
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReductive cyclodimerization of chalcones: exploring the “self-adaptability” of galvanostatic electrosynthesis†
Mauro
Garbini
a,
Andrea
Brunetti
ab,
Riccardo
Pedrazzani
ab,
Magda
Monari
 ab,
Massimo
Marcaccio
ab,
Giulio
Bertuzzi
*ab and
Marco
Bandini
*ab
ab,
Massimo
Marcaccio
ab,
Giulio
Bertuzzi
*ab and
Marco
Bandini
*ab
aDipartimento di Chimica “Giacomo Ciamician”, Alma Mater Studiorum – Università di Bologna, Via P. Gobetti 85, 40129, Bologna, Italy. E-mail: giulio.bertuzzi2@unibo.it; marco.bandini@unibo.it
bCenter for Chemical Catalysis – C3, Dipartimento di Chimica “Giacomo Ciamician”, Alma Mater Studiorum – Università di Bologna, Via P. Gobetti 85, 40129, Bologna, Italy
First published on 28th November 2023
Abstract
The “self-adaptability” of galvanostatic electrolysis was shown to assist a multistage unprecedented chemo- and diastereoselective electrochemically promoted cyclodimerization of chalcones. The process, all involving the reductive events, delivered densely functionalized cyclopentanes featuring five contiguous stereocenters (25 examples, yields of up to 95%, dr values up to >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Dedicated and combined experimental as well as electrochemical investigation revealed the key role of a dynamic kinetic resolution of the aldol intermediate for the reaction mechanism.
:1). Dedicated and combined experimental as well as electrochemical investigation revealed the key role of a dynamic kinetic resolution of the aldol intermediate for the reaction mechanism.
In recent years, the development of effective and sustainable synthetic strategies in organic chemistry has been shown to be tremendously advanced by the diversification of enabling technologies.1 In particular, the highest potential is reached when the application of different techniques dictates reaction outcomes such as chemoselectivity, product distribution and stereoselectivity. This goal extends the shades of the synthetic palette, allowing a multifaceted chemical space, where the outcome can be easily diversified even when drawing from the same pool of starting materials.
The use of synthetic organic electrochemistry (electroChem) not only replaces stoichiometric oxidizing/reducing chemical agents with electrons as traceless equivalents, but also delivers novel reaction pathways and complementary selectivity.2 In this scenario, the peculiar features of galvanostatic electrolysis offer a reaction manifold of unparalleled power and simplicity.3 Self-adapting the reaction potential to the one that is needed for the less demanding process to occur allows for the precise control of cascade processes, even where the subsequent reactive events are energetically more demanding than the previous ones (Fig. 1). This approach has yielded significant landmarks in organic synthesis with applications in non-trivial C–C and C–X bond formations.4 Moreover, by exercising easy and precise control over the quantity and energy of the electrons participating in a redox process, electroChem is an unparalleled protocol for obtaining insights into reaction pathways, in particular by enabling ready access to the isolation of intermediates in a controlled manner. In contrast, traditional stoichiometric or other redox technologies commonly lack the same “live” adaptability and operational tunability during the chemical event.5 On the other hand, the use of harsh chemical reagents, capable of covering all potential ranges of the redox-active functional groups, could be detrimental to the overall process selectivity.
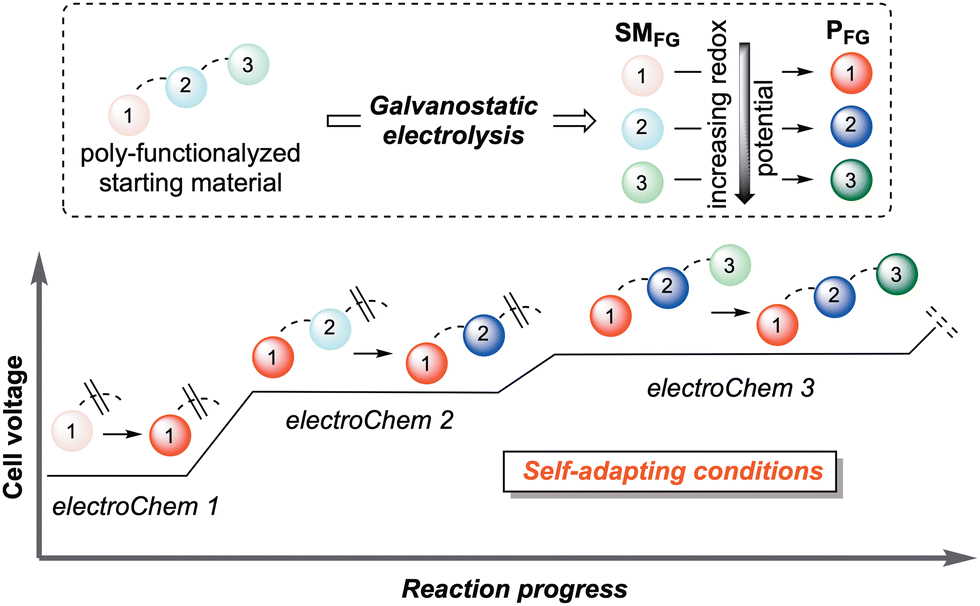 | ||
| Fig. 1 Pictorial representation of the concept of “self-adaptability” of galvanostatic electroChem utilized in organic synthesis. | ||
In continuation with our recent interests in selective radical processes and electrochemical reaction development,6 we tackled the exploitation of galvanostatic electrolysis for achieving rapid access to chemical diversity and efficient monitoring of reaction pathways, by investigating the reductive cyclodimerization of chalcones 1.7 This machinery was used as a platform to improve and expand reductive synthetic methodologies, featuring chemo-, regio-, and diastereoselectivity issues in the preparation of densely functionalized cyclopentanes (Scheme 1).8
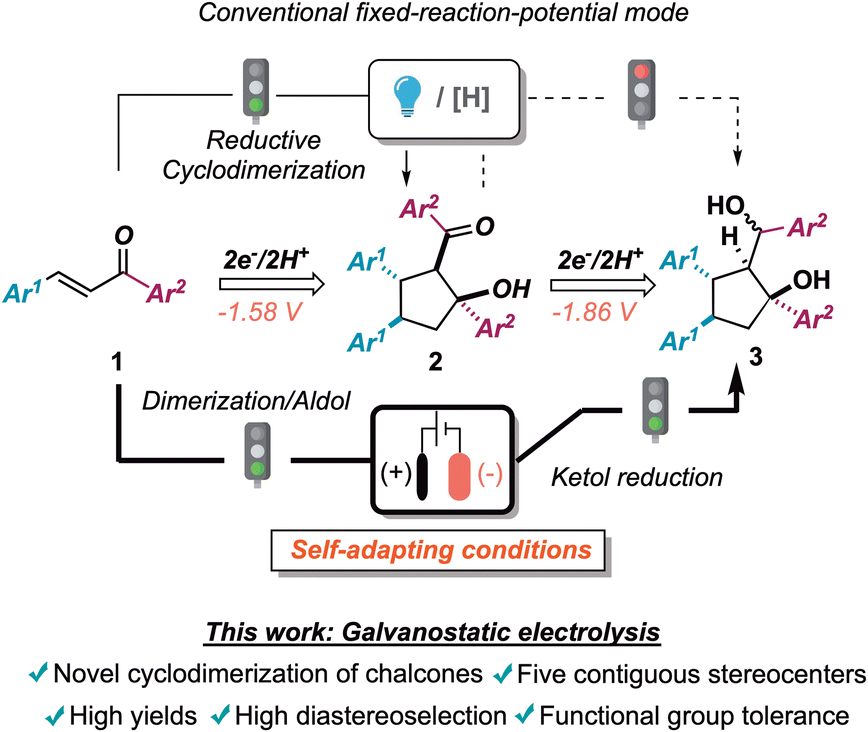 | ||
| Scheme 1 Electroreductive cyclodimerization of chalcones. The state of the art and the present proposal. [H]: stoichiometric chemical reductive agent. | ||
In detail, the 1,3-keto alcohol moiety of cyclodimerization products 2 could undergo further reduction to 1,3-diols 3 (vide infra). This process, creating a further exocyclic stereocenter, was posited to be more energetically demanding than the first monoelectronic reduction of the extended enone system (vide infra for an electrochemical characterization of both starting materials as well as intermediates). Not surprisingly, stoichiometric metal-promoted or photocatalytically assisted reductive conditions led exclusively to aldols 2 (Scheme 1 upper panel). In contrast, self-adaptive galvanostatic electrochemical conditions could yield an elegant way to achieve densely functionalized 1,3-diols 3 featuring five contiguous stereogenic centers (Scheme 1 lower panel).9
Despite the intrinsic synthetic challenges posed by this transformation, namely chemoselectivity (3avs. 2a/2a′ formation) as well as diastereoselectivity (formation of five consecutive stereogenic centers), we were pleased to find that the use of a silver (Ag) cathode and graphite (C) anode, in the presence of triethanolamine (TEOA, 2 equiv.)7j,10 as a sacrificial reductant (constant current electrolysis, I = 4 mA, 4 F mol1a−1), resulted in the fully chemo- and diastereoselective formation of 3a in 84% isolated yield (Table 1, entry 1). Importantly, the supporting electrolyte (TEABF4: tetraethylammonium tetrafluoroborate) could be used in a substoichiometric amount (20 mol%, 0.01 M) without consistent issues concerning the medium resistivity. In addition, the reaction could be implemented to the mmol scale with minimal loss of efficiency (yield = 79%).
|
|
|||
|---|---|---|---|
| Entry | Deviation from optimal | Y 3ab [%] | 2a/2a′/2a′′ |
|
a All reactions were carried out in dry solvents and under nitrogen conditions.
b Isolated yields after flash chromatography. In all cases, 2a was isolated in >20:1 dr as determined from 1H-NMR analysis of the crude reaction products.
c Yields determined via1H-NMR analysis with mesitylene as an internal standard.
d Isolated yield for a 1.0 mmol-scale reaction (see ESI for additional details). CCE: constant current electrolysis; HE: Hantzsch ester.
|
|||
| 1 | None | 84 (79d) | Traces |
| 2 | C graph as cathode | 22 | —/12/— |
| 3 | Ni as cathode | 75 | Traces |
| 4 | LiBF4 as electrolyte | 47 | Traces |
| 5 | TEABF4 (100 mol%) | 82 | Traces |
| 6 | HE as reductant | — | 18/17/— |
| 7 | Zn as anode, no TEOA | — | 33/—/— |
| 8 | CCE I = 2 mA | 74 | —/5/— |
| 9 | CCE I = 6 mA | 69 | Traces |
| 10 | DMSO as solvent | 69 | Traces |
| 11 | CH3CN as solvent | 58 | Traces |
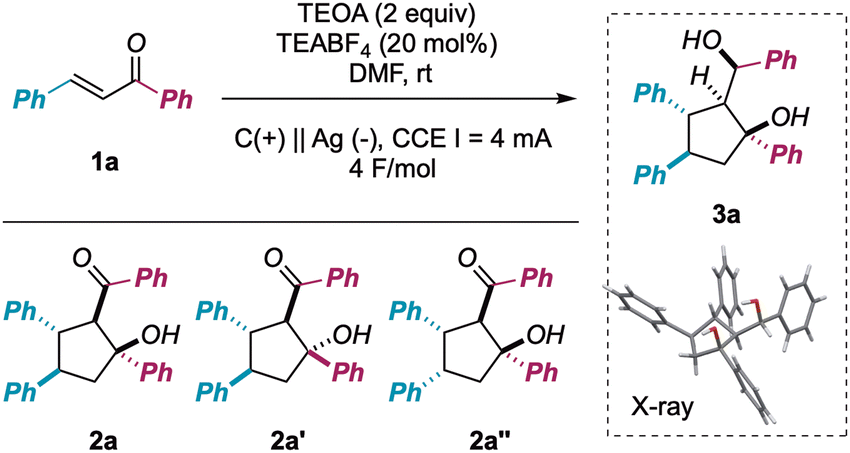
An extensive survey of reaction parameters (see ESI†) revealed that variations from optimal conditions resulted in a significant drop in reaction performance or no formation of desired product at all. In particular, the importance of using a non-Lewis-acid electrolyte and a metal cathode was underscored for entries 2, 3 and 4, with graphite giving a mixture of 2a′′ and 3a in poor yield and Ni behaving similarly to the optimal Ag. Analogously, using different stoichiometric reductants (entry 6) or a sacrificial anode (entry 7) prevented the formation of the desired product, yielding mixtures of aldols 2 and partial reduction of 1a. On the other hand, the outcome of the process was found to be less reliant on the intensity of the current (entries 8 and 9) or the reaction solvent (entries 10 and 11), always furnishing diol 3a, although with diminished efficiency.
Once optimal reaction conditions were established, the generality of the electroreductive cyclodimerization was assessed on a range of densely functionalized chalcones 1b–x (Scheme 2). Apart from a few cases, the target products 3 were isolated as single diastereoisomers in good to excellent yields (up to 95%). In particular, high tolerance toward electronic perturbations of the electron-deficient double bond, via decoration of the adjacent aryl group, was observed. Electron-donating substituents, including alkyl (1b–c), phenyl (1d), methoxy (1h,m) and mercaptomethyl (1i) groups placed at the para or meta positions of the arenes, provided a smooth formation of the products 3 (43–80% yields). Electron-poor arenes bearing CF3 (1e), ester (1f) cyano (1g) and halogen substituents (F, Cl, Br 1k–n) performed equally well (59–84% yields). Finally, an extended π-system (1o), as well as electron-rich heteroarenes (furan and thiophene, 1p, q) and electron-poor pyridine (1r), were also well tolerated in the disclosed protocol(35–95% yields).
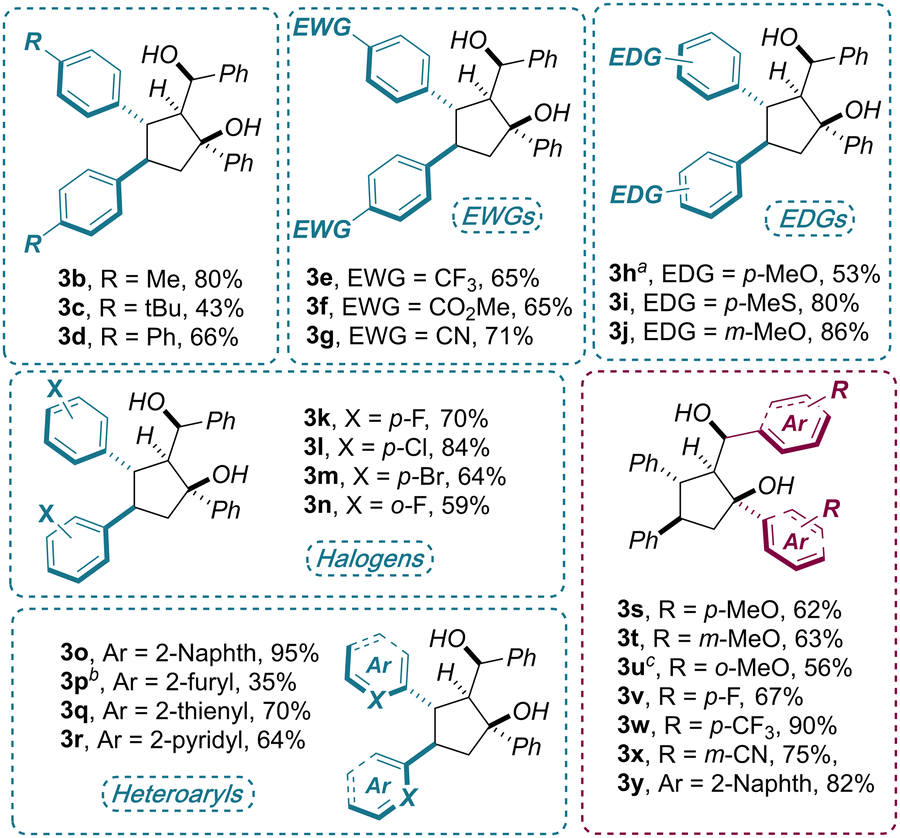 | ||
| Scheme 2 Reaction scope: reaction conditions as in Table 1, entry 1. Isolated yields were determined after flash chromatography. All compounds were isolated essentially as a single diastereoisomer (dr > 20:1) unless otherwise specified: adr = 8.0:1. bdr = 11:1.cdr = 4.4:1. | ||
Variations of the aromatic ketone moiety of 1 were also investigated by means of testing substrates 1s–y. Here, when subjecting electron-rich (1s–u) and electron-poor (1v–x) chalcones to the optimized conditions, good to excellent yields (62–90%) were obtained regardless of the functional group position. In fact, ortho, meta and para substitutions did not affected significantly the outcome of the process, with only a slight drop in diastereoselectivity (4.4:1) with product 3u.
In order to shed light on the mechanism of the whole process—and, in particular, on the key unprecedented late-stage reduction to 3—we monitored the progress of the reaction over time. To our surprise, by simply interrupting the reaction after providing 1 F mol1a−1, an equimolar mixture of diastereoisomers 2a7h–k and 2a′ was observed (68% combined yield). Isomer 2a′ was unambiguously identified as an epimer of 2avia a single-crystal X-ray diffraction analysis (Scheme 3 upper-right), thus showing a lack of diastereoselectivity in the aldol cyclization stage. This evidence resulted in sharp contrast with the highly diastereoselective outcome observed at the end of the cascade (3a, dr >20:1). Interestingly, by re-subjecting either isomer 2a or 2a′ to the optimized conditions independently, 3a was isolated in a diastereoselective manner (dr >20:1) in both cases and no traces of the putative epimer 3a′ (theoretically 2a′ → 3a′) were observed. Thus, both 2a and 2a′ were deemed to be productive intermediates in the formation of 3a and, while 2a was reduced directly to the final target, 2a′ is believed to undergo a retro-aldol ring opening event, responsible for an equilibration between 2a and 2a′.11 Following these experimental results, the tentative mechanism depicted in the lower panel of Scheme 3 is porposed. Accordingly, after a monoelectronic reduction of chalcone 1a (radical anion A), a tail-to-tail Giese addition to a second molecule of 1a (intermediate B) is postulated, giving, after a second cathodic event and protonation, the 1,6-diketone monoenolate C (pathway a). Alternatively, C can be directly produced (after protonation) from a radical-radical coupling of A, given the high concentration of this species at the cathode surface (pathway b). Hereafter, C is thought to undergo a non-diastereoselective intramolecular aldol cyclization, followed by a dynamic kinetic resolution of the intermediates resulting from an irreversible ketone reduction. This hypothesis could be rationalized in terms of higher reactivity of isomer 2a, relative to 2a′, toward the reductive step, probably due to the formation of an activating intramolecular hydrogen bond being possible only in the former isomer. Moreover, note that in the present machinery, TEOA would act both as a sacrificial reductant as well as protic source, whenever needed.7j,10
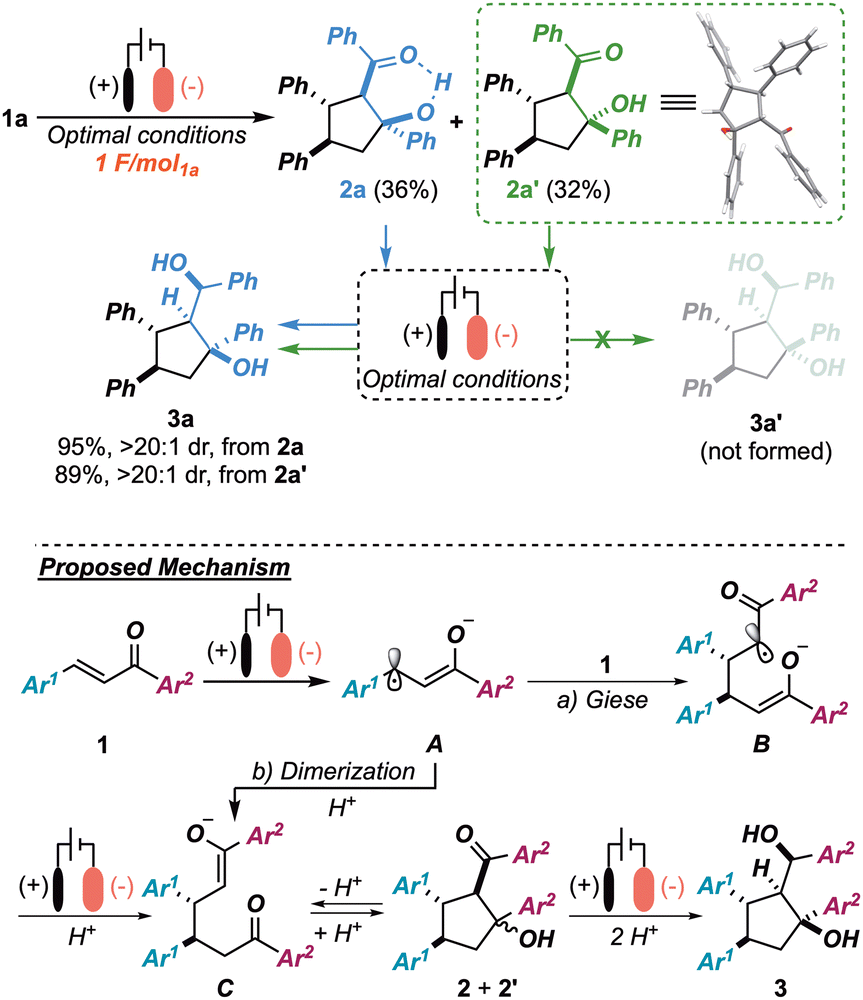 | ||
| Scheme 3 Upper panel: control experiments proving the electrochemical dynamic resolution. Lower panel: overall picture of the tentative operating mechanism. | ||
In this framework, a preliminary electrochemical investigation deploying cyclic voltammetry was carried out on chalcone 1a and intermediate 2a (Fig. S3, ESI†). The analysis of the cyclic voltammetric curve of 1a showed a completely chemically irreversible process (Epc = −1.52 V vs. SCE) followed by a broad and partially reversible voltammetric wave, made of two closely spaced processes, with a height of about half of the first irreversible reduction. To account for the proton exchange stages involved in the mechanism (formation of C, followed by an aldol cyclization), an experiment with a controlled concentration of H2O was carried out.
Here, the second peak was made completely reversible and occurred at E½ = −1.86 V. The cyclic voltammetric curve of 2a was also recorded under the same conditions, and showed a reversible reduction at the same potential (E½ = −1.86 V) as that of the second voltammetric wave of chalcone 1a. Interestingly, the voltammetric analysis supported the first steps of the mechanism depicted in Scheme 3. In particular, the reduction of 1a (formation of intermediate A) was concluded to be followed by a dimerization of the radical anion A and proton exchange (Scheme 3, pathway b).12,13 Finally, the exocyclic ketone moiety of the newly formed product(s) 2 can undergo a reduction process. All these steps agree both with the redox potentials recorded and the half height of the second voltammetric peak with respect to the first one.
In conclusion, we have demonstrated that the “self-adaptability” of galvanostatic electrolysis is a compelling tool for the realization of cascade processes with a high level of chemo- and stereocontrol. Under these conditions, a new route for the reductive cyclodimerization of chalcones was developed, leading to cyclopentane scaffolds, in high yields (up to 95%) and diastereocontrol (dr > 20:1) over five contiguous stereocenters. The tunability of reaction conditions allowed us to shed light on the whole operating electrochemically mediated mechanism, revealing a dynamic kinetic resolution of the intermediate aldol products by means of the final irreversible reduction.
We thank the University of Bologna and Consorzio CINMPIS for their support. GB is also grateful to the PRIN-2022 project (20227Z3BL8) for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) C. O. Kappe, J. Mack and C. Bolm, J. Org. Chem., 2021, 86, 14242 CrossRef PubMed (all dedicated issue); (b) S. G. Newmann, Enabling Tools and Techniques for Organic Synthesis: A Practical Guide for Experimentation Automation, and Computation, Wiley, 2023 CrossRef; (c) L. Grimaud, S. Lakhdar and M. R. Vitale, Curr. Opin. Elctrochem., 2023, 40, 101307 CrossRef CAS.
- (a) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230 CrossRef CAS PubMed; (b) A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594 CrossRef CAS PubMed; (c) C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. Y. Peters, Y. Kawamata and P. S. Baran, Acc. Chem. Res., 2020, 53, 72 CrossRef CAS PubMed; (d) R. D. A. Little, J. Org. Chem., 2020, 85, 13375 CrossRef CAS PubMed; (e) Y. Yuan and A. Lei, Nat. Commun., 2020, 11, 802 CrossRef CAS PubMed; (f) L. F. Novaes, J. Liu, Y. Sgen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941 RSC; (g) M. C. Leech and K. Lam, Nat. Rev. Chem., 2022, 6, 275 CrossRef PubMed; (h) L. Zheng, J. Wang, D. Wang, H. Yi and A. Lei, Angew. Chem., Int. Ed., 2023, e20230962 Search PubMed; (i) R. Franche and Curr Opin, Electrochem., 2023, 36, 101111 Search PubMed; (j) P. K. Baroliya, M. Dhaker, S. Panja, S. A. Al-Thabaiti, S. M. Albukhari, Q. A. Alsulami, A. Dutta and D. Maiti, ChemSusChem, 2023, 16, e202202201 CrossRef CAS PubMed; (k) Y. Li, S. Dana and L. Ackermann, Curr. Opin. Electrochem., 2023, 40, 101312 CrossRef CAS; (l) C. E. Hatch and W. C. Chain, ChemElectroChem, 2023, 10, e202300140 CrossRef CAS.
- For a general review, see: (a) M. Rafiee, M. N. Mayer, B. T. Punchihewa and M. R. Mumau, J. Org. Chem., 2021, 86, 15866 CrossRef CAS PubMed ; See also: ; (b) K. D. Moeller, Tetrahedron, 2000, 56, 9527 CrossRef CAS; (c) B. H. Nguyen, R. J. Perkins, J. A. Smith and K. D. Moeller, Beilstein J. Org. Chem., 2015, 11, 280 CrossRef CAS PubMed; (d) Q.-L. Yang, Y.-Q. Li, C. Ma, P. Fang, X.-J. Zhang and T.-S. Mei, J. Am. Chem. Soc., 2017, 139, 3293 CrossRef CAS PubMed.
- (a) Z.-W. Hou, Z.-Y. Mao, J. Song and H.-C. Xu, ACS Catal., 2017, 7, 5810 CrossRef CAS; (b) S. Blank, Z. Nguyen, D. G. Boucher and S. D. Minteer, Curr. Opin. Electrochem., 2022, 35, 101049 CrossRef CAS; (c) P. L. Norcott, Chem. Commun., 2022, 58, 2944 RSC; (d) Z. Lin, U. Dhawa, X. Hou, M. Surke, B. Yuan, S.-W. Li, Y.-C. Liou, M. J. Johansson, L.-C. Xu, C.-H. Chao, X. Hong and L. Ackermann, Nat. Commun., 2023, 14, 4224 CrossRef CAS PubMed; (e) C. Zhang, D. Chen, J.-P. Wan and Y. Liu, Synthesis, 2023, 2911 CrossRef CAS.
- For selected reviews comparing photo- and electrochemical protocols, see: (a) R. H. Verschueren and W. M. De Borggraeve, Molecules, 2019, 24, 2122 CrossRef PubMed; (b) J. Liu, L. Lu, D. Wood and S. Lin, ACS Cent. Sci., 2020, 6, 1317 CrossRef CAS PubMed; (c) N. E. S. Tai, D. Lehnherr and T. Rovis, Chem. Rev., 2022, 122, 2487 CrossRef PubMed.
- (a) Y. Liu, S. Battaglioli, L. Lombardi, A. Menichetti, G. Valenti, M. Montalti and M. Bandini, Org. Lett., 2021, 23, 4441 CrossRef CAS PubMed; (b) S. Battaglioli, G. Bertuzzi, R. Pedrazzani, J. Benetti, G. Valenti, M. Montalti, M. Monari and M. Bandini, Adv. Synth. Catal., 2022, 364, 720 CrossRef CAS; (c) L. Lombardi, A. Kovtun, S. Mantovani, G. Bertuzzi, L. Favaretto, C. Bettini, V. Palermo, M. Melucci and M. Bandini, Chem. – Eur. J., 2022, 28, e202200333G CrossRef PubMed; (d) L. Lombardi, A. Cerveri, R. Giovanelli, M. Castiñeira Reis, C. Silva López, G. Bertuzzi and M. Bandini, Angew. Chem., Int. Ed., 2022, 61, e202211732 CrossRef CAS PubMed; (e) G. Bertuzzi, G. Ombrosi and M. Bandini, Org. Lett., 2022, 24, 4354 CrossRef PubMed; (f) A. Brunetti, G. Bertuzzi and M. Bandini, Synthesis, 2023, 55, 3047–3055 CrossRef CAS; (g) L. Rapisarda, A. Fermi, P. Ceroni, R. Giovanelli, G. Bertuzzi and M. Bandini, Chem. Commun., 2023, 59, 2664 RSC.
- For stoichiometric low-valent metal induced protocols see: (a) K. Takaki, F. Beppu, S. Tanaka, Y. Tsubaki, T. Jintoku and Y. Fujiwara, J. Chem. Soc., Chem. Commun., 1990, 6, 516 RSC; (b) K. Takaki, K. Nagase, F. Beppu and Y. Fujiwara, Chem. Lett., 1991, 1665 CrossRef CAS; (c) L.-H. Zhou, D.-Q. Shi, Y. Gao, W.-B. Shen, G.-Y. Dai and W.-X. Chen, Tetrahedron Lett., 1997, 38, 2729 CrossRef CAS; (d) L. Zhou and Y. Zhang, Synth. Commun., 2000, 30, 597 CrossRef CAS; (e) L. Moisan, C. Hardouin, B. Rousseau and E. Doris, Tetrahedron Lett., 2002, 43, 2013 CrossRef CAS; (f) K. K. Kapoor, S. Kumar and B. A. Ganai, Tetrahedron Lett., 2005, 46, 6253 CrossRef CAS; (g) Y.-J. Chen, L.-T. Wu, T.-A. Li, M.-Q. Pu, X.-L. Sun, H. Bao and W.-M. Wan, Angew. Chem., Int. Ed., 2023, 62, e202304033 CrossRef PubMed ; for photocatalytic methodologies see: ; (h) G. Zhao, C. Yang, L. Guo, H. Sun, R. Lin and W. Xia, J. Org. Chem., 2012, 77, 6302 CrossRef CAS PubMed; (i) G. K. Hodgson and J. C. Scaiano, ACS Catal., 2018, 8, 2914 CrossRef CAS; (j) B. Kurpil, Y. Markushyna and A. Savateev, ACS Catal., 2019, 9, 1531 CrossRef CAS For seminal electrosynthetic reports, targeting (unselectively) products 2, see: ; (k) F. Fournier, J. Berthelot and J.-J. Basselier, Tetrahedron, 1985, 41, 5667 CrossRef CAS; (l) J. H. P. Hutley, C. Z. Smith and M. Motevalli, J. Chem. Soc., Perkin Trans. 2, 2000, 1053 Search PubMed.
- On the relevance of related cyclopentane scaffolds see: (a) Å. Rosenquist, B. Samuelsson, P.-O. Johansson, M. D. Cummings, O. Lenz, P. Raboisson, K. Simmen, S. Vendeville, H. de Kock, M. Nilsson, A. Horvath, R. Kalmeijer, G. de la Rosa and M. Beumont-Mauviel, J. Med. Chem., 2014, 57, 1673 CrossRef PubMed; (b) L. Zhou, Y. Tuo, Y. Hao, X. Guo, W. Tang, Y. Xue, J. Zeng, Y. Zhou, M. Xiang, J. Zuo, G. Yao and Y. Zhang, Org. Lett., 2017, 19, 3029 CrossRef CAS PubMed.
- L. Wang, X. Zhang, R. Y. Xia, C. Yang, L. Guo and W. Xia, Synlett, 2022, 1302 CrossRef CAS.
- (a) Y. Pellegrin and F. Odobel, C. R. Chim, 2017, 20, 283 CrossRef CAS; (b) S. Mazzanti, B. Kurpil, B. Pieber, M. Antonietti and A. Savateev, Nat. Commun., 2020, 11, 1387 CrossRef CAS PubMed; (c) O. Savateev and Y. Zou, ChemistryOpen, 2022, 11, e202200095 CrossRef CAS PubMed.
- A control experiment carried out on pure 2a′ through exposition to TEOA (2 equiv.) furnished a 1:1 2a:2a′ mixture.
- Pathway a in Scheme 3 is also in accordance with the present voltammetric profile, provided that the reduction of B to C happens at a less negative potential compared to the reduction of chalcone 1a to radical anion A.
- (a) J.-M. Savéant, Elements of molecular and biomolecular electrochemistry, Wiley, 2006 CrossRef; (b) C. Bruno, E. Ussano, G. Barucca, D. Vanossi, G. Valenti, E. A. Jackson, A. Goldoni, L. Litti, S. Fermani, L. Pasquali, M. Meneghetti, C. Fontanesi, L. T. Scott, F. Paolucci and M. Marcaccio, Chem. Sci., 2021, 12, 8048 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2290358 and 2290359. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc04920e |
| This journal is © The Royal Society of Chemistry 2024 |