 Synthetic transformations of product 2a. | ||
Based on the literature reports4,5d,8f and our mechanistic studies,17 we hypothesize that Pd(0) forms a Pd-carboxylate species I upon oxidative addition with the carboxylic acid. An insertion of I into the alkyne of substrate 1a generates species II. A subsequent β-hydrogen elimination releases the allene intermediate III, which, upon hydropalladation with I, forms the active π-allyl species IV. The selective nucleophilic insertion of N–H of indole then releases product 2a and regenerates Pd(0) (Scheme 3).
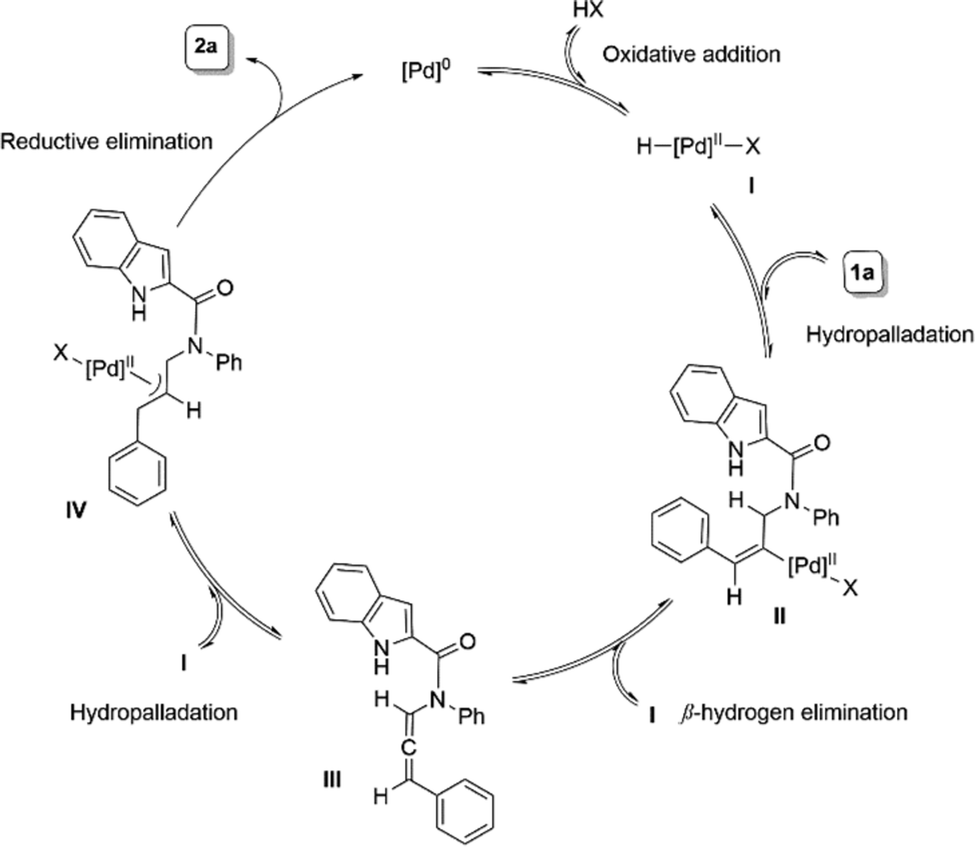 | ||
| Scheme 3 Most probable mechanism. | ||
We have developed the first catalytic intramolecular N-allylation of indoles and pyrroles with alkynes. The reaction employs a Pd(0) and Brønsted acid combined catalytic system. The use of alkynes as allene surrogates enables a direct, completely atom-economic allylation without employing a leaving group, oxidizing agent, or an additional step for the formation of allenes. Biologically important N-fused heterocycles, imidazo-[1,5a]indoles, and pyrrolo-[1,2c]imidazoles could be accessed in high yields under the reaction conditions. The system demonstrates a broad substrate scope and installs a synthetically relevant allyl substituent with the concomitant generation of a stereogenic center. The development of an asymmetric version of the reaction is currently ongoing.
Financial supports by IISER Berhampur and the Science and Engineering Research Board (SERB), Government of India (CRG/2018/000130) are gratefully acknowledged by the authors. C. D. thanks the Ministry of Education for PMRF. The authors are thankful to Dr C. S. Purohit, School of Chemical Sciences, NISER Bhubaneshwar for his help in recording the X-ray data.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) G. Wu, J.-R. Wu, Y. Huang and Y.-W. Yang, Chem. – Asian J., 2021, 16, 1864–1877 CrossRef CAS PubMed; (b) B. M. Trost, Tetrahedron, 2015, 71, 5708–5733 CrossRef CAS PubMed; (c) J. F. Hartwig and L. M. Stanley, Acc. Chem. Res., 2010, 43, 1461 CrossRef CAS PubMed; (d) J. Tsuji, Palladium Reagents and Catalysts: New Perspectives for the 21st Century, John Wiley & Sons, Chichester, 2004, pp. 431–517 CrossRef.
- (a) P. S. Wang and L. Z. Gong, Acc. Chem. Res., 2020, 53, 2841–2854 CrossRef CAS PubMed; (b) A. M. Kazerouni, Q. A. McKoy and S. B. Blakey, Chem. Commun., 2020, 56, 13287–13300 RSC; (c) F. Liron, J. Oble, M. M. Lorion and G. Poli, Eur. J. Org. Chem., 2014, 5863–5883 CrossRef CAS.
- (a) J. Liu, Q. Zhou, Y. Lin, Z.-L. Wu, T. Cai, W. Wen, Y. Huang and Q. Guo, ACS Catal., 2023, 13, 6013–6022 CrossRef CAS; (b) H. Xu, M. Wang, Y. Liang, Y. Zhao and Z. Shi, Nat. Synth., 2022, 1, 227–234 CrossRef; (c) R. Blieck, M. Taillefer and F. Monnier, Chem. Rev., 2020, 120, 13545–13598 CrossRef CAS PubMed; (d) P. Koschker and B. Breit, Acc. Chem. Res., 2016, 49, 1524–1536 CrossRef CAS PubMed; (e) T. Lu, Z. Lu, Z.-X. Ma, Y. Zhang and R. P. Hsung, Chem. Rev., 2013, 113, 4862–4904 CrossRef CAS PubMed; (f) E. Beccalli, A. Bernasconi, E. Borsini, G. Broggini, M. Rigamonti and G. Zecchi, J. Org. Chem., 2010, 75, 6923–6932 CrossRef CAS PubMed; (g) Y. Yamamoto, M. Al-Masum and N. Asao, J. Am. Chem. Soc., 1994, 116, 6019–6020 CrossRef CAS.
- (a) G. Cera and G. Maestri, Chemcatchem, 2022, 14, e202200295 CrossRef CAS; (b) A. M. Haydl, B. Breit, T. Liang and M. J. Krische, Angew. Chem., Int. Ed., 2017, 56, 11312–11325 CrossRef CAS PubMed; (c) B. M. Trost, Chem. – Eur. J., 1998, 4, 2405–2412 CrossRef CAS.
- For selected examples of allylation of carbon nucleophile with alkynes, see: (a) W. Ren, Q.-M. Zuo, Y. Niu and S. Yang, Org. Lett., 2019, 21, 7956–7960 CrossRef CAS PubMed; (b) G. Cera, M. Lanzi, D. Balestri, N. D. Ca’, R. Maggi, F. Bigi, M. Malacria and G. Maestri, Org. Lett., 2018, 20, 3220–3224 CrossRef CAS PubMed; (c) F. A. Cruz and V. M. Dong, J. Am. Chem. Soc., 2017, 139, 1029–1032 CrossRef CAS PubMed; (d) S. Gao, Z. Wu, X. Fang, A. Lin and H. Yao, Org. Lett., 2016, 18, 3906–3909 CrossRef CAS PubMed; (e) N. T. Patil, D. Song and Y. Yamamoto, Eur. J. Org. Chem., 2006, 4211–4213 CrossRef CAS; (f) N. T. Patil, I. Kadota, A. Shibuya, Y. S. Gyoung and Y. Yamamoto, Adv. Synth. Catal., 2004, 346, 800–804 CrossRef CAS.
- For selected examples of allylation of oxygen nucleophile with alkynes, see: (a) Y. Ogiwara, K. Sato and N. Sakai, Org. Lett., 2017, 19, 5296–5299 CrossRef CAS PubMed; (b) Z. Li and B. Breit, Angew. Chem., Int. Ed., 2016, 128, 8580–8583 CrossRef; (c) W. Zhang, A. R. Haight and M. C. Hsu, Tetrahedron Lett., 2002, 43, 6575–6578 CrossRef CAS; (d) B. M. Trost, W. Brieden and K. H. Baringhaus, Angew. Chem., Int. Ed. Engl., 1992, 31, 1335–1336 CrossRef.
- For selected examples of allylation of sulphur nucleophile with alk-ynes, see: (a) C. Lu, H. Chen, D. Chen, H. Wang, Z. Yang, J. Gao and H. Jin, Org. Biomol. Chem., 2016, 14, 10833–10839 RSC; (b) K. Xu, V. Khakyzadeh, T. Bury and B. Breit, J. Am. Chem. Soc., 2014, 136, 16124–16127 CrossRef CAS PubMed.
- For selected examples of intermolecular allylation of nitrogen nucle-ophile with alkynes, see: (a) C.-J. Lu, X. Yu, D. Chen, H. Wang, Q. Song and J. Gao, Org. Biomol. Chem., 2019, 17, 3545–3551 RSC; (b) D. Berthold and B. Breit, Org. Lett., 2018, 20, 598–601 CrossRef CAS PubMed; (c) C.-J. Lu, D. Chen, H. Chen, H. Wang, H. Jin, X. Huang and J. Gao, Org. Biomol. Chem., 2017, 15, 5756–5763 RSC; (d) A. M. Haydl, L. J. Hilpert and B. Breit, Chem. – Eur. J., 2016, 22, 6547–6551 CrossRef CAS PubMed; (e) Q.-A. Chen, Z. Chen and V. M. Dong, J. Am. Chem. Soc., 2015, 137, 8392–8395 CrossRef CAS PubMed; (f) I. Kadota, A. Shibuya, L. M. Lutete and Y. Yamamoto, J. Org. Chem., 1999, 64, 4570–4571 CrossRef CAS PubMed . For selected examples of intramolecular allylation of nitrogen nucleophile with alkynes, see: ; (g) M. Narsireddy and Y. Yamamoto, J. Org. Chem., 2008, 73, 9698–9709 CrossRef CAS PubMed; (h) N. T. Patil, H. Wu and Y. Yamamoto, J. Org. Chem., 2007, 72, 6577–6579 CrossRef CAS PubMed; (i) Y. Yamamoto, et al. , Tetrahedron Lett., 2005, 46, 2101–2103 CrossRef; (j) L. M. Lutete, I. Kadota and Y. Yamamoto, J. Am. Chem. Soc., 2004, 126, 1622–1623 CrossRef CAS PubMed.
- D. J. Faulkner, Nat. Prod. Rep., 2001, 1–49 RSC.
- (a) I. Ngantchou, B. Nyasse, C. Denier, C. Blonski, V. Hannaert and B. Schneider, Bioorg. Med. Chem. Lett., 2010, 20, 3495 CrossRef CAS PubMed; (b) V. Sharma, P. Kumar and D. Pathak, J. Heterocycl. Chem., 2010, 47, 491–502 CrossRef CAS; (c) L. S. Fernandez, M. S. Buchanan, A. R. Carroll, Y. J. Feng, R. J. Quinn and V. M. Avery, Org. Lett., 2009, 11, 329 CrossRef CAS PubMed; (d) M. Nettekoven, J.-M. Plancher, H. Richter, O. Roche and S. Taylor, WO US 2007/0135416 A1, 2007; (e) J.-F. Liu, Z.-Y. Jiang, R.-R. Wang, Y.-T. Zheng, J.-J. Chen, X.-M. Zhang and Y.-B. Ma, Org. Lett., 2007, 9, 4127 CrossRef CAS PubMed.
- (a) W. B. Wright, US Pat., 3565902, 1971; Chem. Abstr., 1971, 75, 36033 Search PubMed; (b) M. Varasi, F. Heidempergher, C. Caccia and P. Salvati, PCT Int., Appl., WO 9532209, 1995; Chem. Abstr., 1995, 124, 232456.
- M. E. Voss, P. H. Carter, A. J. Tebben, P. A. Scherle, G. D. Brown, L. A. Thompson, M. Xu, Y. C. Lo, G. Yang, R.-Q. Liu, P. Strzemienski, J. G. Everlof, J. M. Trzaskos and C. P. Decicco, Bioorg. Med. Chem. Lett., 2003, 13, 533–538 CrossRef CAS PubMed.
- F. Micheli, R. Di Fabio, A. Giacometti, A. Roth, E. Moro, G. Merlo, A. Paio, E. Merlo-Pich, S. Tomelleri, F. Tonelli, P. Zarantonello, L. Zonzini and A. M. Capelli, Bioorg. Med. Chem. Lett., 2010, 20, 7308–7311 CrossRef CAS PubMed.
- (a) W. Kong, X. Chen, M. Wang, H. Dai and J. Yu, Org. Lett., 2017, 20, 284–287 CrossRef PubMed; (b) I. Yamawaki, Y. Matsushita, N. Asaka, K. Ohmori, N. Nomura and K. Ogawa, Eur. J. Med. Chem., 1993, 28, 481–498 CrossRef CAS.
- (a) L. A. Oparina, N. A. Kolyvanov, I. A. Ushakov, L. P. Nikitina, O. V. Petrova, L. N. Sobenina, K. B. Petrushenko and B. A. Trofimov, Int. J. Mol. Sci., 2023, 24, 3404 CrossRef CAS PubMed; (b) B. Banerji, S. Chatterjee, K. Chandrasekhar, S. Ghosh, K. Mukherjee and C. Mandal, J. Org. Chem., 2018, 83, 13011–13018 CrossRef CAS PubMed.
- A diene was isolated under standard reaction conditions. Please see ESI† for more details. For example of Pd catalyzed 1,3-diene formation from propargylic alkynes, see: N. T. Patil, Z. Huo, G. B. Bajracharya and Y. Yamamoto, J. Org. Chem., 2006, 71, 3612–3614 CrossRef CAS PubMed.
- For mechanistic studies, please see ESI†.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, 1 mmol synthesis, NMR data. CCDC 2259848. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc05023h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |