
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcceleration of hydrolytic ring opening of N7-alkylguanine by the terminal carbamoyl group of glycidamide†
Shigenori
Iwai
 *a,
Yuta
Hayashi
b,
Tomohiro
Baba
a and
Yasutaka
Kitagawa
b
*a,
Yuta
Hayashi
b,
Tomohiro
Baba
a and
Yasutaka
Kitagawa
b
aDivision of Chemistry, Graduate School of Engineering Science, Osaka University, 1-3 Machikaneyama, Toyonaka, Osaka 560-8531, Japan. E-mail: iwai.s.es@osaka-u.ac.jp
bDivision of Chemical Engineering, Graduate School of Engineering Science, Osaka University, 1-3 Machikaneyama, Toyonaka, Osaka 560-8531, Japan
First published on 1st April 2024
Abstract
Hydrolytic ring opening of guanine N7-adducts with compounds containing an oxacyclopropane ring, namely glycidamide, glycidol and 1,2-epoxybutane, was analyzed, and the reaction of the glycidamide adduct was the fastest. The differences in the reaction rates were confirmed by theoretical calculations.
Acrylamide (AA) is produced by heating food at high temperature.1 One of the pathways of AA formation is the Maillard reaction between asparagine and a reducing sugar,2 and AA is metabolically converted to glycidamide (GA, Scheme 1) by cytochrome P450 2E1 (CYP2E1) in cells.3 The oxacyclopropane ring of GA undergoes nucleophilic reactions, and the main product formed initially in DNA is the N7 adduct of 2′-deoxyguanosine (N7-GA-dG), as shown in Scheme 1.4 Since N7-alkylguanine is positively-charged, the glycosidic bond of this modified nucleoside is labile, and in the case of adduct formation with GA, N7-(2-carbamoyl-2-hydroxyethyl)guanine (N7-GA-Gua) is released as a stable final product, leaving an abasic site in the DNA strand.4 N7-Alkylguanine in DNA also undergoes hydrolytic opening of the imidazole ring to yield a so-called formamidopyrimidine (FAPy) lesion (GA-FAPy-dG).5 AA is classified as group 2A (probably carcinogenic to humans) by the International Agency for Research on Cancer, and GA formation is considered to be the route to the genotoxicity and carcinogenicity of AA.6 On the other hand, glycidyl esters are formed during the refining process of vegetable oils7 and the thermal processing of food using oils,8 and are hydrolyzed to glycidol (GO, Fig. 1a) and fatty acid after ingestion. GO is another genotoxic carcinogen in group 2A,9 but compared to GA, the information available for GO is limited. In this study, we have analyzed the ring-opening reactions of the guanine adducts with these compounds in oligonucleotides and found that the terminal functional groups affect the reaction rates.
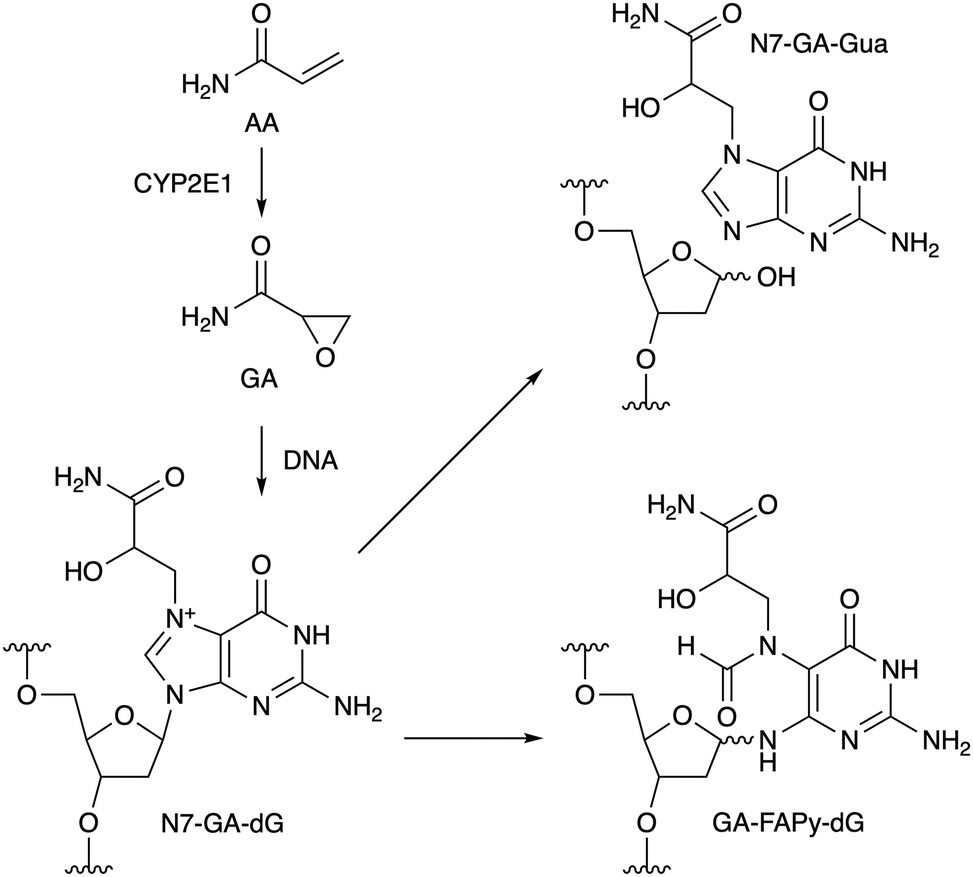 | ||
| Scheme 1 Reaction of GA with guanine in DNA. | ||
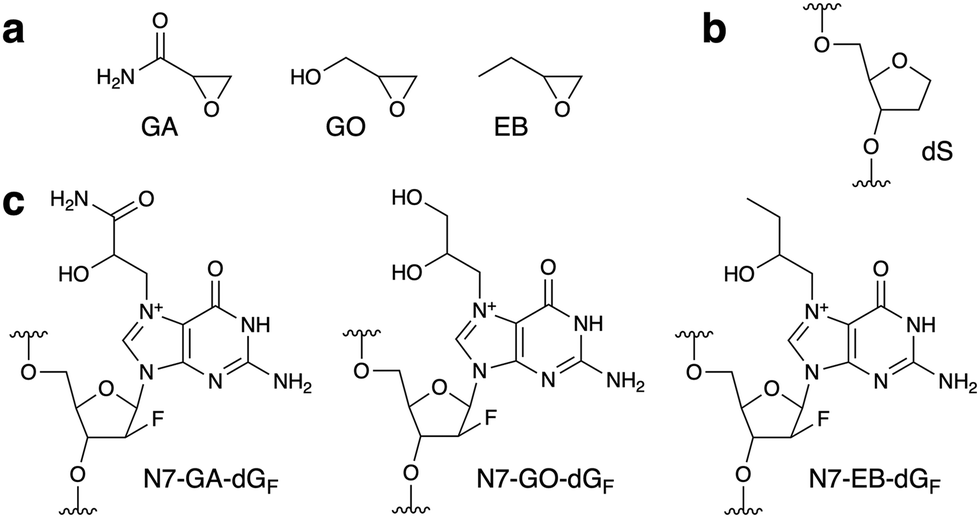 | ||
| Fig. 1 (a) Structures of the compounds used in this study. (b) and (c) Structures of the abasic site analog (dS) and the N7-adducts of dGF in oligonucleotides. | ||
Since the glycosidic bond of N7-alkylguanine is labile, we used 9-(2-deoxy-2-fluoro-β-D-arabinofuranosyl)guanine (dGF) for adduct formation in oligonucleotides. The fluorine atom at the upper position of C2′ stabilizes the glycosidic bond due to its large electronegativity and maintains the DNA-type sugar conformation, as reported for N7-methylguanine.10 Prevention of the glycosidic bond cleavage by the 2′-F modification was confirmed by analyzing the reaction of dGF with GA. LC-MS analysis revealed that the product of this reaction was the N7-GA adduct of dGF, and the depurination product (N7-GA-Gua) was not detected (Fig. S1, ESI†). Prior to the oligonucleotide preparation, we performed two types of preliminary experiments. One was to detect the formation of the N7-GA adduct of dGF (N7-GA-dGF, Fig. 1c) and its conversion to the FAPy derivative. To facilitate the identification of the modified base in an oligonucleotide by a spectroscopic method, a 9-mer containing dGF in a stretch of abasic site analogs ((2R,3S)-2-(hydroxymethyl)tetrahydrofuran-3-ol, abbreviated as dS, Fig. 1b), d(SSSSGFSSSS), was designed. Since dS does not absorb UV, we expected that the product could be identified by measuring the UV-absorption spectrum. According to the method for N7-GA-Gua preparation,4 the oligonucleotide was treated with a large excess of GA in 50 mM sodium phosphate (pH 7.0) at 60 °C for 4 h, and the product was purified by reversed-phase HPLC. The product had a shorter retention time than the starting 9-mer in an anion-exchange HPLC analysis (Fig. S2b, ESI†), attributable to the positive charge of the N7-GA adduct, and its absorption maximum was 259 nm, which is identical to that of N7-methylguanosine.11 This oligonucleotide was incubated in 0.1 M sodium phosphate (pH 8.0) at 37 °C for 20 h, and a new product peak with an absorption maximum of 274 nm, which is close to that of Me-FAPy-G (273 nm),11 was detected at a longer retention time (Fig. S2c, ESI†). These results indicated that the adduct-containing oligonucleotides could be obtained by these procedures. The other experiment was to analyze the reaction of GA with nucleosides other than 2′-deoxyguanosine. As reported previously,12 2′-deoxyadenosine and 2′-deoxycytidine reacted with GA (Fig. S3, ESI†), and thus we decided to use only thymidine in the sequences flanking dGF for the reaction with GA.
A 5′-phosphorylated 9-mer, p-d(TTTTGFTTTT), was employed in this study. The phosphate moiety at the 5′ end, as well as the hydroxy group at the 3′ end, can be used in DNA ligase reactions to prepare sequence-defined longer oligonucleotides for utilization in biochemical experiments. For the preparation of the GA-adducted oligonucleotide, this 9-mer was treated with GA in 50 mM sodium phosphate (pH 7.0) at 60 °C, and to increase the yield, the incubation time was extended to 12 h. The reversed-phase HPLC analysis revealed a product peak at a retention time shorter than that of the starting 9-mer (Fig. S4a, ESI†). This product (GA 9-mer) was isolated using a semi-preparative column, and after further purification (described in ESI†), its purity and the adduct formation were confirmed by mass spectrometry (Fig. S6a, ESI†). The 9-mers containing the GO and 1,2-epoxybutane (EB) adducts (Fig. 1c), designated as GO 9-mer and EB 9-mer, respectively, were prepared in the same manner (Fig. S5, S6b and c, ESI†).
To confirm that the GO-adducted guanine base undergoes hydrolytic ring opening in the same manner as the GA adduct, GA and GO 9-mers dissolved in 0.1 M sodium phosphate (pH 8.0) were incubated at 37 °C. Although the molecular weights of the products were 18 mass-units larger than the starting 9-mers (Fig. S7, ESI†), which demonstrated that the reaction was hydrolysis in both cases, the peak of the product obtained from GO 9-mer was unexpectedly smaller than that observed for GA 9-mer (Fig. S8, ESI†). Therefore, the reactions were analyzed carefully using GA, GO and EB 9-mers. As shown in Fig. 2, there were large differences in the reaction rates. The GA-adducted guanine base was hydrolyzed much faster than the GO and EB adducts, and EB 9-mer was hydrolyzed most slowly. When the temperature was raised to 60 °C, the reaction of EB 9-mer became faster (Fig. S9, ESI†).
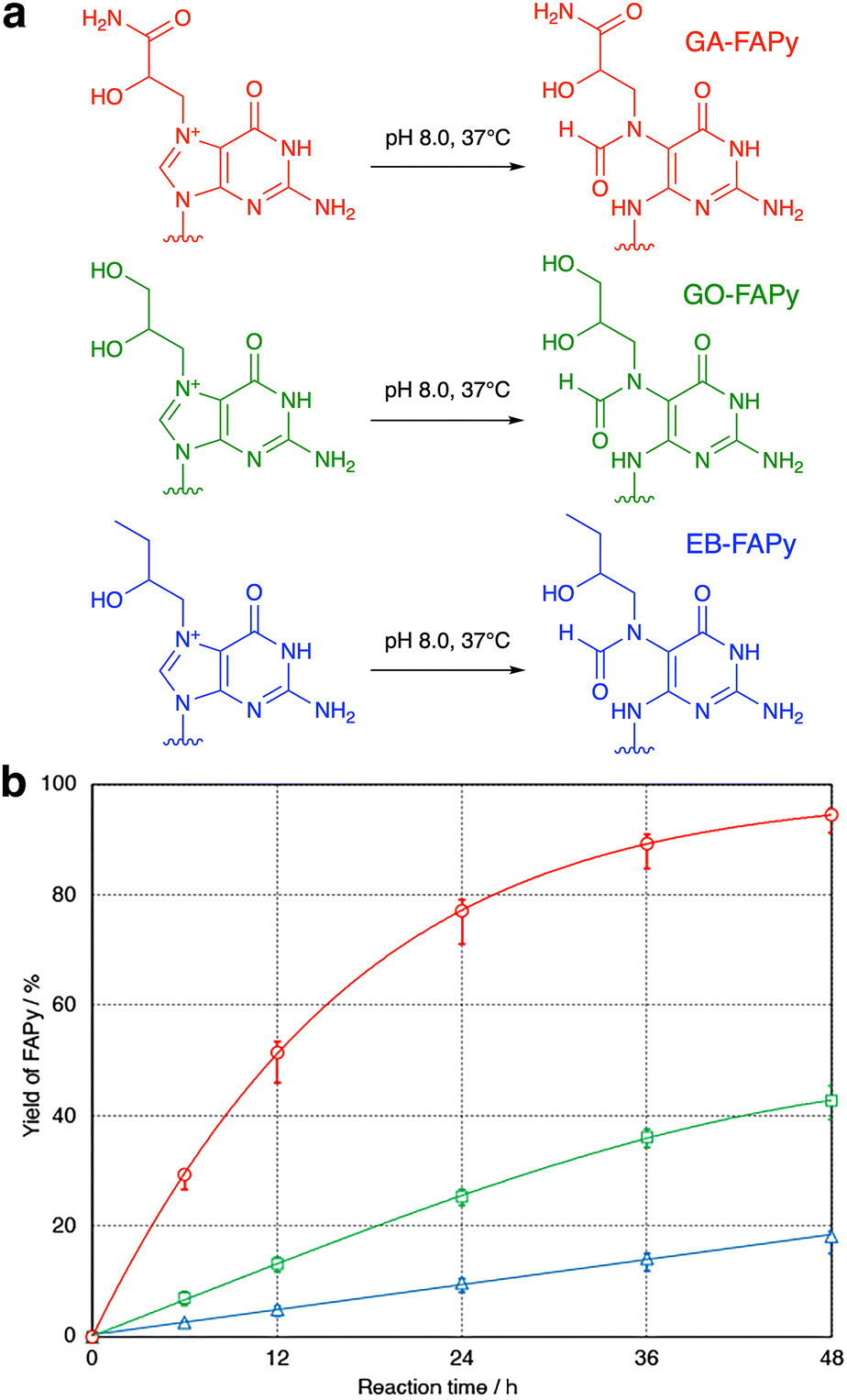 | ||
| Fig. 2 (a) Hydrolytic ring opening to form the FAPy derivatives. (b) Formation of GA-FAPy 9-mer (red circles), GO-FAPy 9-mer (green squares) and EB-FAPy 9-mer (blue triangles) from GA 9-mer, GO 9-mer and EB 9-mer, respectively. | ||
To explain the difference in the reaction rates among the GA, GO and EB adducts, the mechanism was examined by the density functional theory (DFT) method with the B3LYP/6-31G* level of theory.13 To analyze the ring-opening process shown in Fig. 2a, reaction coordinates were obtained by using the model structure shown in Fig. S10a (ESI†). One of the enantiomers at the chiral carbon (the S isomer of the GA adduct) was used for the calculation. As depicted in Fig. 3, the results suggested two intermediates (IMs) and two transition states (TSs). The first step of the reaction is the nucleophilic attack of OH− to the C8 atom of the purine moiety in the guanine adduct with no TS, followed by IM1 production. The second step is the ring-opening reaction to produce IM2 through TS1. The final step is proton transfer to form a formyl group through TS2. The estimated relative energies of MI1, MI2, TS2 and the product structures were in the order of GA ≪ GO ≈ EB, consistent with the experimental results. However, an appropriate transition state between IM1 and IM2 (i.e., TS1) was not found. Therefore, the potential energy curve between IM1 and IM2 was examined by the nudged elastic band (NEB) method,14 as illustrated in Fig. S10b (ESI†). Although the potential energy curve suggested some reaction steps between IM1 and IM2, the expected transition energy was much lower than that of TS2. For this reason, we focused on TS2, which is considered to be the rate-determining step. Among the three compounds, the TS2 energy of the GA adduct was lower than those of the others by about 3 kcal mol−1, and thus the ring-opening reaction is more likely to occur in the GA adduct. Finally, the frontier orbital energies of the guanine N7-adducts were also examined to explain the difference. As illustrated in Fig. S10c (ESI†), LUMOs are distributed on the purine moiety, and the GA adduct shows lower LUMO energy in comparison with the others, suggesting that it reacts more easily with nucleophiles. In addition, an intramolecular hydrogen bond was found in the proton migration reaction from IM2 to TS2, and it is suggested that its strength is considered to contribute to the difference in the reaction barrier of TS2 (Table S1, ESI†).
 | ||
| Fig. 3 Reaction coordinates obtained by DFT calculations. The numbers represent zero-point energy (ZPE)-corrected relative energies. The energy of TS1 was examined by NEB, as explained in Fig. S10b, (ESI†). | ||
The positively-charged N7 adduct of guanine in DNA undergoes two types of reactions, glycosidic bond cleavage and imidazole ring opening, as shown in Scheme 1. In the cases of the GO and EB adducts, the former reaction may occur predominantly, because the latter reaction is slow, as revealed in this study. This possibility was tested by treating unmodified 2′-deoxyguanosine with GA, GO and EB. In our previous study,15 we identified N7-GA-Gua and GA-FAPy-dG in a reaction mixture of 2′-deoxyguanosine with GA by UV-absorption spectra and LC-MS analyses, as shown in Fig. 4a. The same experiment was performed using GO and EB. As shown in Fig. 4b and c, the N7 adducts of the guanine base without the sugar, which had the same absorption maximum as 7-methylguanine (284 nm),11 were obtained, but the products corresponding to GA-FAPy-dG (peak ii in Fig. 4a) were not detected. GA reacts with adenine and cytosine in addition to guanine, as reported previously12 and as shown in Fig. S3 (ESI†). Therefore, the mutation spectrum caused by GA is complicated. In our previous study, we showed that GA-FAPy-dGF preferentially induced a G:C-to-A:T transition mutation, followed by G:C-to-T:A and G:C-to-C:G transversions,15 which can contribute to GA's broad mutation spectrum revealed by next-generation sequencing.16 In the cases of GO17 and EB,18 information about the adducts formed in DNA and mutations caused by them is much less. In this study, we focused on the properties of their N7 adducts of guanine and found a difference that would have consequences for the mutation. Since FAPy derivatives are more stable and mutagenic than their parental N7-dG adducts,19 the N7-GA-dG's distinctive property that the terminal carbamoyl group facilitates the hydrolytic ring opening offers insights into mutagenesis induced by the N7-dG adducts.
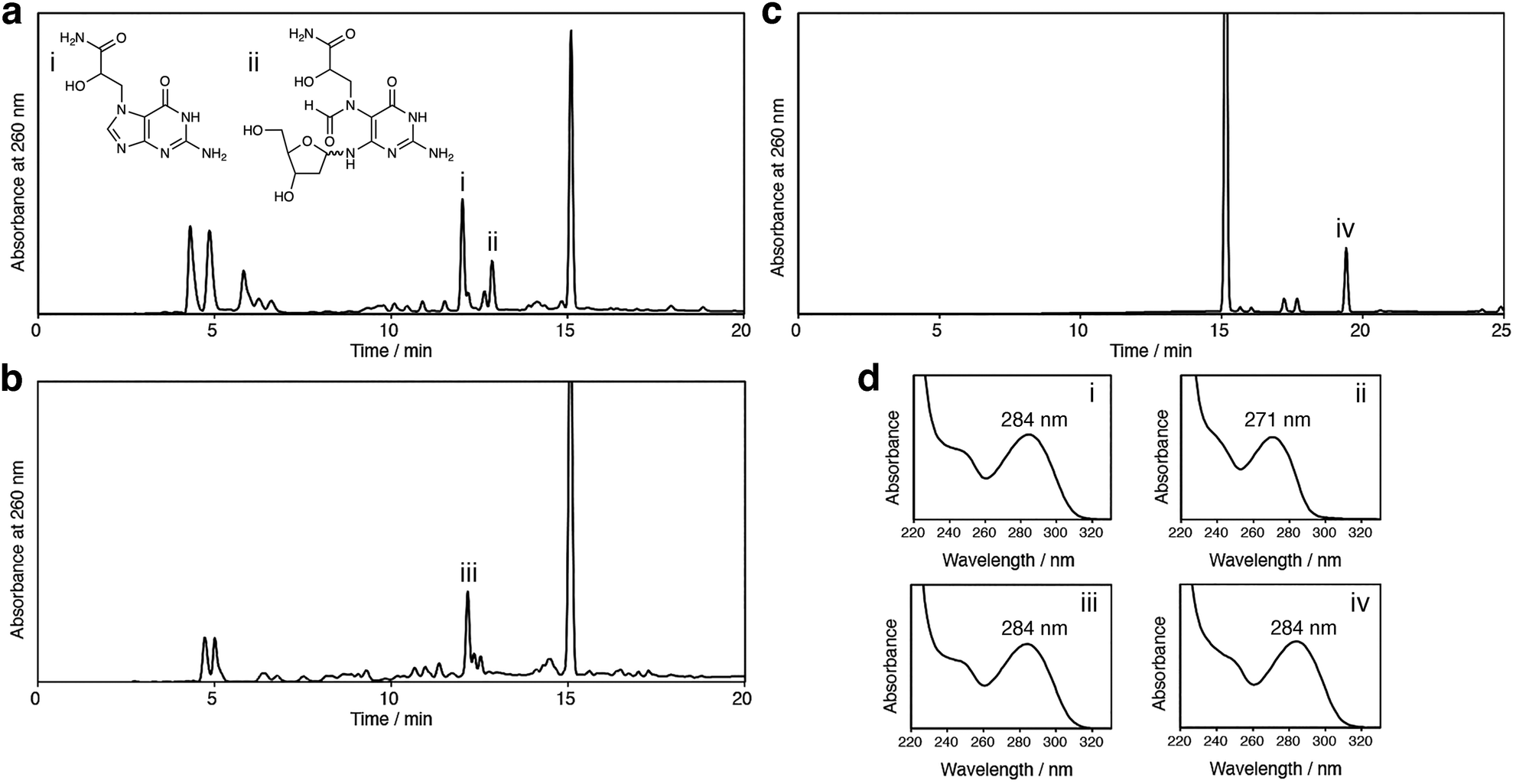 | ||
| Fig. 4 Analyses of the reactions of 2′-deoxyguanosine with GA (a), GO (b) and EB (c). The mixtures of 2′-deoxyguanosine and GA, GO or EB in 0.1 M sodium phosphate (pH 7.0) were incubated at 37 °C for 48 h and then analyzed by reversed-phase HPLC. The largest peak at 15.1 min is remaining 2′-deoxyguanosine, and the UV absorption spectra and absorption maxima of peaks i, ii, iii and iv are shown in (d). | ||
In summary, we have found that the terminal functional groups affect the hydrolytic ring opening of the N7 adducts of guanine with oxacyclopropane-containing compounds. Our calculations showed that the carbamoyl group in the GA adduct stabilizes the intermediates in the ring-opening reaction and lowers the energy of the transition state at the rate-determining step. When 2′-deoxyguanosine was treated with GO or EB, the glycosidic bond cleavage occurred predominantly. Accordingly, the GO- and EB-adducted guanine bases would probably not remain in DNA, and the mutation profile induced by GA should be different from those by GO and EB.
SI performed all the experiments using nucleosides and oligonucleotides. TB analyzed N7-GA-dGF by LC-MS. YH and YK performed the theoretical calculations and analyzed the results. SI and YK wrote the paper.
We thank Dr Jun-ichi Akagi (National Institute of Health Sciences) for helpful discussion on the biological relevance of this study. Y. K. acknowledges JSPS KAKENHI Grant Number 22H02050.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Semla, Z. Goc, M. Martiniaková, R. Omelka and G. Formicki, Physiol. Res., 2017, 66, 205–217 CAS.
- Y. Xu, B. Cui, R. Ran, Y. Liu, H. Chen, G. Kai and J. Shi, Food Chem. Toxicol., 2014, 69, 1–12 CrossRef CAS PubMed.
- S. C. J. Sumner, T. R. Fennell, T. A. Moore, B. Chanas, F. Gonzalez and B. I. Ghanayem, Chem. Res. Toxicol., 1999, 12, 1110–1116 Search PubMed.
- D. Segerbäck, C. J. Calleman, J. L. Schroeder, L. G. Costa and E. M. Faustman, Carcinogenesis, 1995, 16, 1161–1165 CrossRef PubMed.
- S. N. Bamberger, C. K. Malik, M. W. Voehler, S. K. Brown, H. Pan, T. L. Johnson-Salyard, C. J. Rizzo and M. P. Stone, Chem. Res. Toxicol., 2018, 31, 924–935 Search PubMed.
- EFSA Panel on Contaminants in the Food Chain, EFSA J., 2015, 13, 4104 Search PubMed.
- N. Bakhiya, K. Abraham, R. Gürtler, K. E. Appel and A. Lampen, Mol. Nutr. Food Res., 2011, 55, 509–521 CrossRef CAS PubMed.
- K. M. Goh, Y. H. Wong, C. P. Tan and K. L. Nyam, Curr. Res. Food Sci., 2021, 4, 460–469 CrossRef CAS PubMed.
- EFSA Panel on Contaminants in the Food Chain, EFSA J., 2016, 14, 4426 Search PubMed.
- S. Lee, B. R. Bowman, Y. Ueno, S. Wang and G. L. Verdine, J. Am. Chem. Soc., 2008, 130, 11570–11571 CrossRef CAS PubMed.
- K. Yamauchi and M. Kinoshita, J. Chem. Soc., Perkin Trans. 1, 1978, 762–767 RSC.
- G. Gamboa da Costa, M. I. Churchwell, L. P. Hamilton, L. S. Von Tungeln, F. A. Beland, M. M. Marques and D. R. Doerge, Chem. Res. Toxicol., 2003, 16, 1328–1337 Search PubMed; J. Backman, R. Sjöholm and L. Kronberg, Chem. Res. Toxicol., 2004, 17, 1652–1658 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- O.-P. Koistinen, V. Ásgeirsson, A. Vehtari and H. Jónsson, J. Chem. Theory Comput., 2019, 15, 6738–6751 CrossRef CAS PubMed.
- J. Akagi, M. Yokoi, Y. Miyake, T. Shirai, T. Baba, Y.-M. Cho, F. Hanaoka, K. Sugasawa, S. Iwai and K. Ogawa, J. Biol. Chem., 2023, 299, 105002 CrossRef CAS PubMed.
- M. Zhivagui, A. W. T. Ng, M. Ardin, M. I. Churchwell, M. Pandey, C. Renard, S. Villar, V. Cahais, A. Robitaille, L. Bouaoun, A. Heguy, K. Z. Guyton, M. R. Stampfer, J. McKay, M. Hollstein, M. Olivier, S. G. Rozen, F. A. Beland, M. Korenjak and J. Zavadil, Genome Res., 2019, 29, 521–531 CrossRef CAS PubMed; L. Hölzl-Armstrong, J. E. Kucab, S. Moody, E. P. Zwart, L. Loutkotová, V. Duffy, M. Luijten, G. Gamboa da Costa, M. R. Stratton, D. H. Phillips and V. M. Arlt, Arch. Toxicol., 2020, 94, 4173–4196 CrossRef PubMed.
- J. Aasa, D. Vare, H. V. Motwani, D. Jenssen and M. Törnqvist, Mutat. Res., 2016, 805, 38–45 CAS.
- E. W. Vogel and M. J. M. Nivard, Mutat. Res., 1998, 405, 259–271 CrossRef CAS PubMed; R. Thier, F. A. Wiebel and H. M. Bolt, Arch. Toxicol., 1999, 73, 489–492 CrossRef PubMed.
- K. S. Gates, T. Nooner and S. Dutta, Chem. Res. Toxicol., 2004, 17, 839–856 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and supplementary figures. See DOI: https://doi.org/10.1039/d3cc04997c |
| This journal is © The Royal Society of Chemistry 2024 |