
DMSO–KOH mediated stereoselective synthesis of Z-enamides: an expeditious route to Z-enamide bearing natural products†
Showkat Ahmad
Bhat
 ac,
Qazi Naveed
Ahmed
*bc and
Khursheed Ahmad
Bhat
*ac
ac,
Qazi Naveed
Ahmed
*bc and
Khursheed Ahmad
Bhat
*ac
aBioorganic Chemistry Division, Indian Institute of Integrative Medicine (CSIR), Srinagar, Jammu & Kashmir 190005, India. E-mail: kabhat@iiim.res.in
bNatural Product and Medicinal Chemistry Division, CSIR-Indian Institute of Integrative Medicine, Jammu 180001, India
cAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad 201002, India
First published on 22nd November 2023
Abstract
An efficient strategy towards stereoselective amidation of alkynes is reported. The given method features operational simplicity, excellent functional group tolerance, broad substrate scope and fast kinetics to furnish Z-enamides. Moreover, the method was successfully applied for the facile synthesis of the natural products lansiumamide A, lansiumamide B and Z-alatamide. Notably, DMSO plays two vital roles: hydrogen source and solvent.
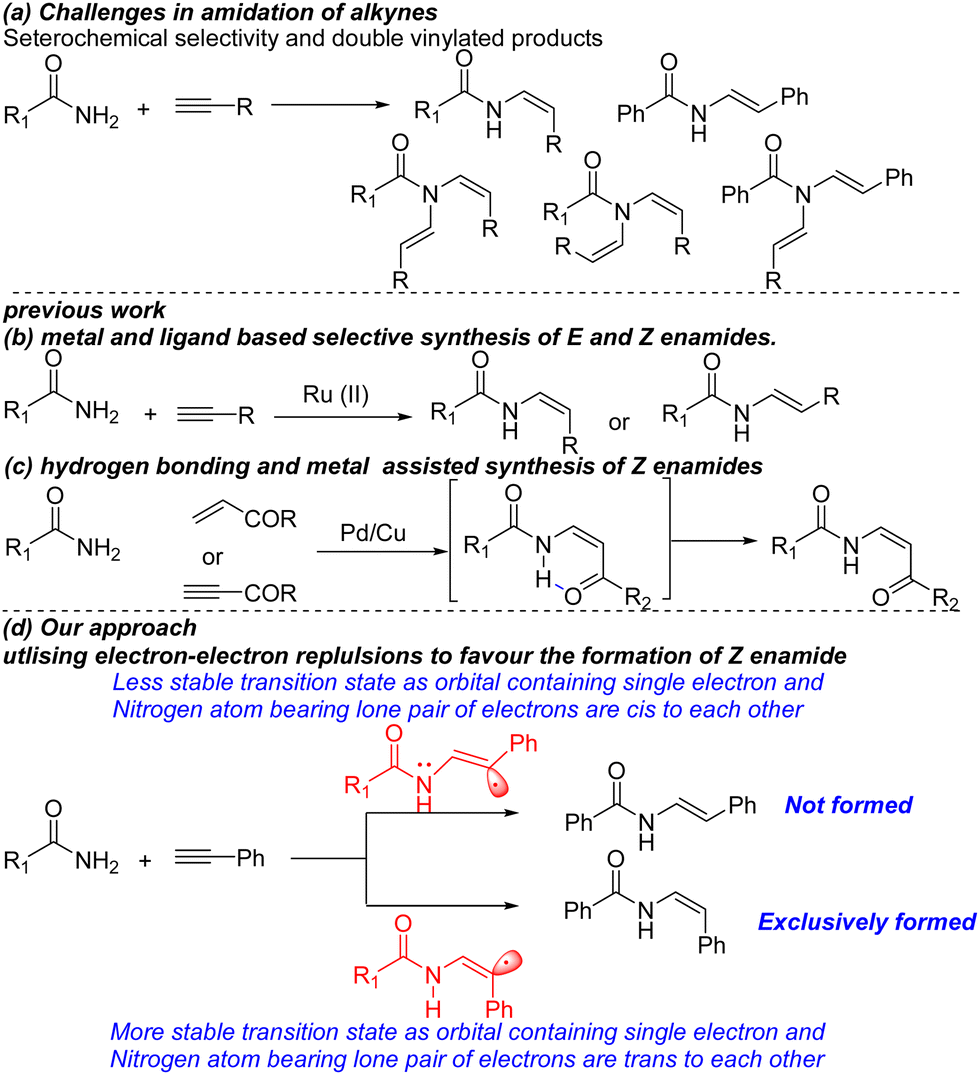
Enamides serve as privileged skeletons in a number of natural products, such as apicularen A, crocacins, lansiumamide A, storniamides, enamidoin, terpeptin, palytoxin, aspergillamides, chondriamides, salicylihalamides, TMC-95A-DD and alatamide (Fig. 1).1 Furthermore, they serve as vital synthetic intermediates in cross couplings, Heck reactions, and asymmetric and heterocyclic synthesis. As a result, a number of methods have been developed for their synthesis.2 Conventionally, they are synthesized by condensation of carbonyl derivatives with amides or acylation of imines leading to product mixtures with the E isomer as a major product.3 This thermodynamically favoured isomer is also synthesized by oxidative amidation of alkenes, and codimerisation of N-vinyl amides with alkenes.4 However, stereoselective synthesis of thermodynamically disfavoured Z-enamide has rarely been explored. A few methods, such as the elimination of β-hydroxy-α-silylamides (Peterson reaction), and catalytic cross-couplings of amides with vinyl halides, pseudohalides, or alkenyl trifluoroborate salts, have been developed for the stereoselective synthesis of enamides.5 However, these methods often require anhydrous as well as harsh reaction conditions in addition to the complex starting materials, which often limits their practical applications. On the other hand, the addition of amides to alkynes has recently attracted remarkable attention in enamide synthesis. The addition of alkynes to amides poses some challenges, like the formation of double vinylated products and a mixture of E and Z enamides (Scheme 1(a)). Wantanbe and co-workers reported ruthenium–complex mediated hydroamidation for the stereoselective synthesis of E-enamides.6 Gooβen and co-workers developed ligand-mediated, ruthenium catalysed synthesis of either E or Z enamides using amides or terminal alkynes (Scheme 1(b)).7 Chang and co-workers followed by Mothkuri's group reported Pd-catalysed oxidative amidation of conjugated olefins and alkynes for the synthesis of Z-enamides (Scheme 1(c)).8 In both these methods the notable preference for Z-enamide arises due to an intramolecular hydrogen bonding between the amido proton and carbonyl carbon. Despite these advances, there are still some limitations such as limited substrate scope, lower yields, harsh reaction conditions and longer reaction time that need to be addressed. Due to our interest in DMSO-promoted organic synthesis,9 we developed transition metal-free, stereoselective synthesis of Z-enamides using benzamide, phenylacetylene, DMSO and KOH. When the reaction was carried out between benzamide and phenylacetylene in DMSO at 100 °C using KOH as a base, we observed that Z-enamide was formed exclusively with excellent yield in 15 minutes. However, when the reaction was carried out for a longer time (24 hours), it was observed that the Z-enamide slowly undergoes isomerisation to form almost a 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of E and Z enamides. This isomerisation was proved by obtaining E and Z isomers from synthetically prepared pure Z-enamide when reacted under standard reaction conditions for 24 hours. The reaction of benzamide with phenylacetylene in the DMSO–KOH system is proposed to proceed through a radical mechanism. The orbital on the carbon atom containing the single electron and the nitrogen atom bearing a lone pair of electrons are trans in the (Z)-N-styrylbenzamide radical, unlike in the (E)-N-styrylbenzamide radical. The reaction therefore proceeds exclusively through the (Z)-N-styrylbenzamide radical leading to the formation of the corresponding Z-enamides (Scheme 1(d)). This kinetically fast protocol was further used for the one step synthesis of natural product lansiumamide A. The conventional protocol for the synthesis of this natural product involves multistep, harsh reagents and dry conditions. Another natural product, lansiumamide B, was also synthesized by performing the N-methylation of lansiumamide A. Furthermore, the Z-isomer of the natural product alatamide was also synthesized using the standard protocol.
:1 mixture of E and Z enamides. This isomerisation was proved by obtaining E and Z isomers from synthetically prepared pure Z-enamide when reacted under standard reaction conditions for 24 hours. The reaction of benzamide with phenylacetylene in the DMSO–KOH system is proposed to proceed through a radical mechanism. The orbital on the carbon atom containing the single electron and the nitrogen atom bearing a lone pair of electrons are trans in the (Z)-N-styrylbenzamide radical, unlike in the (E)-N-styrylbenzamide radical. The reaction therefore proceeds exclusively through the (Z)-N-styrylbenzamide radical leading to the formation of the corresponding Z-enamides (Scheme 1(d)). This kinetically fast protocol was further used for the one step synthesis of natural product lansiumamide A. The conventional protocol for the synthesis of this natural product involves multistep, harsh reagents and dry conditions. Another natural product, lansiumamide B, was also synthesized by performing the N-methylation of lansiumamide A. Furthermore, the Z-isomer of the natural product alatamide was also synthesized using the standard protocol.
 | ||
| Fig. 1 Some bioactive enamides. | ||
 | ||
| Scheme 1 Challenges, previous work and reaction design. | ||
To begin our study, reaction conditions including the temperature, type and concentration of base and concentration of reactants were optimised (Table 1). Initially, when benzamide (1a) and phenylacetylene (2a) each 1 mmol and NaOH (0.5 mmol) were reacted in 3 mL of DMSO at 70 °C for 15 minutes. The desired product 3a was isolated with a 15% yield (entry 1). In order to improve the yield of 3a, reactions with different bases were conducted (entries 2 and 3). As evident, the best yield of 3a was obtained when KOH was used (entry 3). Furthermore, upon changing the concentration of KOH (entries 4–6), it was observed that using 1 mmol of KOH gave the best results, whereas further increase in the concentration of KOH had no profound effect on the yield of the desired product. Next, the concentration of 1a was optimised (entries 6–9), and it was evident that using 1 mmol of 1a gave better results. The effect of temperature was also studied (entries 10–13), and it was revealed that the reaction at 100 °C with 1 mmol of each reactant and base produced the best yield of desired product 3a. The effect of time (entries 14–16), showed that the best yield of product was obtained when the reaction was carried out in 15 minutes. When the reaction was left for 24 hours, a mixture of E and Z enamides was obtained in the ratio of 1:1.
|
|
||||||
|---|---|---|---|---|---|---|
| S. no. | 1a (mmol) | 2a (mmol) | Base (mmol) | Temp. (°C) | Time | Yielda [%] |
| Reaction conditions: the reaction of 1a (1 mmol) and 2a (1 mmol) and KOH (1 mmol) in 3 mL of DMSO at 100 °C for 15 minutes.a a Isolated yield. | ||||||
| 1 | 1.0 | 1.0 | NaOH (0.5) | 70 | 15 | 15 |
| 2 | 1.0 | 1.0 | KOtBu (0.5) | 70 | 15 | 20 |
| 3 | 1.0 | 1.0 | KOH (0.5) | 70 | 15 | 55 |
| 4 | 1.0 | 1.0 | KOH (1.0) | 70 | 15 | 64 |
| 5 | 1.0 | 1.0 | KOH (1.5) | 70 | 15 | 65 |
| 6 | 1.0 | 1.0 | KOH (2.0) | 70 | 15 | 66 |
| 7 | 0.5 | 1.0 | KOH (1.0) | 70 | 15 | 58 |
| 8 | 1.5 | 1.0 | KOH (1.0) | 70 | 15 | 59 |
| 9 | 2.0 | 1.0 | KOH (1.0) | 70 | 15 | 52 |
| 10 | 1.0 | 1.0 | KOH (1.0) | rt | 15 | 0 |
| 11 | 1.0 | 1.0 | KOH (1.0) | 80 | 15 | 54 |
| 12 | 1.0 | 1.0 | KOH (1.0) | 100 | 15 | 69 |
| 13 | 1.0 | 1.0 | KOH (1.0) | 120 | 15 | 69 |
| 14 | 1.0 | 1.0 | KOH (1.0) | 100 | 15 | 69 |
| 15 | 1.0 | 1.0 | KOH (1.0) | 100 | 1 h | 68 |
| 16 | 1.0 | 1.0 | KOH (1.0) | 100 | 24 h | Mixture |

Having the optimised reaction conditions in hand, the substrate scope of the present methodology was subsequently assessed. It was found that under optimised conditions, various substituted amides and phenylacetylenes reacted smoothly to afford the desired Z-enamides, as shown in Scheme 2 and 3. The electronic properties of the substituent on the aromatic rings of the amides and alkynes appeared to have a negligible effect on the efficiency of the reaction. Amides bearing electron donating groups (1a–1e) (–Me, –OMe, –OEt) reacted smoothly to afford the corresponding Z-enamides. The reaction of 2-methylbenzamide (1b) occurred smoothly with phenylacetylene (2a) to form 3b with 67% yield. Similarly the reaction of 4-methoxybenzamide (1c) with phenylacetylene (2a) furnished 3c with a yield of 67%. The reaction of 2-ethoxybenzamide (1d) with phenylacetylene led to the formation of the corresponding Z-enamide 3d with a yield of 66%. Similarly the reaction of 2,3-dimethoxybenzamide (1e) led to the formation of 3e with 65% yield. When benzamides containing electron withdrawing groups (–F, –Cl, Br) were used (1f–1k), the corresponding products were formed in good yields. The reaction of 2-fluorobenzamide (1f) & 3-fluorobenzamide (1g) with phenylacetylene afforded 3f & 3g with 80% yield. A similar reaction between 3-chlorobenzamide (1h) & 4-chlorobenzamide (1i) with phenylacetylene yielded 3h & 3i with 73% yield. Various disubstituted amides such as 2,6-difluorobenzamide (1j) & 2,6-dichlorobenzamide (1k) reacted smoothly with phenylacetylene to afford 3j & 3k with a yield of 85% & 78%, respectively. Interestingly, when 2-aminobenzamide (1l) & 3-aminobenzamide (1m) were reacted with phenylacetylene, we observed exclusive amidation of alkynes. The amino group remained intact during the reaction. The corresponding Z-enamides 3l and 3m were formed with a yield of 68% and 67%, respectively. Heterocyclic and aliphatic substrates such as thiophene-2-carboxamide (1n) & cyclohexanecarboxamide (1o) also reacted smoothly to afforded the desired products 3n & 3o with yields of 64% and 63%, respectively.
 | ||
| Scheme 2 Generality of the reaction: all the reactions were carried out with 1a (1 mmol) and 2a (1 mmol) and KOH (1 mmol) in 3 mL of DMSO at 100 °C for 15 minutes. | ||
The scope of this method was further extended to different alkynes (Scheme 3). A number of phenylacetylenes bearing electron donating groups (2b–2d) such as (–Me, –Et, –Ph) were tested, it was observed that the corresponding products (4a–4c) were formed in good yields. The reaction of 3-methylphenylacetylene (2b) and 4-n-propylphenylacetylene (2c) with benzamide (1a) afforded the desired products 4a and 4b with a yield of 67% and 65%, respectively. When 4-ethynyl-1,1′-biphenyl (2d) was reacted with benzamide, the corresponding Z-enamide (4c) was isolated with a yield of 61%. Various phenylacetylenes bearing electron withdrawing groups (2e–2j) such as –F, Cl, –Br and –CF3 reacted smoothly to afford the corresponding Z-enamides. The reaction of 2-fluorophenylacetylene (2e), 2-chlorophenylacetylene (2f), 3-fluorophenylacetylene (2g), 4-fluorophenylacetylene (2h), 4-bromophenylacetylene (2i), and 4-(trifluoromethyl)phenylacetylene (2j) with benzamide furnished 4d, 4e, 4f, 4g, 4h and 4i with the yields of 78%, 73%, 78%, 78%, 71%, and 75%, respectively.
 | ||
| Scheme 3 Generality of the reaction: all the reactions were carried out with 1a (1 mmol) and 2a (1 mmol) and KOH (1 mmol) in 3 mL of DMSO at 100 °C for 15 minutes. | ||
In order to demonstrate the practical application of this method, we next achieved gram scale synthesis of 3a and the synthesis of natural products lansiumamide A, lansiumamide B and the Z-isomer of natural product alatamide (Scheme 4). The reaction of cinnamamide (5) with phenylacetylene (2a) under optimised conditions resulted in the synthesis of lansiumamide A (6a) with a yield of 63%. To the best of our knowledge, this is the first transition metal free procedure for such synthesis. Lansiumamide A was further alkylated with methyl iodide to form another natural product lansiumamide B with 97% yield. The reaction of benzamide with 4-methoxyphenylacetylene under standard conditions led to the synthesis of (Z)-N-(4-methoxystyryl)benzamide (7) (yield 66%), which is the Z isomer of the natural product alatamide. The conventional methods for the synthesis of these molecules involve metal catalysts, anhydrous moisture sensitive reagents and multistep procedures.10
 | ||
| Scheme 4 Gram scale & natural product synthesis. | ||
In order to understand the mechanism of the reaction, some control experiments (Scheme 5) were executed. Initially (control experiment 1) benzamide (1a), phenylacetylene (2a) and TEMPO were reacted under standard reaction conditions; it was observed that no product was formed under such conditions, which indicates that the reaction proceeds through a radical mechanism. In control experiment 2, benzamide (1a), and phenylacetylene (2a) were reacted in DMSO-d6 using KOH as a base, and it was observed that (Z)-N-(2-phenylvinyl-2-d)benzamide (8) was isolated with a yield of 64%. The structure of 8 was confirmed by 1H NMR, which confirms DMSO as a source of hydrogen in the reaction. By using the known procedure, 3a was further isomerised into E-enamide 9 with a yield of 85%.
 | ||
| Scheme 5 Some control experiments. | ||
Based on control experiments and literature reports, a plausible mechanism is shown in Scheme 6. Initially KOH abstracts a proton from DMSO to form a dimesyl anion, which further undergoes single electron transfer (SET) to produce a dimesyl radical.11 This radical species abstracts hydrogen from benzamide (1a) to produce an amidoyl radical species.12 The radical species b reacts with phenylacetylene (2a) to produce more stable intermediate c, as the radical electron and nitrogen atom are trans to each other. The intermediate c abstracts a hydrogen from DMSO to furnish the final product 3a. The configuration of the olefinic bond in the final products was further proved by single crystal X-ray and NOESY studies.
 | ||
| Scheme 6 Plausible mechanism. | ||
In summary, we have developed an efficient method for the stereoselective synthesis of Z-enamides using benzamides and phenylacetylenes. The operationally simple reaction shows a broad substrate scope and good functional group tolerance. Moreover, the method was used to synthesize some natural products such as lansiumamide A, lansiumamide B and the Z-isomer of natural product alatamide. Currently, we are exploring the reagent system for the synthesis of different exigent structures.
This research was generously supported by the SERB, DST India project (File No. CRG/2022/001920). The institutional publication number of the manuscript is CSIR-IIIM/IPR/00670. Showkat Ahmad Bhat thanks CSIR for providing a fellowship and AcSIR for PhD registration.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J.-H. Lin, Phytochemistry, 1989, 28, 621–622 CrossRef CAS; (b) A. Fürstner, C. Brehm and Y. Cancho-Grande, Org. Lett., 2001, 3, 3955–3957 CrossRef PubMed.
- (a) X. Wang and J. A. Porco, J. Org. Chem., 2001, 66, 8215–8221 CrossRef CAS; (b) T. Guan, J.-Y. Guo, Q.-H. Zhang, X.-W. Xu, X.-Y. Yu, Y. Zhang and K. Zhao, Green Chem., 2022, 24, 6524–6530 RSC; (c) J.-Y. Tao, Y.-X. Wang, Q.-H. Zhang, K. Ni, T.-H. Zhu and K. Zhao, Green Chem., 2022, 24, 4004–4011 RSC; (d) J.-Y. Guo, Z.-Y. Zhang, T. Guan, L.-W. Mao, Q. Ban, K. Zhao and T.-P. Loh, Chem. Sci., 2019, 10, 8792–8798 RSC; (e) K. Zhao, Z.-Y. Zhang, X.-L. Cui, Y.-X. Wang, X.-D. Wu, W.-M. Li, J.-X. Wu, L.-L. Zhao, J.-Y. Guo and T.-P. Loh, Org. Lett., 2020, 22, 9029–9035 CrossRef CAS PubMed.
- (a) R. K. Boeckman, S. W. Goldstein and M. A. Walters, J. Am. Chem. Soc., 1988, 110, 8250–8252 CrossRef CAS; (b) T. Hosokawa, M. Takano, Y. Kuroki and S.-I. Murahashi, Tetrahedron Lett., 1992, 33, 6643–6646 CrossRef CAS.
- (a) K. Sorimachi and M. Terada, J. Am. Chem. Soc., 2008, 130, 14452–14453 CrossRef CAS PubMed; (b) R. Brettle and A. J. Mosedale, J. Chem. Soc., Perkin Trans. 1, 1988, 2185–2195, 10.1039/P19880002185.
- (a) A. Fürstner, C. Brehm and Y. Cancho-Grande, Org. Lett., 2001, 3, 3955–3957 CrossRef; (b) D. J. Wallace, D. J. Klauber, C.-Y. Chen and R. P. Volante, Org. Lett., 2003, 5, 4749–4752 CrossRef CAS PubMed; (c) Y. Bolshan and R. A. Batey, Angew. Chem., Int. Ed., 2008, 47, 2109–2112 CrossRef CAS.
- T. Kondo, A. Tanaka, S. Kotachi and Y. Watanabe, J. Chem. Soc., Chem. Commun., 1995, 413–414, 10.1039/C39950000413.
- L. J. Gooßen, K. S. M. Salih and M. Blanchot, Angew. Chem., Int. Ed., 2008, 47, 8492–8495 CrossRef PubMed.
- J. M. Lee, D.-S. Ahn, D. Y. Jung, J. Lee, Y. Do, S. K. Kim and S. Chang, J. Am. Chem. Soc., 2006, 128, 12954–12962 CrossRef CAS.
- (a) S. A. Bhat, M. Y. Bhat, S. A. Rather, I. Gani, K. A. Bhat and Q. N. Ahmed, Org. Biomol. Chem., 2022, 20, 8197–8202 RSC; (b) S. A. Bhat, M. Y. Bhat, S. A. Rather, S. Jameel, K. A. Bhat and Q. N. Ahmed, Org. Lett., 2023, 25, 2382–2387 CrossRef CAS.
- A. E. Pasqua, F. D. Ferrari, J. J. Crawford and R. Marquez, Tetrahedron Lett., 2014, 55, 6042–6043 CrossRef CAS.
- C. L. Øpstad, T.-B. Melø, H.-R. Sliwka and V. Partali, Tetrahedron, 2009, 65, 7616–7619 CrossRef.
- J. H. Horner, O. M. Musa, A. Bouvier and M. Newcomb, J. Am. Chem. Soc., 1998, 120, 7738–7748 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2247294. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc04642g |
| This journal is © The Royal Society of Chemistry 2024 |