
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modified minimal-size fragments of heparan sulfate as inhibitors of endosulfatase-2 (Sulf-2)†
Alice
Kennett
a,
Sven
Epple
a,
Gabriella
van der Valk
a,
Irene
Georgiou
a,
Evelyne
Gout
b,
Romain R.
Vivès
 b and
Angela J.
Russell
*ac
b and
Angela J.
Russell
*ac
aDepartment of Chemistry, University of Oxford, Oxford OX1 3TA, UK. E-mail: angela.russell@chem.ox.ac.uk
bUniv. Grenoble Alpes, CNRS, CEA, IBS, Grenoble, France
cDepartment of Pharmacology, University of Oxford, Oxford OX1 3QT, UK
First published on 7th November 2023
Abstract
Sulf-2 has been identified as a putative target for anticancer therapies. Here we report the design and synthesis of sulfated disaccharide inhibitors based on IdoA(2S)-GlcNS(6S). Trisulfated disaccharide inhibitor IdoA(2S)-GlcNS(6Sulfamate) demonstrated potent Sulf-2 inhibition. The IC50 value was determined to be 39.8 μM ± 18.3, which is comparable to a tetrasaccharide inhibitor of HSulf-1 reported in the literature. We propose that the disaccharide IdoA(2S)-GlcNS(6S) is the shortest fragment size required for effective inhibition of the Sulfs.
Endosulfatases (Sulf-1 and Sulf-2) are located in the extracellular matrix and are responsible for the selective desulfation of the sulfate group on the glucosamine 6-O-sulfate residues within heparan sulfate (HS) proteoglycans and have a strong substrate specificity for the [Glc/IdoA(2S)-GlcNS(6S)] trisulfated disaccharide (Fig. 1).1a,b The trisulfated disaccharide [Glc/IdoA(2S)-GlcNS(6S)] has a low abundance within HS, and therefore seemingly subtle modifications by Sulf activity result in major functional consequences.2 This highlights the importance of Sulf activity and indicates how targeting the Sulfs could have significant downstream effects on HS-mediated processes. Sulf-2 inhibitors are putative anticancer therapeutics because the sulfs have been linked to the regulation of signalling pathways such as Wnt and FGF via the modulation of the 6-O-sulfation status of HS.3 Sulf-2 expression is induced or upregulated in in various cancers and its role has been identified as being pro-tumourgenic, with Sulf-2 gene silencing or knock-out leading to decreased tumour formation. Therefore, Sulf-2 inhibition has been identified as a potential therapeutic target for many cancers.4a,b For this reason, the development of endosulfatase inhibitors has gained attention over the past decade.
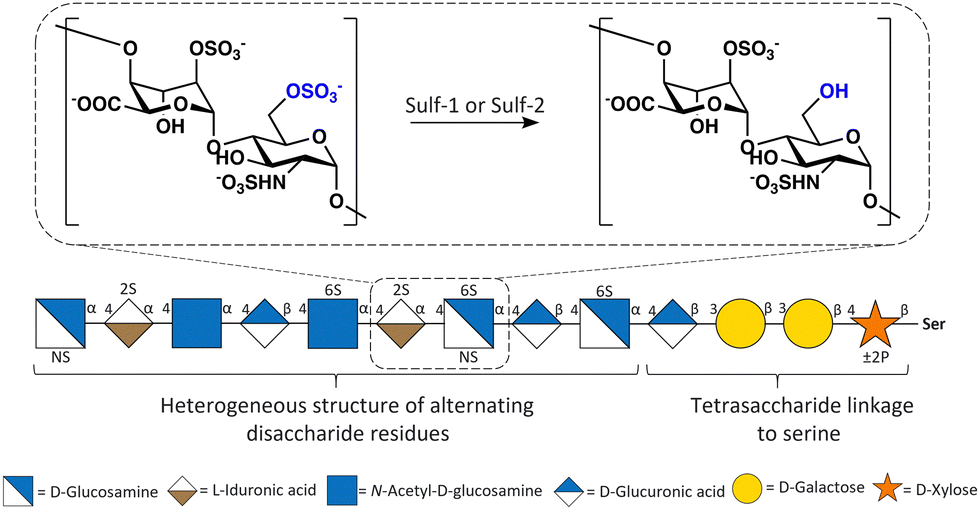 | ||
| Fig. 1 Structure of HS highlighting the disaccharide residue, IdoA(2S)-GlcNS(6S), that Sulfs have a preference for. | ||
Scheilwies et al. reported glucosamine-based small molecule inhibitors substituting the 6-O-sulfate (–OSO3−) with the sulfamate motif (–OSO2NH2). This preliminary work utilised the smallest, most relevant unit of HS, α-GlcNS(6S) to template inhibitor design.5 The biochemical characterisation of this compound in a competition assay with fluorogenic substrate 4-methylumbelliferyl sulfate (4-MUS), revealed that the sulfamate inhibitor had an IC50 values of 95 μM against HSulf-1 and 130 μM against HSulf-2, and importantly was more selective for the Sulfs than other sulfatases investigated. In 2015, Miller et al. aimed to replicate the inhibitory activity of the glucosamine-6-sulfamate inhibitors and develop a structure activity relationship. All compounds synthesised were found to have minimal inhibition of Sulf-2 at 1 mM.6 However, there were some discrepancies in assay protocol between the two papers that may explain the different inhibition potencies reported, so the question remains of whether 1 is a true inhibitor of Sulfs. Recently, Chiu et al. reported the design and synthesis of di-, tri- and tetra-saccharide fragments of HS with the sulfamate modification as inhibitors of Sulf-1.7 The disaccharide, GlcNS(6Sulfamate)-IdoA(2S) only caused 20% Sulf-1 inhibition at 0.7 mM (IC50 value not determined), and the trisaccharide and tetrasaccharide analogues were more potent with IC50 values of 0.53 and 29.6 μM, respectively.
We propose that the IdoA(2S)-GlcNS(6Sulfamate) disaccharide will have superior potency as a minimal fragment Sulf inhibitor compared to the beforementioned reported disaccharide because the substrate specificity studies of the Sulfs point towards this disaccharide unit as being the most frequently desulfated among HS.8 In this study, we report the re-evaluation of monosaccharide glucosamine-6-O-sulfamates as inhibitors of Sulf-2, and the synthesis of putative disaccharide inhibitors using the IdoA(2S)-GlcNS(6S) scaffold as a guide in order to determine whether this fragment size is the shortest effective moiety for HS inhibition.
Putative inhibitors 1 and 2 (Fig. 2) were synthesised according to literature protocols.5,6 Additionally, a non-hydrolysable analogue 3, bearing a methylene sulfonamide in the 6-position, was designed and prepared. The key step was the installation of the methylsulfonamide functional group which was achieved via nucleophilic substitution of an orthogonally-protected triflate 9, which was synthesized from glucosamine hydrochloride according to Scheme 1. The 3- and 4-hydroxyls of intermediate 76 were protected by reaction with 2,3-butanedione, trimethyl orthoformate and catalytic sulfonic acid in refluxing methanol, to give 8 as a single diastereomer in 83% yield.
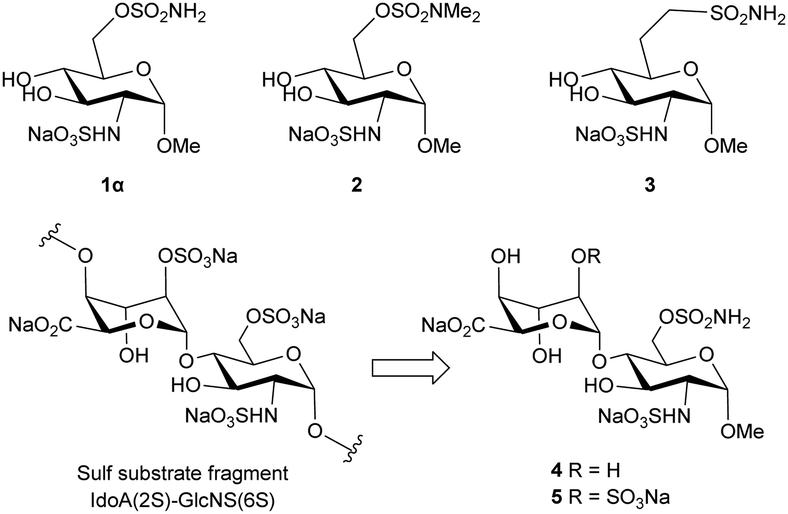 | ||
| Fig. 2 Structure of putative disaccharide inhibitors. | ||
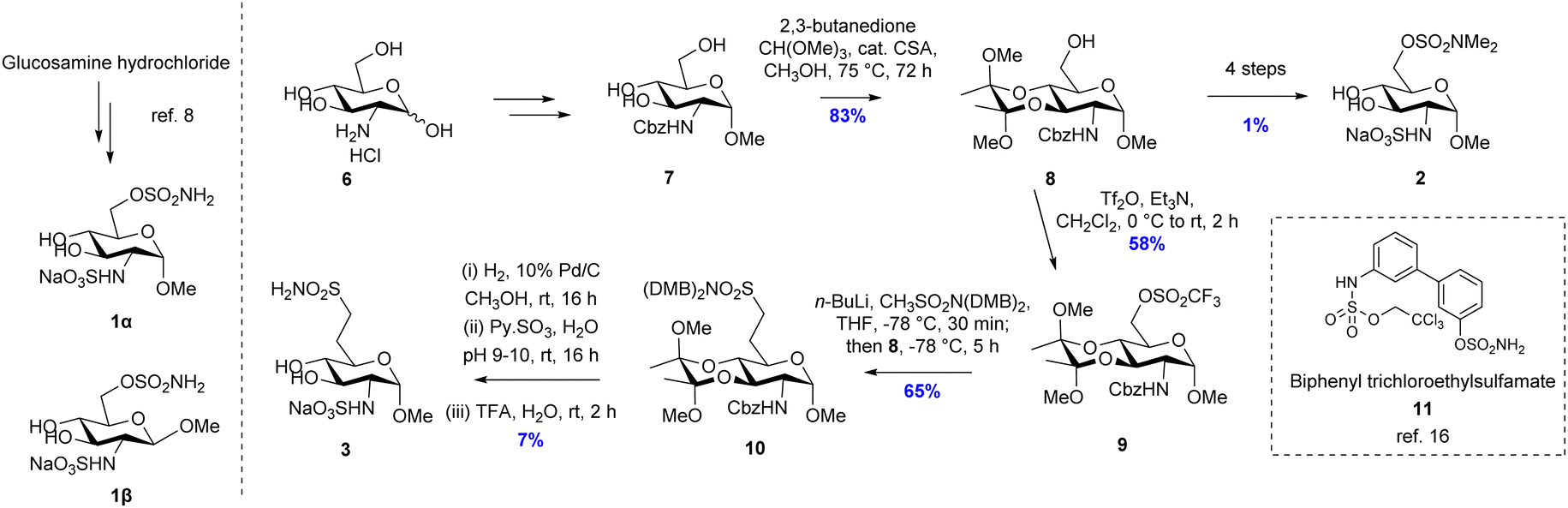 | ||
| Scheme 1 Synthesis of inhibitors 1α, 1β, 2 and 3. | ||
Triflation of 8 was achieved with triflic anhydride in the presence of Et3N at 0 °C to give 9 in 58% yield. The nucleophile LiCH2SO2(DMB)2 was generated in situ by deprotonation of methylsulfonamide CH3SO2(DMB)2 with n-BuLi at −78 °C.9 The methylene sulfonamide unit was then introduced by nucleophilic substitution of triflate 9 with LiCH2SO2N(DMB)2 to give fully protected intermediate 10 in 65% yield. Finally, a three-reaction sequence was performed: (1) the carboxybenzyl group was deprotected in 46% yield by palladium-catalyzed hydrogenation; (2) addition of sulfur trioxide pyridine complex to the amine intermediate in water at pH 9–10 resulted in the sulfation of the amino group in 37% yield; (3) deprotection of the two N-2,4-dimethoxybenzyl and 3,4-bisacetal units was achieved using TFA in water in 52% yield. Following purification the putative non-hydrolysable inhibitor 3 was isolated as a sodium salt in 7% yield over three steps.
Two disaccharide inhibitors, 4 and 5, were designed based on the trisulfated disaccharide fragment of HS identified by Sulf substrate specificity studies, incorporating the 6-O-sulfamate group, Fig. 2. Retrosynthetic analysis of inhibitor 4 identified key intermediates p-tolyl 2,4,6-tri-O-acetyl-3-O-benzyl-1-thio-L-idopyranoside 12 prepared from diacetone D-glucose, 6 steps, 22% yield10 and glucosamine glycosyl acceptor 16, which was synthesized according to Scheme 2. First, the 4-OH and 6-OH of intermediate 7 were protected using a benzylidene acetal, which formed 13 as a single diastereoisomer in 87% yield. Next, the 3-OH was protected using sodium hydride and benzyl bromide in DMF to obtain 14 in 58% yield. Benzylidene acetal 14 was hydrolysed to 15 using 70% acetic acid at 65 °C (63% yield) and finally, silyl ether formation using tert-butyldiphenylchlorosilane gave acceptor 16 in 90% yield.
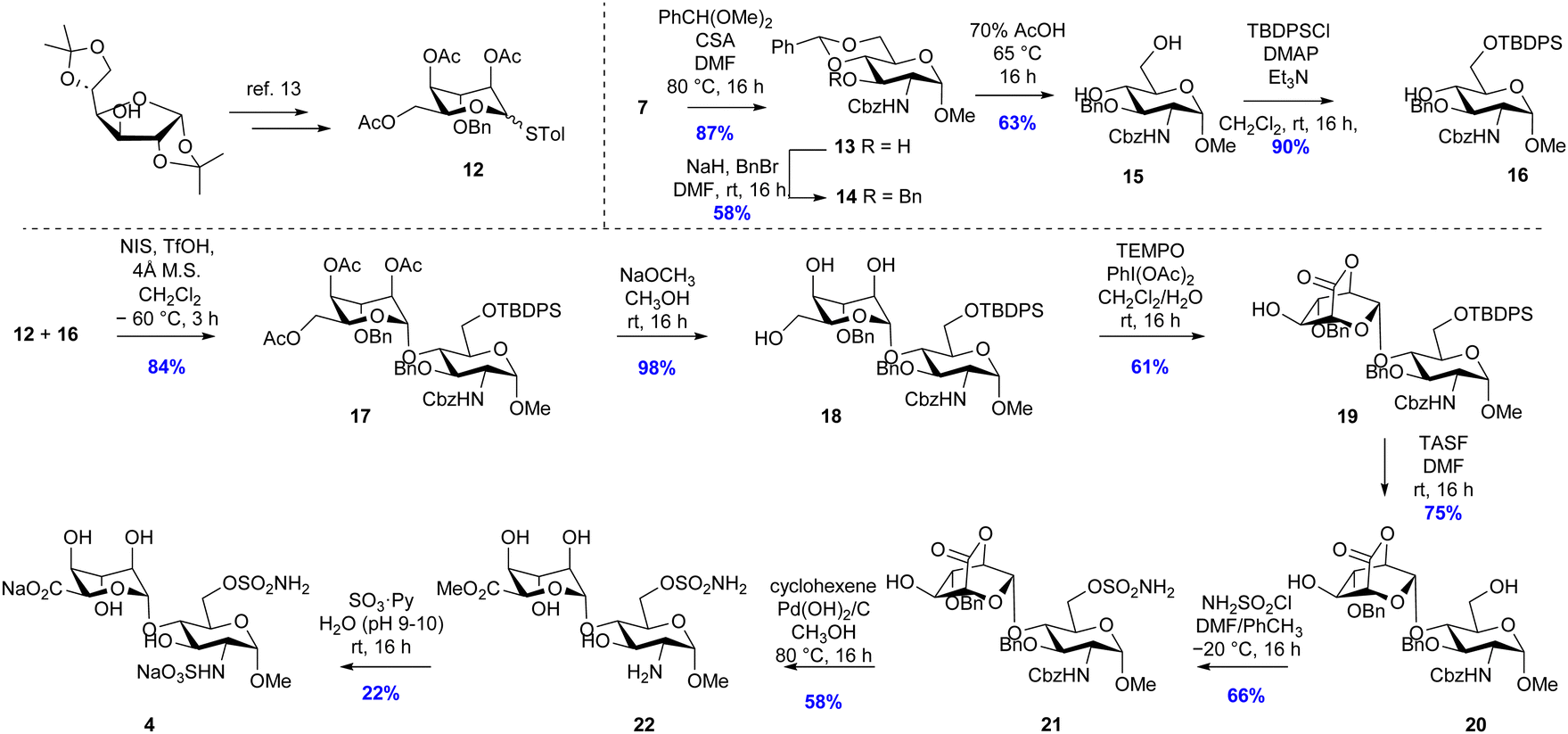 | ||
| Scheme 2 Synthesis of inhibitor 4. | ||
The glycosylation reaction between 12 and 16 was achieved using the NIS/TfOH reagent system to activate the thioglycoside donor to give the desired disaccharide intermediate 17 isolated in 84% yield as a single (alpha) anomer (Scheme 3). Anomeric stereochemistry was assigned by 1H NMR spectroscopy. Next, the acetate esters were removed under Zemplén conditions to give triol 18 in 98% yield. The primary alcohol was oxidised using catalytic TEMPO and stoichiometric PIDA which produced lactone 19 in 61% yield. Desilylation of compound 19 initially proved challenging due to the instability of the lactone with nucleophiles causing low yields under TBAF deprotection conditions. Even when buffered with acetic acid, only a low yield (45%) of 20 was isolated. The optimised conditions used tris(dimethylamino)sulfonium difluorotrimethylsilicate (TASF) that gave an isolated yield of 75%. Next, regioselective sulfamoylation of 20 was achieved under conditions reported by Miller et al., to give sulfamate 21 in 66% yield. Multiple conditions were trialled for the global debenzylation and deprotection of the amino-Cbz group, and the optimal conditions were found to be catalytic transfer hydrogenation using cyclohexene as the hydrogen donor with 20% Pd(OH)2 in refluxing methanol.11 Under these conditions, methyl ester 22 was isolated in 58% yield. Finally, the primary amine was sulfated using sulfur trioxide–pyridine complex in basic aqueous medium. Purification by ion-paired reverse-phase HPLC using 2 M triethylammonium bicarbonate and acetonitrile gradient, followed by elution through a Dowex® 50WX8 Na+-form column, gave 4 in 22% isolated yield.
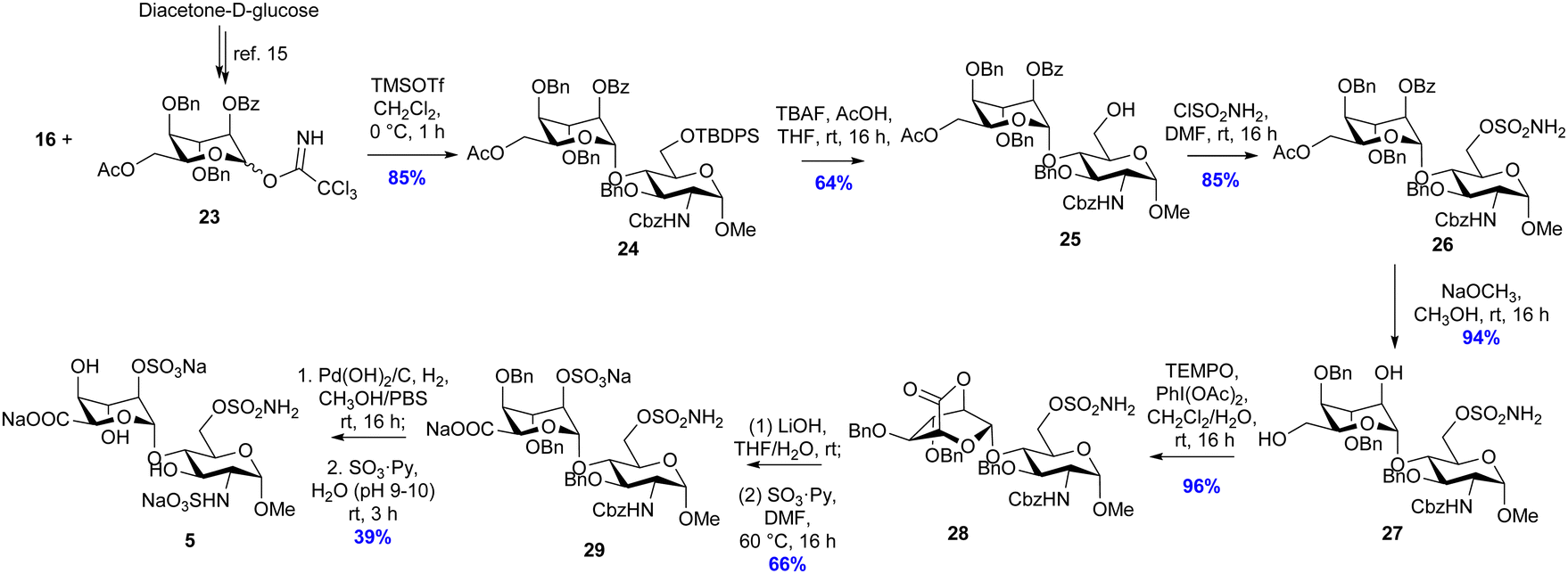 | ||
| Scheme 3 Synthesis of inhibitor 5. | ||
It was originally envisioned that the synthesis of putative inhibitor 5 could diverge from the synthesis of 4, via benzylation of intermediate alcohol 19. However, all attempts at benzyl protection of the ido 4-OH of 19 were unsuccessful and therefore idose glycosyl donor 23 was synthesised according to Hu et al. (Scheme 3).12 With the alternative ido-glycosyl donor in hand, the glycosylation reaction between 23 and glycosyl acceptor 16 was activated using TMSOTf and proceeded effectively to afford the desired disaccharide 24 in 85% yield. Desilyation of the 6-O-TBDPS group of 24 using TBAF buffered in acetic acid proceeded to afford 25 in 64% yield. Subsequent sulfamoylation of the primary alcohol to 26 was achieved in 74% yield by altering the previous conditions to use 2 equivalents of sulfamoyl chloride at 0 °C. Subsequently, the base-labile protecting groups of compound 26 were removed by catalytic NaOCH3 in CH3OH to produce diol 27 in 94% yield. The resulting diol 27 was then subjected to oxidation with the TEMPO/PIDA reagent system to afford lactone 28 in 58% yield. 28 was immediately hydrolysed in basic aqueous medium, and the resulting 2-OH moiety was treated with sulfur trioxide–pyridine complex under microwave irradiation. After elution through a Dowex® 50WX8 Na+-form column, 29 was isolated in 66% yield over two steps. Finally, the hydrogenolysis-labile protecting groups of 29 were cleaved by Pd(OH)2/C catalysed hydrogenation in methanol and aqueous phosphate buffered saline (20 mM, pH 7.4), to give a primary amine intermediate, which was successively subjected to sulfur trioxide pyridine complex in basic aqueous medium to afford final compound 5 in 39% over two steps as the tri-sodium salt.
The inhibition of HSulf-2 with inhibitors 1, 2, 3, 4 and 5 was determined using a competition assay with purified, recombinant HSulf-2 and a fluorogenic substrate 4-methylumbelliferyl sulfate (4-MUS) (Fig. 3). HSulf-2 shows pro-tumoral behaviour and therefore is a prime target for the design of inhibitors. Compounds were tested at a single concentration (500 μM) to compare inhibitory activity and the IC50 value was determined for the most potent inhibitor. Biphenyl trichloroethylsulfamate 1113 (Scheme 4), was included as a benchmark Sulf-2 inhibitor. Inhibition of sulfatase from Aerobacter aerogenes, a bacterial sulfatase was used to assess selectivity of the compounds.
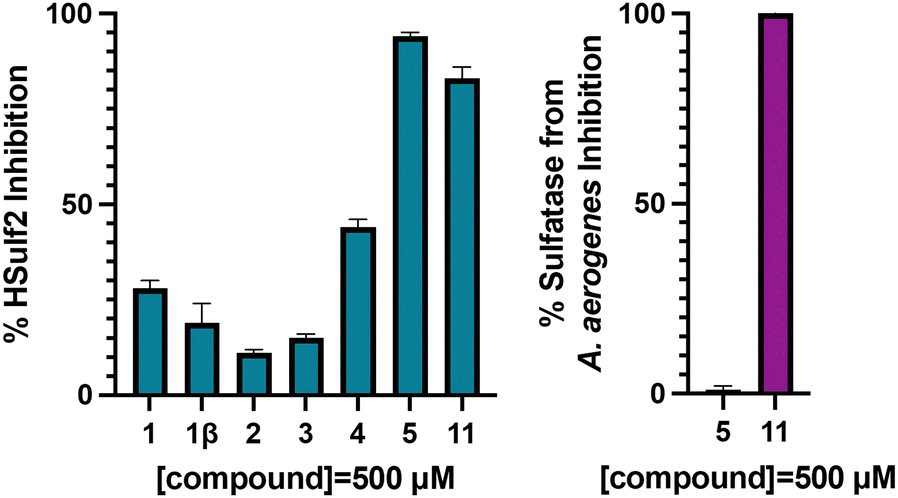 | ||
| Fig. 3 (left) Sulf-2 and (right) sulfatase from A. aerogenes inhibition data for glucosamine-based inhibitors and biphenyl trichloroethylsulfamate 11. Data represented as the mean ± SD, (n = 2). Inhibition values are reported as percentages of the uninhibited control values. | ||
In the monosaccharide series, parent glucosamine-6-O-sulfamate 1 was found to display weak inhibition of 28% at 500 μM, and 1β, 2 and 3 inhibited Sulf 2 by <15% at 500 μM. As predicted, the extension of fragment size to the disaccharide scaffold led to an increased inhibition at 500 μM. Disulfated disaccharide 4 inhibited Sulf-2 by 44% and trisulfated disaccharide 5 inhibited Sulf-2 almost completely (95%) at 500 μM.
The inhibition of Sulf-2 by compound 11 was evaluated over a concentration range and the IC50 was found to be 39.8 μM ± 17.6 (Fig. S1, ESI†). In comparison, the best biphenyl inhibitor reported by Reuillon et al., compound 11, was reported of having an IC50 value of 167 ± 5 μM against Sulf-2. In the present study, compound 11 was used as a benchmark compound and it was found to be less potent than compound 11 (80% vs. 95% inhibition of Sulf-2 at 500 μM, Fig. 3). Furthermore, at this single concentration compound 11 exhibited potent inhibition of sulfatase from A. aerogenes (100%) compared to compound 5 (1% ± 1). This shows that compound 5 is more potent and more selective than the previous best inhibitor of Sulf-2 reported in the literature.
A small library of saccharide-based endosulfatase inhibitors was prepared incorporating a 6-sulfamate group in place of the glucosamine 6-O-sulfate. The presented study supports previous findings that the replacement of the glucosamine-6-O-sulfate with the 6-sulfamate group leads to effective inhibition of HSulf-2 activity. The putative inhibitors were evaluated in a competition assay with recombinant HSulf-2 and a fluorogenic substrate 4-methylumbelliferyl sulfate (4-MUS). Trisulfated 5 was found to be superior to the other inhibitors investigated and is more potent against Sulf-2 and more selective for Sulf-2 vs. other sulfatases than a biphenyl trichloroethylsulfamate inhibitor reported in the literature.13 We propose that compound 5, and consequently the disaccharide IdoA(2S)-GlcNS(6S), may represent the minimal-size fragment of HS required for effective inhibition of the endosulfatases. The disaccharide IdoA2S-GlcNS(6S) is not a substrate of the Sulfs,14 however this fragment-size does efficiently bind to the active site (evidenced by inhibition in the 4-MUS assay), making it a good scaffold for inhibitor design. While inhibitor 5 displays effective inhibition, the fate of 5 in the presence of Sulf-2 remains unknown: whether the C(6)O–S bond is hydrolyzed or 5 simply binds to the active site and functions as a competitive inhibitor requires further investigation.
A. K. is grateful to the EPSRC and OxStem for financial support. S. E. thanks EPSRC SBM CDT (EP/L015838/1) for a studentship. This work was also supported by the “Investissements d’avenir” program Glyco@Alps (ANR-15-IDEX-02) and a grant from the Agence Nationale de la Recherche (ANR-17-CE11-0040). I. B. S. acknowledges integration into the Interdisciplinary Research Institute of Grenoble (IRIG, CEA).
Conflicts of interest
There are no conflicts to declare.References
- (a) E. H. Pempe, T. C. Burch, C. J. Law and J. Liu, Glycobiology, 2012, 22, 1353–1362 CrossRef CAS PubMed; (b) A. Seffouh, et al. , FASEB J., 2013, 27, 2431–2439 CrossRef CAS PubMed.
- A. Seffouh, et al. , Cell. Mol. Life Sci., 2019, 76, 1807–1819 CrossRef CAS PubMed.
- P. C. Billings and M. Pacifici, Connect. Tissue Res., 2015, 56, 272–280 CrossRef CAS PubMed.
- (a) E. Hammond, A. Khurana, V. Shridhar and K. Dredge, Front. Oncol., 2014, 4, 195 Search PubMed; (b) S. D. Rosen and H. Lemjabbar-Alaoui, Expert Opin. Ther. Targets, 2010, 14, 935–949 CrossRef CAS PubMed.
- M. Schelwies, et al. , ChemBioChem, 2010, 11, 2393–2397 CrossRef CAS PubMed.
- D. C. Miller, et al. , Org. Biomol. Chem., 2015, 13, 5279–5284 RSC.
- L. T. Chiu, et al. , J. Am. Chem. Soc., 2020, 142, 5282–5292 CrossRef CAS PubMed.
- X. B. Ai, et al. , J. Cell Biol., 2003, 162, 341–351 CrossRef CAS PubMed.
- L. Navidpour, W. Lu and S. D. Taylor, Org. Lett., 2006, 8, 5617–5620 CrossRef CAS PubMed.
- T. H. Li, et al. , ChemMedChem, 2014, 9, 1071–1080 CrossRef CAS PubMed.
- K. M. Sureshan, et al. , J. Med. Chem., 2012, 55, 1706–1720 CrossRef CAS PubMed.
- Y. P. Hu, et al. , Nat. Chem., 2011, 3, 557–563 CrossRef CAS PubMed.
- T. Reuillon, et al. , Chem. Sci., 2016, 7, 2821–2826 RSC.
- O. M. Saad, et al. , Glycobiology, 2005, 15, 818–826 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc02565a |
| This journal is © The Royal Society of Chemistry 2024 |