
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDivergent process for the catalytic decarboxylative thiocyanation and isothiocyanation of carboxylic acids promoted by visible light†
Jordan
Vigier
a,
Mélissa
Gao
a,
Philippe
Jubault
 a,
Hélène
Lebel
*b and
Tatiana
Besset
*a
a,
Hélène
Lebel
*b and
Tatiana
Besset
*a
aNormandie Univ, INSA Rouen, UNIROUEN, CNRS, COBRA (UMR 6014), 76000 Rouen, France. E-mail: tatiana.besset@insa-rouen.fr
bDepartment of Chemistry and Center in Green Chemistry and Catalysis (CGCC), Université de Montréal, P.O. Box 6128, Station Downtown, Montréal, QC H3C 3J7, Canada. E-mail: helene.lebel@umontreal.ca
First published on 4th December 2023
Abstract
A divergent photoinduced selective synthesis of thiocyanate and isothiocyanate derivatives from readily available carboxylic acids was developed using N-thiocyanatosaccharin and a catalytic amount of base or acid. This molecular editing strategy allowed the functionalization of bioactive compounds. A mechanism for the transformation was proposed based on control experiments.
Organothiocyanates1 are interesting linchpin residues,2 often used to access a wide variety of sulfur-containing molecules.3 They are also found in a number of agrochemical and pharmaceutical compounds and natural products.1 Molecules bearing a SCN residue on a C(sp3) center are of great interest as exemplified by 9-thiocyanatopupukeanane, fasicularin and psammaplin B. Although synthetic routes for the preparation of SCN-containing molecules are available, there remains a demand for the development of sustainable chemical transformations that meet environmental challenges (e.g. circular economy, sustainability, valorization of raw materials). Recently, significant advances have been achieved in direct thiocyanation of C(sp3)–H bonds, in particular for the functionalization of benzylic C–H positions and redox active esters.4 The decarboxylative thiocyanation of carboxylic acids seems a promising complementary approach to selectively introduce a thiocyanate moiety in the presence of benzylic C–H positions. Given the plethora of carboxylic acids readily available, the development of such synthetic route will be appealing for both academia and industry, and will expand the chemical space of this class of compounds.5 The direct molecular editing strategy6 starting from a simple CO2H group, and its conversion into various functional groups by decarboxylation7 has offered a synthetic manifold in organic chemistry with a limited amount of waste, especially when using a photocatalyst. However, the use of such a strategy to form a C–SR bond from a C–CO2H bond is limited compared to other transformations. In particular, for the thiocyanation reaction, only a few examples have been reported to date (Scheme 1). The decarboxylative thiocyanation reaction of cinnamic acid derivatives using either electrochemistry8a or photocatalysis has been achieved, to afford the corresponding vinyl thiocyanates.8b The α-chlorination of β-keto acids with N-chlorosaccharin (SaccharinCl) followed by decarboxylative thiocyanation has also been reported to yield the corresponding 2-chloro-2-thiocyanato ketones.8c To the best of our knowledge, no general decarboxylative thiocyanation reaction has been reported with a wide range of carboxylic acids containing a variety of functional groups. Recently, we have shown a strong interest in developing new catalytic processes to synthesize thiocyanate compounds.9
 | ||
| Scheme 1 State of the art and this work. PhthSCN = N-thiocyanato-phthalimide. | ||
Herein, we demonstrate the photoinduced decarboxylative thiocyanation and isothiocyanation reaction on C(sp3) centers of carboxylic acids using an organic photocatalyst. The study started with the functionalization of the α,α-dimethylphenyl acetic acid 1a (0.2 mmol) in the presence of the N-thiocyanatosaccharin I. After the screening of various parameters, the thiocyanation of 1a selectively afforded the desired product 2a in 72% yield after 45 min, when using 2,4,6-triphenylpyrylium tetrafluoroborate (TPT) as the photocatalyst, in the presence of the source I, at 50 °C, in chlorobenzene under blue LED irradiation (Table 1, entry 1). The use of the 9-mesityl-10-methylacridinium salt (entry 2) or the N-thiocyanatophthalimide (entry 3) for 16 h was detrimental to the reaction. In the latter case, traces of the isothiocyanate product 3a4,10 were also observed. When other photocatalysts, light sources, electrophilic and nucleophilic thiocyanate reagents were evaluated, low or no product was observed (see ESI†). Replacing chlorobenzene with other solvents (entries 4 and 5) was not beneficial for the formation of 2a. Changing the additive (20 mol% of acid or base) and the reaction time (entries 1, 6–9) had a significant effect on the outcome of the reaction, and a selective and divergent process to selectively access 2a (45 min, 72%, entry 1) or 3a (4 h, 91%, entry 9) was achieved. In the absence of photocatalyst or light, no reaction occurred (entries 10 and 11) (see ESI†). After establishing the best reaction conditions, the reaction scope was explored (Scheme 2). Tertiary (1a–1d) and secondary (1e–1j) benzylic carboxylic acids were efficiently converted to the corresponding thiocyanates in good yields (2a–2j). Interestingly, a scale up was conducted in the case of 1f and the reaction was also possible under flow conditions affording 2f in a quantitative yield (see ESI†). Primary benzylic carboxylic acids (1k–1ai) were also suitable substrates for this reaction, demonstrating that any substitution pattern for the carbon bearing the carboxylic acid function was tolerated. Furthermore, the reaction was compatible with various functional groups, including halides, azides, or cyanos. Chemoselective functionalization was also observed with substrates containing active benzylic C–H bond, such as 1o, 1p, 1z and 1ac affording the desired products in good yields. In addition, 2p was isolated in 69% yield without any nucleophilic substitution reaction with the alkyl bromide, illustrating the complementarity of this approach to existing strategies. Carboxylic acids containing pyridine (1ag), indole (1ah) and pyrene (1ai) residues provided the corresponding products 2ag2ah and 2ai in 59%, 29% and 82% yields, respectively. The functionalization of alkyl tertiary carboxylic acids was also achieved. When the reaction was performed with 1-adamantanecarboxylic acid or 2,2-dimethyl-3-phenylpropanoic acid, 2aj and 2ak were isolated in 39% and 57% yields, highlighting the added-value of the approach compared with existing routes. Finally, thiophenoxy- and phenoxyacetic acids were suitable substrates, affording 2al and 2am in 55% and 85%, respectively. This molecular editing strategy allowed the site-selective late-stage functionalization of several bioactive and complex molecules (1an–1as, Scheme 2), showing tolerance for halogen (diclofenac 1aq, indometacin 1as) and ketone (ketoprofen 1ap and isoxepac 1ar). Here again, a significant benefit of this decarboxylative strategy was further demonstrated with the control of the site selectivity of the thiocyanation reaction in the presence of highly reactive benzylic C–H centers as illustrated with the functionalization of ibuprofen 1an and isoxepac 1ar. The selective synthesis of isothiocyanate derivatives was possible by using a catalytic amount of acetic acid and extending the reaction time to 4 hours (Table 1, entry 9). Hence, the phenyl isobutyric acid 1a was easily converted to the corresponding isothiocyanate 3a in 93% yield. Tertiary and secondary carboxylic acids were suitable for this reaction as illustrated by the synthesis of compounds 3a–3c, 3h, 3at and NCBz-phenylglycine 3av.
|
|
|||
|---|---|---|---|
| Entry | Variation from standard conditions | Yield 2aa (%) | Yield 3aa (%) |
| 1H NMR yields were determined using 1,1,2,2-tetrachloroethane as internal standard.a Isolated yields were given in parenthesis.b 16 h.c TPT (5 mol%). n.r. = no reaction. | |||
| 1 | None | 87 (72) | — |
| 2 | Mes-Acr+ BF4− (5 mol%) | 10 | — |
| 3bc | PhthSCN | 8 | 3 |
| 4bc | 1,2-Dichloroethane | 5 | 33 |
| 5bc | CH2Cl2 | 21 | 36 |
| 6bc | Et3N (20 mol%) | 16 | 9 |
| 7bc | Cs2CO3 (20 mol%) | 56 | 28 |
| 8c | AcOH (20 mol%) | 29 | 48 |
| 9 | AcOH (20 mol%), 4 h | — | (91) |
| 10bc | No TPT | n.r. | n.r. |
| 11bc | No light | n.r. | n.r. |

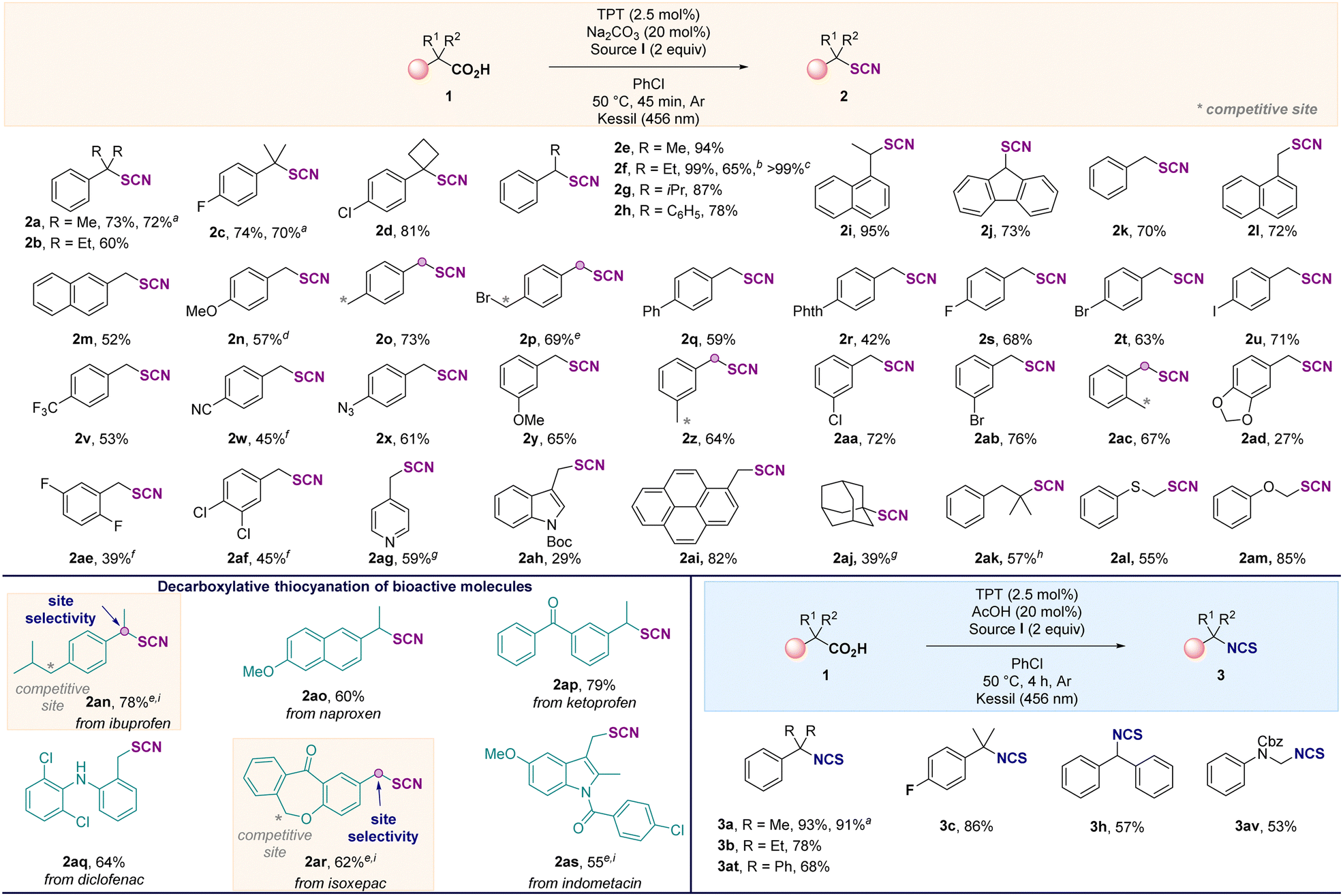 | ||
Scheme 2 0.3 mmol of 1 was used unless otherwise stated. Isolated yields are provided. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 0.2 mmol scale. b3 mmol scale. cUnder flow conditions. dProduct isolated with an inseparable impurity. eNo base was added. f4 h. g16 h. h2ak was isolated with 2g in a 4:1 ratio. i20 min. 0.2 mmol scale. b3 mmol scale. cUnder flow conditions. dProduct isolated with an inseparable impurity. eNo base was added. f4 h. g16 h. h2ak was isolated with 2g in a 4:1 ratio. i20 min. | ||
Since organocyanates are known to be potent molecular platform, the SCN residue was readily converted into high value-added moieties (Scheme 3A). In the presence of TMSCF3 under basic conditions, a Langlois-type nucleophilic substitution afforded trifluoromethylthiolate compound 4 in 66% yield.8a,11a By reaction with NaN3, a [3+2]-cycloaddition reaction afforded 5 in 74% yield.11b Furthermore, by replacing reagent I with the N-selenocyanatosaccharin, the corresponding seleno adduct 6 was isolated in 87% yield.12
 | ||
| Scheme 3
A. Post-functionalization reactions & extension to the SeCN group: On 0.3 mmol scale. Isolated yields were given. B. Mechanistic experiments: aOn 0.2 mmol scale. bDetermined by 1H NMR using 1,1,2,2-tetrachloroethane as an internal standard. cAdduct detected by HRMS. C. Plausible mechanism. | ||
Mechanistic studies were then performed to further understand the transformation (Scheme 3B). First, radical trapping experiments were performed in the presence of 3,5-di-tert-4-butylhydroxytoluene (BHT), 2,2,6,6-tetramethyl-piperidine-N-oxyl (TEMPO) and 1,1-diphenylethylene (Scheme 3B(a)). In all cases, 2a was observed in a significantly lower yield compared to the standard reaction conditions and in the last two cases, the TEMPO-adduct and 2,2-diphenylethenyl thiocyanate were detected by HRMS (see ESI†). A radical clock experiment was then conducted starting from 1aw; the ring-opening product 7 along with the isothiocyanate compound 8 were isolated in 45% and 22% yields, respectively (Scheme 3B(b)). All these results suggested that a radical pathway might be involved in the process. A light ON/OFF experiment (see ESI†) (no reaction in the dark) and the determination of the quantum yield Φ = 23 (see ESI†) supported a photoinduced radical chain process.
To further understand the initiation step of the process, cyclic voltammetry measurements revealed two reduction waves for N-thiocyanatosaccharin I (Ep1 = −0.08 V vs. SCE & Ep2 = −1.6 V vs. SCE), while 1a (with and without base) showed an oxidative potential (Ep = +2.4 V vs. SCE) (see ESI†). Considering the redox properties of the organophotocatalyst (TPT) at the excited state (Et(PC*/PC˙−) = +2.3 V vs. SCE),13 a first reaction of the photocatalyst with the carboxylic acid appears favorable. To elucidate the formation of the isocyanate derivative, additional experiments were performed.12 When 2a was used as the starting material, a full conversion to 3a was observed, whereas no reaction occurred starting from 3a (Scheme 3B(c)), indicating that 3a might result from the isomerization of 2a (see kinetic study in the ESI†). In addition, both light irradiation and acidic conditions were required for the efficient synthesis of isothiocyanates.12 Based on these experiments and literature reports,14 a plausible mechanism is shown in Scheme 3C. After irradiation, the excited state of TPT participates in a single-electron oxidation with the aliphatic carboxylate (formed by the deprotonation of the carboxylic acid with sodium carbonate or Ib) or the carboxylic acid under the isothiocyanation reaction conditions, to afford the corresponding carbon-centered radical (R˙) after decarboxylation. The reaction with the source I yields product 2 together with the radical residue (Ia) derived from saccharin (I). To complete the catalytic cycle, the latter is then reduced by the radical anion of the photocatalyst (PC˙−) to regenerate the photocatalyst together with the saccharin anion Ib (path a). Alternatively (path b), and taking into account the measured quantum yield (Φ = 23), a radical chain propagation is likely. Therefore, Ia could also react with the carboxylic acid according to a HAT event to form the corresponding carboxylic acid radical, which upon decarboxylation produces the alkyl radical R˙. With respect to the formation of 3, under acidic conditions and light irradiation, 2 could be fully converted to 3.
In summary, a transition metal-free photoinduced process has been developed for the divergent and selective synthesis of thiocyanates (45 examples, up to 99% yield) and isothiocyanates (6 examples, up to 93% yield) from a variety of primary, secondary, and tertiary alkyl carboxylic acids including at benzylic and on non-activated tertiary positions as well as on (thio)phenoxyacetic acids. A large number of functional groups including halides, ethers, cyanos, azides, as well as heterocycles are compatible with the reaction conditions. Note that this approach requires no pre-activation steps for the carboxylic acids, and relies on a simple catalytic system using a catalytic amount of additives (base or acid) and organophotocatalyst. The thiocyanation reaction was completed in only 45 min. Various analogs of bioactive drugs were obtained in an efficient manner thanks to this molecular editing strategy, and the method showed a complementary site-selectivity compared to existing methods. Mechanistic studies were performed and a photoinduced radical chain process was proposed for this transformation.
This work has been partially supported by University of Rouen Normandy, INSA Rouen Normandy, the Centre National de la Recherche Scientifique (CNRS), European Regional Development Fund (ERDF), Labex SynOrg (ANR-11-LABX-0029), Carnot Institute I2C, the graduate school for research XL-Chem (ANR-18-EURE-0020 XL CHEM), and Region Normandie. T. B. thanks the European Research Council under the European Union's Horizon 2020 research and innovation program (758710). J. V. thanks the Region Normandy for a doctoral fellowship. H. L. thanks the Natural Science and Engineering Research Council of Canada (RGPIN-2022-04168), the Canada Foundation for Innovation (Leaders Opportunity Funds - 225006), the Université de Montréal and the Centre in Green Chemistry and Catalysis (FRQNT-2020-RS4-265155-CCVC).
Conflicts of interest
There are no conflicts to declare.Notes and references
- For reviews, see: (a) A. W. Erian and S. M. Sherif, Tetrahedron, 1999, 55, 7957 CrossRef CAS; (b) T. Castanheiro, J. Suffert, M. Donnard and M. Gulea, Chem. Soc. Rev., 2016, 45, 494 RSC; (c) Q. Xu, L. Zhang, G. Feng and C. Jin, Chin. J. Org. Chem., 2019, 39, 287 CrossRef CAS; (d) M. Gulea and M. Donnard, Curr. Green Chem., 2020, 7, 201 CrossRef CAS; (e) S. Rezayati and A. Ramazani, Tetrahedron, 2020, 76, 131382 CrossRef CAS; (f) M. Gao, M. Vuagnat, M.-Y. Chen, X. Pannecoucke, P. Jubault and T. Besset, Chem. – Eur. J., 2021, 27, 6145 CrossRef PubMed; (g) H. Chen, X. Shi, X. Liu and L. Zhao, Org. Biomol. Chem., 2022, 20, 6508 RSC; (h) F. Buttard and T. Besset, SynOpen, 2023, 7, 117 CrossRef CAS.
- (a) R. G. Guy, The Chemistry of Cyanates and Their Thio Derivatives, ed. S. Patai, John Wiley & Sons, New York, 1977, ch. 18, vol. 2, pp. 819–886 Search PubMed; (b) B. Bayarmagnai, C. Matheis, K. Jouvin and L. J. Gooβen, Angew. Chem., Int. Ed., 2015, 54, 5753 CrossRef CAS PubMed; (c) H.-Y. Xiong, A. Bayle, X. Pannecoucke and T. Besset, Angew. Chem., Int. Ed., 2016, 55, 13490 CrossRef CAS PubMed.
- E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832 CrossRef CAS PubMed.
- For benzylic C–H thiocyanation, see: (a) C. Jiang, P. Chen and G. Liu, CCS Chem., 2020, 3, 1884 CrossRef; (b) D. Liu, Z. Zhang, J. Yu, H. Chen, X. Lin, M. Li, L. Wen and W. Guo, Org. Chem. Front., 2022, 9, 2963 RSC; (c) B. Maeda, Y. Aihara, A. Sato, T. Kinoshita and K. Murakami, Org. Lett., 2022, 24, 7366 CrossRef CAS PubMed; (d) D. Wu, Y. Duan, K. Liang, H. Yin and F.-X. Chen, Chem. Commun., 2021, 57, 9938 RSC ; From redox active esters: ; (e) J. Wu, C. Shu, Z. Li, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2023, e202309684 CAS.
- L. J. Gooßen, N. Rodríguez and K. Gooßen, Angew. Chem., Int. Ed., 2008, 47, 3100 CrossRef PubMed.
- C. Hui, Z. Wang, S. Wang and C. Xu, Org. Chem. Front., 2022, 9, 1451 RSC.
- (a) J. Xuan, Z.-G. Zhang and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632 CrossRef CAS PubMed; (b) L. Li, Y. Yao and N. Fu, Eur. J. Org. Chem., 2023, e202300166 CrossRef CAS and references cited therein: ; (c) A. Reichle and O. Reiser, Chem. Sci., 2023, 14, 4449–4462 RSC.
- (a) S.-M. Yang, T.-J. He, D.-Z. Lin and J.-M. Huang, Org. Lett., 2019, 21, 1958 CrossRef CAS PubMed; (b) D. Jaiswal, J. Tiwari, S. Singh, Kartikey, J. Singh and J. Singh, Catal. Lett., 2021, 151, 1738 CrossRef CAS; (c) D. Wu, C. Li, Y. Duan, H. Yin and F.-X. Chen, Org. Chem. Front., 2021, 8, 3724 RSC.
- (a) M. Gao, M.-Y. Chen, X. Pannecoucke, P. Jubault and T. Besset, Chem. – Eur. J., 2020, 26, 15497 CrossRef CAS PubMed; (b) M.-Y. Chen, X. Pannecoucke, P. Jubault and T. Besset, J. Org. Chem., 2019, 84, 13194 CrossRef CAS PubMed.
- J. Qiu, D. Wu, L. Yuan, P. Long, H. Yin and F.-X. Chen, J. Org. Chem., 2019, 84, 7917 CrossRef CAS PubMed.
- (a) K. Jouvin, C. Matheis and L. J. Goossen, Chem. – Eur. J., 2015, 21, 14324 CrossRef CAS PubMed; (b) S. Vorona, T. Artamonova, Y. Zevatskii and L. Myznikov, Synthesis, 2014, 781 Search PubMed.
- The following paper describing the formation of C(sp3)–SeCN bonds from alkyl carboxylic acids appeared during the writing of this manuscript: Y. Gao, R. Hua, H. Yin and F.-X. Chen, Org. Chem. Front., 2023, 10, 2538 RSC.
- https://macmillan.princeton.edu/photoredox-3/ .
- (a) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed; (b) M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426 RSC; (c) L. Candish, L. Pitzer, A. Gómez-Suárez and F. Glorius, Chem. – Eur. J., 2016, 22, 4753 CrossRef CAS PubMed; (d) T. Brégent, J.-P. Bouillon and T. Poisson, Org. Lett., 2020, 22, 7688 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc04624a |
| This journal is © The Royal Society of Chemistry 2024 |