
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic conformational equilibria in the active states of KRAS and NRAS†
Enrico
Rennella
b,
Chrystèle
Henry
a,
Callum J.
Dickson
 c,
Florian
Georgescauld‡
d,
Thomas E.
Wales
d,
Dirk
Erdmann
a,
Simona
Cotesta
a,
Michel
Maira
a,
Richard
Sedrani
a,
Saskia M.
Brachmann
a,
Nils
Ostermann
a,
John R.
Engen
d,
Lewis E.
Kay
b,
Kim S.
Beyer
*a,
Rainer
Wilcken§
*a and
Wolfgang
Jahnke
*a
c,
Florian
Georgescauld‡
d,
Thomas E.
Wales
d,
Dirk
Erdmann
a,
Simona
Cotesta
a,
Michel
Maira
a,
Richard
Sedrani
a,
Saskia M.
Brachmann
a,
Nils
Ostermann
a,
John R.
Engen
d,
Lewis E.
Kay
b,
Kim S.
Beyer
*a,
Rainer
Wilcken§
*a and
Wolfgang
Jahnke
*a
aNovartis Biomedical Research, Basel, Switzerland. E-mail: wolfgang.jahnke@novartis.com; rwilcken@flaretx.com; kim.beyer@novartis.com
bUniversity of Toronto, Department of Biochemistry, Toronto, Canada
cNovartis Biomedical Research, Cambridge, MA, USA
dNortheastern University, Department of Chemistry and Chemical Biology, Boston, MA, USA
First published on 25th November 2024
Abstract
The design of potent RAS inhibitors benefits from a molecular understanding of the dynamics in KRAS and NRAS and their oncogenic mutants. Here we characterize switch-1 dynamics in GTP-state KRAS and NRAS by 31P NMR, by 15N relaxation dispersion NMR, hydrogen–deuterium exchange mass spectrometry (HDX-MS), and molecular dynamics simulations. In GMPPNP-bound KRAS and NRAS, we see the co-existence of two conformational states, corresponding to an “inactive” state-1 and an “active” state-2, as previously reported. The KRAS oncogenic mutations G12D, G12C and G12V only slightly affect this equilibrium towards the “inactive” state-1, with rank order wt < G12C < G12D < G12V. In contrast, the NRAS Q61R oncogenic mutation shifts the equilibrium fully towards the “active” state-2. Our molecular dynamics simulations explain this by the observation of a transient hydrogen bond between the Arg61 side chain and the Thr35 backbone carbonyl oxygen. NMR relaxation dispersion experiments with GTP-bound KRAS Q61R confirm a drastic decrease in the population of state-1, but still detect a small residual population (1.8%) of this conformer. HDX-MS indicates that higher populations of state-1 correspond to increased hydrogen–deuterium exchange rates in some regions and increased flexibility, whereas low state-1 populations are associated with KRAS rigidification. We elucidated the mechanism of action of a potent KRAS G12D inhibitor, MRTX1133. Binding of this inhibitor to the switch-2 pocket causes a complete shift of KRAS G12D towards the “inactive” conformation and prevents binding of effector RAS-binding domain (RBD) at physiological concentrations, by signaling through an allosteric network.
Introduction
KRAS, HRAS and NRAS are important proteins in many signal transduction pathways, and dysregulation of their activity is often related to cancer.1 KRAS is somatically mutated in ∼10% of all cancers, including ∼90% of pancreatic cancers.2,3 KRAS mutations most commonly occur at positions 12, 13 and 61, with resulting mutations such as G12D, G12V, or G12C.4 KRAS is a molecular switch that cycles between an active GTP-bound state and an inactive GDP-bound state.5 Mutations in KRAS often lead to an imbalance of GTP- and GDP-binding of KRAS, and thus to constitutively active signal transduction and uncontrolled cell proliferation. Mutated KRAS has been recognized for a long time as an important cancer target,6 but only the last decade has seen significant advancements in targeting KRAS with small molecules.7–9 Notably, several small molecule KRAS inhibitors are approved or in the clinic that target the G12C, G12D or other mutations.10–15Protein dynamics are at the heart of protein function, and understanding of protein dynamics can greatly enhance drug discovery.16,17 In fact, the main binding sites on KRAS, the switch-1/2 and switch-2 pockets, are “cryptic”, meaning that the binding sites are not available in the energetically most favored and thus most populated “ground state”, and only transiently open through dynamic processes. Previous work has shown that members of the RAS family are highly dynamic proteins. Most notably, a region near the N-terminus called switch-1 was identified as a highly dynamic loop in the GTP-state of HRAS.18 Using 31P NMR, Kalbitzer and colleagues found that in GTP-state RAS, switch-1 can adopt an open conformation (termed state-1) that is not able to bind effector proteins and thus represents an inactive state. However, switch-1 can also adopt a closed conformation (termed state-2) that binds effector proteins and is thus active in signal transduction.19–22 The notion that GTP-bound RAS is in an active “ON-state” while GDP-bound RAS is in an inactive “OFF-state” is thus oversimplified. Several additional studies have corroborated and further characterized these dynamic processes in GTP-state RAS.23–31 However, while KRAS and NRAS have been recognized as the most therapeutically relevant RAS isoforms, and NMR studies on the conformation and dynamics of NRAS32 and KRAS30,33 have been carried out, most 31P NMR studies have been carried out on HRAS or MRAS and on mechanistic mutants, rather than oncogenic mutants. Only very recently, a 31P NMR study on KRAS and its oncogenic mutants was published.34 Our paper adds additional and complementary information on conformational dynamics of KRAS and NRAS wildtype and mutants and clarifies the mechanism of action of a clinical KRAS inhibitor.
Experimental
Protein production for 31P NMR (unlabeled GMPPNP-loaded KRAS/NRAS)
DNA sequences coding for the G-domains of different RAS variants (amino acids 1–169) were inserted in a plasmid which allows protein expression in E. coli under control of the T7 promoter. Expression was induced by addition of 1 mM IPTG and performed at 18 °C overnight.Bacterial pellets were resuspended in lysis buffer (20 mM Tris pH 8.0, 500 mM NaCl, 5 mM imidazole, 2 mM TCEP, 10% glycerol, Complete protease inhibitor (Roche) and 40 U ml−1 Turbonuclease (Sigma)). Cells were lysed with a high-pressure homogenizer (Avestin Emulsiflex C3) and the lysate was clarified by ultracentrifugation at 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 40 minutes at 4 °C. Clarified lysate was loaded on a Ni–sepharose HP column (Cytiva). The column was washed and the protein was eluted with a 5–200 mM imidazole gradient in IMAC buffer (20 mM Tris pH 8.0, 500 mM NaCl, 2 mM TCEP, 10% glycerol).
000g for 40 minutes at 4 °C. Clarified lysate was loaded on a Ni–sepharose HP column (Cytiva). The column was washed and the protein was eluted with a 5–200 mM imidazole gradient in IMAC buffer (20 mM Tris pH 8.0, 500 mM NaCl, 2 mM TCEP, 10% glycerol).
The fusion-tag of the eluted protein was cleaved overnight at 4 °C by either His-tagged HRV 3C or TEV protease. Cleaved protein was re-loaded onto the nickel column (reverse-IMAC step) and the flow through containing the target protein was collected. The protein was concentrated with a 10 kDa MWCO ultrafiltration system (Millipore).
Nucleotide exchange was performed by incubating the RAS proteins during 1 hour at RT with a 24-fold molar excess of nucleotide (GMPPNP, Jena Bioscience) in presence of 25 mM EDTA. The mixture buffer was then exchanged using a PD-10 column (Cytiva) against nucleotide loading buffer (40 mM Tris pH 8.0, 200 mM (NH4)2SO4, 0.1 mM ZnCl2). A fresh 24-fold molar excess of nucleotide was again added. 40 U of Shrimp Alkaline Phosphatase (New England Biolabs) was also added. After an incubation for 1 hour at 4 °C MgCl2 was added to a concentration of 30 mM.
The sample was then loaded on a HiLoad 16/600 Superdex 75 pg size-exclusion column pre-equilibrated with SEC buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 5 mM MgCl2, 2 mM TCEP). Fractions containing the protein of interested were pooled, concentrated with 10 kDa molecular weight cut-off ultrafiltration system and snap frozen.
31P NMR
31P NMR experiments were performed at 280 K (7 °C) on a Bruker NMR spectrometer operating at a 31P frequency of 243 MHz, equipped with a BBO cryoprobe tuned and matched on 1H and 31P. GMPPNP-loaded KRAS or NRAS proteins in the presence or absence of additional ligands were used at concentrations around 5 mg ml−1 in a buffer consisting of 20 mM HEPES, pH 7.5, 150 mM NaCl, 5 mM MgCl2, 2 mM TCEP. All 31P spectra were processed with an exponential window function using LB = 10.Relaxation dispersion NMR and preparation of GTP-loaded 15N-KRAS
15N-labeled proteins were produced as His6-SUMO-KRAS(1–169) constructs via expression in E. Coli BL21 (DE3) using M9 media supplemented with 15N-labeled ammonium chloride. The cell cultures (1 L for each variant) were induced at OD ∼ 0.6–0.8 with 0.25 mM IPTG and shaken overnight at 18 °C. The day after, the cells were pelleted by centrifugation at 3000 g for 20’, resuspended in lysis buffer (50 mM TrisHCl pH 7.8, 500 mM NaCl, 30 mM imidazole, 2 mM DTT, traces of DNase I) and sonicated. Insoluble fractions were removed as pellets from lysates by centrifugation at 14000g for 20’. The soluble fractions were then loaded onto prepacked NiNTA resin to purify the His6 labeled KRAS proteins which were eluted using elution buffer (50 mM TrisHCl pH 7.8, 100 mM NaCl, 500 mM imidazole, 2 mM DTT). The His6-SUMO tag was removed by incubation with Ulp1 protease overnight under dialysis conditions (50 mM TrisHCl pH 7.8, 100 mM NaCl, 0.5 mM EDTA, 2 mM DTT). The next day the dialysed solution was loaded onto NiNTA resin to purify KRAS (as flow-through) from the residual uncleaved His6 labeled protein, the His6-SUMO tag, and Ulp1. The flow-through solution was concentrated to 1–2 mL using centrifugal concentrators (with a 10K cutoff) and then loaded onto a prepacked Superdex 75 column equilibrated with SEC buffer (50 mM Hepes pH 7.8, 150 mM NaCl, 10 mM EDTA, 2 mM DTT) and fractions were collected corresponding to the correctly folded monomeric protein. For the preparation of NMR samples, and with slight modifications from the protocol reported by Hansen et al.30 the protein solution was buffer exchanged using centrifugal concentrators first in 20 mM Hepes pH 7.5, 15 mM EDTA, 2 mM DTT (overall dilution factor > 250), afterwards in 20 mM Hepes pH 7, 5 mM TCEP (NMR buffer, overall dilution factor > 500) and then stored at 4 °C until sample preparation. For the final sample preparation, to reach an NMR sample volume of 500 μL, the protein was diluted with NMR buffer, 10 μL D2O, 50 μL of GTP solution (from 100 mM stock purchased from Thermo and adjusted to pH 7 with 24 mM Hepes acid) and 5 μL MgCl2 (from a 0.5 M stock solution). Final concentrations of KRAS, GTP and Mg were 400 μM, 10 mM and 5 mM, respectively. All proteins were stable in the GTP-bound form for at least two days, so that CPMG experiments could be acquired.
All NMR experiments were recorded at 25 °C with a Bruker AVANCE III HD 14.1 T NMR spectrometer, equipped with a cryogenically cooled, triple-resonance probe. 15N CPMG experiments were recorded using a constant-time CPMG element during which the 15N magnetization is in-phase,35 using a 16.7 kHz 1H continuous-wave decoupling field, with 0013 phase-cycling of the CPMG pulses.36,37 Assignments from BMRB entry 52021 were used.30 The datasets for 1H CPMG experiments for the WT protein were downloaded from Hansen et al. via Dryad,30 and re-analysed to get the chemical shift values for amides of state-1, as shown in Fig. 3A for G12 and G13.
Molecular dynamics
The X-ray crystal structures of KRAS (GMPPNP bound) and NRAS (GMPPNP bound) were obtained from the RCSB database, with PDB codes 6god38 and 5uhv39 respectively. These were then prepared for molecular dynamics simulation using CCG MOE protein preparation workflow,40 assigning standard amino acid protonation states at pH 7.4 and optimization of the hydrogen bonding network. CCG MOE was also used to create the KRAS G12D and NRAS Q61R mutants, by mutating glycine-12 of KRAS to aspartic acid and mutating glutamine-61 of NRAS to arginine, followed by a short side-chain minimization.The four proteins (KRAS wt, KRAS G12D, NRAS wt and NRAS Q61R) were then used to prepare a simulation box with Amber20.41 GMPPNP molecules were modelled as GTP using Amber polyphosphate parameters,42 and the Mg2+ ion using 12–6 LJ divalent ion parameters for TIP3P water.43 Amber tleap was used to embed each system into a cubic water box with 12 Å buffer to any protein residue and sodium ions added to charge neutralize the system. The protein was modelled using ff19SB,44 water with TIP3P,45 and sodium ions using JC parameters.46 Each of the four proteins underwent 10 independent replicates of 1 μs production simulation (10 μs total sampling per protein) using the following protocol. First, the system was minimized using sander and the steepest descent method for 5000 steps before switching to the conjugate gradient method for a further 5000 steps. The system was then heated from 0 to 100 K over a 5 ps constant volume run, employing Langevin dynamics,47 with 5 kcal mol−1 Å−2 restraints on all heavy atoms using Amber20 PMEMD CUDA48 on GPU cards.41,49 Next, the volume was allowed to change freely, and the temperature raised to 298 K with Langevin dynamics (γ = 1 ps−1) applied for 100 ps with the Berendsen barostat to maintain pressure around 1 atm by coupling the periodic box with a time constant of 2 ps50 while maintaining the 5 kcal mol−1 Å−2 restraints on all heavy atoms. All restraints were then removed and the system transitioned to 50 ns NPT simulation using Langevin dynamics (γ = 1 ps−1), a 2 fs time-step, the Monte Carlo barostat51 and a cut-off of 9 Å applied to electrostatics and dispersion interactions – beyond this distance electrostatic interactions were treated with PME52 and an analytical correction applied for dispersion interactions. Finally the system was transitioned to 1 μs production NPT simulation using identical settings except for the use of Hydrogen Mass Repartitioning53 to allow a 4 fs time-step. Trajectory frames were written every 10 ps.
Distance, RMSD and hydrogen bond analyses were performed on the resulting trajectories using CPPTRAJ54 and figures generated using python matplotlib.55 For RMSD analysis, switch I was defined as residues 25–40 and switch II as residues 60–76.
Results
Conformational equilibria in KRAS and NRAS detected by 31P NMR
When GMPPNP is bound to KRAS, the 31P signal of its gamma-phosphate, and to a lesser extent the 31P signal of its alpha-phosphate are split into two peaks (Fig. 1A). This is similar to the situation in HRAS22 and reflects the co-existence and dynamic interconversion of two conformational states of similar free energy in the GTP-state of KRAS. “State-1” has been attributed to an inactive conformation of switch-1 and the beginning of switch-2 which is incompetent of binding the RAS binding domains (RBD) of effector proteins such as RAF. “State-2” corresponds to the active conformation of switch-1 and the beginning of switch-2 that can bind RBD and thus transmit RAS-mediated signalling. Accordingly, we refer to state-1 and state-2 as conformations of switch-1 and the beginning of switch-2. Conformational changes involving other regions of KRAS or NRAS are explicitly mentioned. | ||
| Fig. 1 (A) 31P NMR spectra of RAS isoforms and mutants as discussed in this paper and indicated in the Figure. (B) Structural differences between RAS state-1 and state-2 conformations, illustrated by the crystal structures of GMPPNP-bound HRAS in state-2 conformation (green, 5p21) and in state-1 conformation (white, 4efl). The green sphere represents a Mg2+ ion. The side chains of Tyr32 and Thr35 are shown. The transparent blue surface and cartoon illustrate the position of bound RBD and its steric clash with Tyr32 in state-1. | ||
It is known that the non-hydrolyzable GTP analog GMPPNP does not perfectly mimic GTP and that it affects the dynamic equilibrium of KRAS.21,56–58 However, the rank order of the fraction of KRAS in state-1 is almost conserved between GMPPNP and GTP:34 When bound to GMPPNP, it is G12V > G12D > G12C > wt, whereas when bound to GTP, it is G12V > G12D > wt > G12C. We therefore performed all 31P NMR experiments with GMPPNP complexes of KRAS, while the relaxation dispersion experiments described below were performed with the GTP complex.
Structurally, both GTP-states of RAS have been thoroughly characterized by Xray and NMR. In state-2, the active site Mg2+ is coordinated by the β- and γ-phosphates of GTP and the side chains of switch-1 residues Ser17 and Thr35, while two water molecules complete the octahedral coordination sphere. The side chain of Tyr32 is often located over the nucleotide to create a “closed” active site.28 In contrast, in state-1, Thr35 is not interacting with the Mg2+ ion and switch-1 samples a range of more “open” conformations.23,59 Characteristic side chains are Thr35 with its side chain pointing inward to coordinate Mg2+ in the active state-2, and Tyr32 which folds over the bound nucleotide in state-2, but rotates out and sterically interferes with RBD binding in the inactive state-1 (Fig. 1B). The ring current shift from Tyr32 folding over the nucleotide in state-2 may be one of the causes for the upfield shifted 31P resonance of the γ-phosphate of bound nucleotide, although other factors such as hydrogen bonding may also contribute.27
Given the clinical importance of KRAS oncogenic mutants, we set out to investigate their dynamic conformational equilibria. Fig. 1A displays 31P spectra, and Fig. 2A summarizes the populations of state-1 and state-2 in KRAS and selected mutants. Of particular interest are the oncogenic gain-of-function mutants G12D, G12C and G12V which are currently prime targets for a new generation of cancer therapeutics.7 Interestingly, those oncogenic mutations do not exert drastic effects on the switch-1 conformational equilibrium. Instead, they only have mild effects on the equilibrium, and surprisingly, they shift the equilibrium slightly towards a more inactive state, consistent with recent findings by Sharma et al.34 For example, KRAS G12D, the most prevalent oncogenic KRAS mutation and a prime target for drug discovery and development, re-distributes the populations of state-1 and state-2 from 30/70 observed for wt KRAS to 50/50. The oncogenic KRAS G12V mutant behaves similarly to G12D, and tilts the equilibrium from 30/70 to 55/45 towards the inactive state-1, whereas another oncogenic mutant, G12C, has little effect on the conformational equilibrium and retains the populations of state-1 and state-2 similar to wildtype KRAS (Fig. 1A and 2A). The slight re-distribution of populations towards the inactive state-1 may seem counter-intuitive given the oncogenic potential of these KRAS mutants, and it illustrates that oncogenicity is a multifactorial parameter in which other factors, including the nucleotide loading state and the cellular localization are dominant over the conformational equilibrium in the GTP-state.
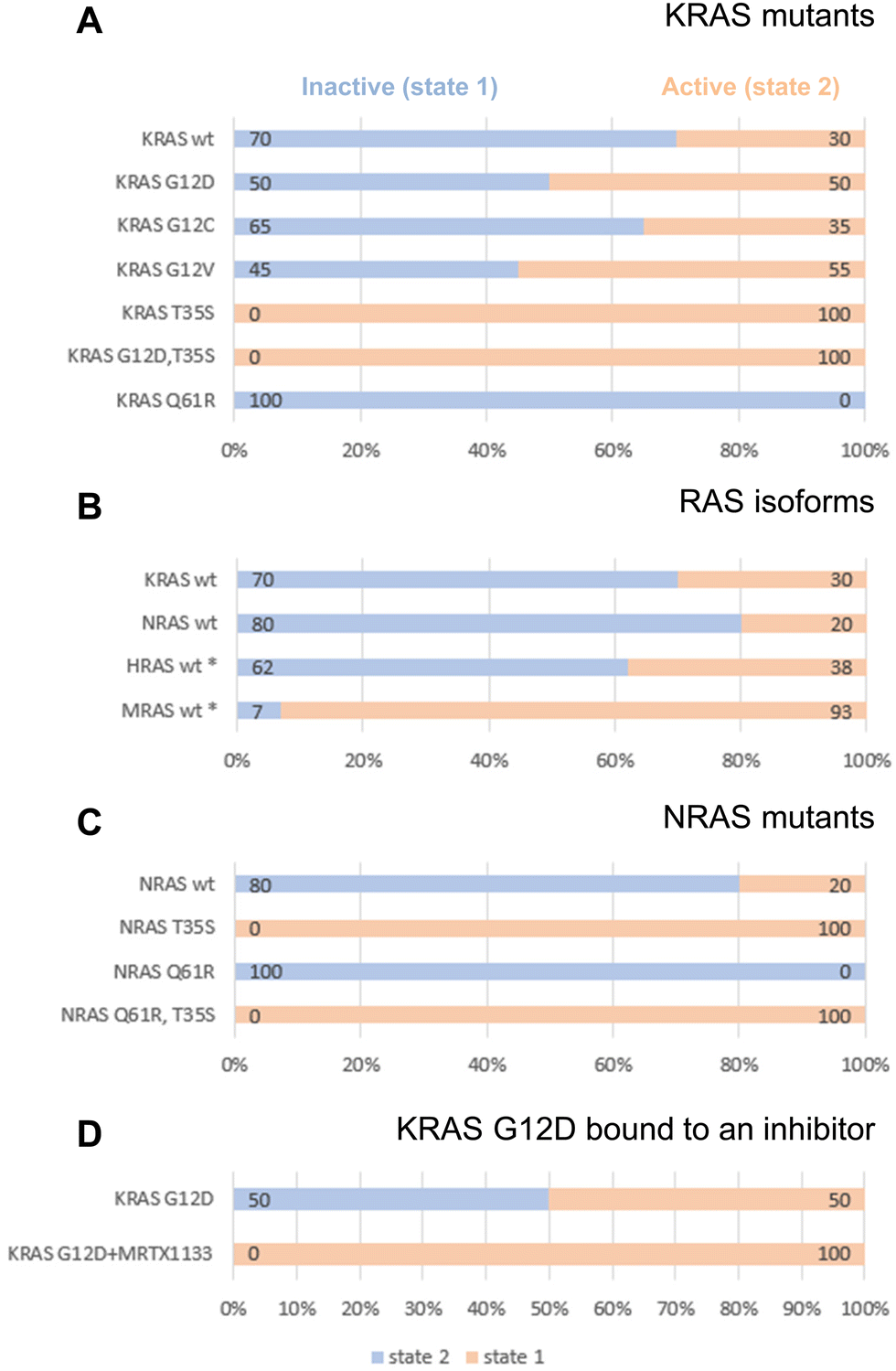 | ||
| Fig. 2 Populations of state-1 (blue) and state-2 (peach) as determined by 31P NMR in (A) KRAS mutants, (B) RAS isoforms, (C) NRAS mutants, (D) KRAS G12D complexed with an inhibitor. All corresponding 31P spectra are shown in Fig. 1A. All RAS proteins were bound to GMPPNP and measurements were carried out at 7 °C. The asterisk (*) refers to previously published data on HRAS22 and MRAS.25,29 | ||
The KRAS T35S mutant is a mechanistic mutant. In contrast to the oncogenic mutants discussed above, the T35S mutant is not found in patients but was introduced into KRAS to better understand protein behavior and dynamics. T35 coordinates the Mg2+ ion in GMPPNP-bound KRAS, and this interaction stabilizes switch-1 in the closed or active state. When T35 is mutated to Alanine or even to Serine, this interaction gets weakened, thus reducing the population of active state-2 in HRAS.20 Our experiments show that also in KRAS, the T35S mutation (corresponding to the loss of a single methyl group) leads to a complete shift of the dynamic equilibrium towards the inactive state-1. Furthermore, a double mutant combining the oncogenic mutation G12D with the mechanistic mutation T35S shows that switch-1 in this KRAS double mutant is in the fully inactive state-1 (Fig. 1A).
Other RAS isoforms play important roles in cancer as well. In particular, NRAS and associated codon 61 mutations play an important role in melanoma, but little is known about conformational equilibria in NRAS. We therefore investigated NRAS and its mutants by 31P NMR.
GMPPNP-bound wildtype NRAS populates the active state-2 by 80% and the inactive state-1 by only 20% (Fig. 1A and 2B). Thus, there is a slight shift from 70% to 80% active conformation going from KRAS to NRAS. Taken together with previously published 31P NMR data, it becomes apparent that the three classical RAS isoforms (KRAS, HRAS, NRAS) behave rather similarly with 60–80% of the GTP-state RAS being in an active state-2 conformation. In contrast, MRAS, a related GTPase, behaves markedly differently and populates predominantly the inactive state-1 conformation (Fig. 2B).
Interestingly, the oncogenic NRAS mutation Q61R leads to a complete shift towards the active state-2. This mutation thus behaves drastically differently from the oncogenic mutations at codon 12.60,61 In order to confirm this result, we also prepared the Q61R mutant of KRAS, and saw the same behavior (Fig. 1A and 2A). This observation demonstrates the stabilization of state-2 by the Q61R mutation. This is surprising but not completely unexpected, given the fact that in both available crystal structures of KRAS Q61R (6xgu)62 and NRAS Q61R (6ziz),63 the side chain of Arg61 points towards switch-1 and fills an empty space surrounded by Pro34, Ile36 and Tyr64. Both of these crystal structures show KRAS and NRAS in state-2, consistent with the 31P NMR observations. This is in stark contrast to the Q61H mutant that was characterized by 31P NMR by Sharma et al.34 and was found to populate state-1 to a higher degree than wildtype KRAS, both in its GMPPNP- and GTP-bound forms. The Arg side chain is obviously critical in stabilizing switch-1 in a closed state, and this could be rationalized by molecular dynamics simulations (see below). Of note, Q61 is a catalytic residue of the GTPase function of RAS proteins. Although the Q61R mutant adopts a higher population of state-2, it cannot hydrolyze GTP due to the absence of the Q61 side chain. Furthermore, we combined the mechanistic mutant T35S, which shifts the equilibrium fully to the inactive state-1 with the oncogenic mutant Q61R, which shifts the equilibrium fully to the active state-2. Intriguingly, in this NRAS T35S,Q61R double mutant we saw the equilibrium fully in the inactive state-1, like in the T35S single mutant. This result shows that the T35S mutation dominates over the Q61R mutation, and that the loss of a single methyl group has a stronger effect on the conformational dynamics than the introduction of a positively charged bulky residue in place of a Glutamine (Fig. 1A and 2C).
Conformational exchange in GTP-bound KRAS_Q61R detected by relaxation dispersion NMR
Motivated by the surprising finding based on 31P NMR studies of the Q61R mutant of KRAS in the GMPPNP-bound form, showing a complete shift to state-2, we studied a number of GTP-bound KRAS variants (WT, T35S and Q61R) by recording first 15N,1H HSQC spectra (Fig. 3A) and subsequently 15N Carr–Purcell–Meiboom–Gill relaxation dispersion experiments (Fig. 3B). CPMG experiments are especially sensitive for detecting low populations of sparsely populated protein conformers.64,65 For WT GTP-bound KRAS, amide signals from switch-1 and switch-2, as well as from other regions in the protein, produced ‘non-flat’ relaxation dispersion profiles (blue data points in Fig. 3B), that are a hallmark of conformational exchange, in this case reflecting the switch-1 interconversion between state-1 and state-2, as previously reported.30,31 In the WT protein, state-2 is the highest populated state (ground state), whereas state-1 is a low-populated ‘excited’ state. Fits of the relaxation dispersion experiments recorded on the WT KRAS protein to a two-site exchange model yield an exchange rate of 414 ± 3 s−1 and a state-1 population of 9.6 ± 0.1%, slightly higher than the 6.1% population detected by 31P NMR experiments.30 | ||
| Fig. 3 NMR data for 15N labeled WT (blue), T35S (green) and Q61R (red) KRAS bound to GTP. (A) Overlay of the glycine spectral region from HSQC spectra, 25 °C. The ‘×’ sign indicates the position of G12 and G13 for Q61R, indicating to a good approximation the positions of the cross-peaks for these residues in ‘pure’ state-2, while the ‘+’ sign indicates the expected cross-peak positions of the ‘pure’ state-1 using chemical shift differences calculated from fits of 1HN and 15N CPMG data for the WT protein. (B) 15N CPMG profiles for five selected residues. Circles indicate experimental data, while lines indicate fits. (C) Position of the two mutated residues (T35 and Q61), of the five 15N probes shown in panel (B), and of GMPPNP·Mg. | ||
The CPMG profiles recorded for the Q61R mutant (red data points in Fig. 3B) are much smaller (more ‘flat’) than the corresponding curves recorded for the WT protein (blue), indicating on a qualitative level, a change in the overall dynamics. Fits of the data establish a reduction of the state-1 population to a value of 1.8% (from 9.6% for WT). That the same exchange process is observed in WT and Q61R is confirmed by the fact that the values of chemical shift changes between ground and excited states are very similar between the two KRAS variants. The effect of the Q61R mutant, therefore, is simply to shift the switch-1 equilibrium towards state-2.
Given the very low population of state-1 in Q61R KRAS, the chemical shift values measured in the 15N,1H HSQC spectra of Fig. 3A are a good approximation of shifts for a ‘pure’ state-2; these ‘pure’ shifts are marked ‘×’ for G12 and G13 within the P-loop region of the protein in Fig. 3A. Using the fitted values of chemical shift changes for 1H and 15N atoms30 obtained from analysis of dispersion profiles recorded on the WT protein, the expected positions of crosspeaks for a pure state-1 conformer can be calculated, indicated by the ‘+’ symbols in the figure. The predicted positions of the resonances for both G12 and G13 of state-1 are highly correlated with peak positions measured for the T35S mutant (compare + with green peaks in Fig. 3A), confirming the previous observation that the T35S mutation results in a protein that mostly populates the open, inactive state-1 conformation.23 CPMG experiments recorded on the T35S mutant indicate the presence of an additional dynamic process within the switches which seems unrelated to the state-1/state-2 equilibrium observed for WT and Q61R KRAS. The kinetics are faster (∼1700 s−1) for this second interconversion and the fitted chemical shift changes for both G12 and G13 are unrelated to those of the state-1/state-2 equilibrium. In order to confirm that the poor agreement of chemical shift changes is not the result of the mutation at position 35, we have also analyzed dispersion profiles from residues D54 and T74 that are farther away from the mutation point (>15 Å between T35 Cγ2 and the backbone nitrogen atom of either D54 and T74). Again, the extracted shift changes between the interconverting sites are not compatible with those from state-1/state-2 exchange, indicating the presence of an additional dynamic process within the poorly structured switch regions. This process indicates that regions beyond switch-1 and the beginning of switch-2 do not have a well-defined conformation in state-1, but an equilibrium between multiple conformations.
Molecular dynamics simulations
In order to further investigate the surprising observations with the oncogenic G12D and Q61R mutants, we performed molecular dynamics (MD) simulations on these mutants and compared them with wildtype KRAS and NRAS, respectively. To this end, we started from wt structures for KRAS and NRAS in state-2 (6god38 and 5uhv,39 respectively) and computationally created the KRAS G12D and NRAS Q61R mutations, to end up with 4 structures that were subjected to molecular dynamics simulations (10 replicate runs each of 1 μs). Fig. 4 shows a heatmap for each of the 4 proteins, separated by the mobility (shown as carbon-alpha RMSD to the starting structure) of switch-1 and switch-2. Since the starting structures were active state-2 structures, structures with RMSDs up to 1.3 Å after the MD simulations were considered active state-2 structures, whereas structures with RMSDs much larger than 1.3 Å were considered inactive state-1 structures. | ||
| Fig. 4 Molecular dynamics (MD) simulations of KRAS wt (A), KRAS G12D (B), NRAS wt (C) and NRAS Q61R (D). The mobilities, seen as carbon-alpha RMSDs relative to the state-2 starting structures, of switch-1 and switch-2 are plotted on the horizontal and vertical axis, respectively. The typical RMSD of 1.3 Å after minimization of starting X-ray structures is shown as a cyan marker. | ||
Fig. 4A and C show that KRAS wt and NRAS wt behave rather similarly in these MD simulations, both sampling similar regions in conformational space. The sampled regions include conformational space near the active state (small RMSD) and away from the active state (large RMSD), reflecting the experimental evidence of a dynamic equilibrium between state-1 and state-2. KRAS G12D samples significantly more conformational space compared to KRAS wt (Fig. 4B), in line with previous computational results66,67 and our experimental data showing an increased propensity of KRAS G12D to adopt the inactive state-1 in which switch-1 is in an open and more flexible conformation. In contrast, for NRAS Q61R, our simulations show that this mutant preferentially samples conformations which are much more confined to the active state-2, where switch-1 is kept in a closed state, again correlating well with our 31P NMR observation of a shift to the active state-2. Importantly, the MD simulations offer an explanation why the NRAS Q61R mutant only samples conformations near the active state-2: with a frequency of about 33%, this mutant forms a hydrogen bond between its Arg61 side chain and the backbone carbonyl oxygen of Thr35 (see ESI† Fig. S1). The formation of this hydrogen bond exclusively in the Q61R mutant probably causes the stabilization of switch-1 in the closed conformation to adopt the active state-2.
Additional analysis and characterization of the KRas WT, KRas G12D, NRas WT and NRas Q61R systems based on the molecular dynamics simulations are shown in the ESI† (Fig. S2–S5).
Hydrogen/deuterium exchange mass spectrometry
In order to gain further insights into the dynamics and flexibility of KRAS and NRAS mutants, we carried out hydrogen/deuterium exchange measurements by mass spectrometry (HDX-MS).68,69 This method can be used to compare two similar proteins (wildtype versus mutant, mutant versus mutant, or apo versus bound, etc.) by the calculation of differential incorporation of deuterons (2H) at exchangeable backbone amide proton (1H) positions after labeling for a measured amount of time with D2O. The spatial resolution is typically stretches of a few to several amino acid residues, depending on the peptides that are produced from the proteolytic cleavage of the analyte protein under quench conditions.In HDX-MS, we compared several of the mechanistic and oncogenic mutants from the 31P NMR study, in order to investigate their dynamic behavior by an independent yet complementary method. Starting with the mechanistic mutant NRAS T35S, which was found by 31P NMR to be fully in the inactive state-1, and comparing to NRAS wt, all bound to GMPPNP, we see by HDX-MS clear, localized increases in deuterium incorporation, which we attribute to increased protein flexibility and backbone de-protection, especially in parts of α1 and the P-loop, and the lower part of the nucleotide pocket which is spanned by the loop between β5 and α4 as well as the first residues of α5 (Fig. 5 and supplemental HDX data file, ESI†). Insofar as the nucleotide pocket is concerned, this observed de-protection could also be interpreted as a potentially weaker affinity of GMPPNP to the T35S mutant protein, leading to less protection from deuterium incorporation for that region of the protein compared to wildtype. There are no comparative HDX data on differences in the switch-1 (amino acids 23–40) region for the T35S or T35S/Q61R double mutants because of the sequence differences between wildtype and the T35S mutation. The same applies for peptides that contain residue 61 for both the Q61R and T35S/Q61R constructs.
 | ||
| Fig. 5 Hydrogen–deuterium exchange mass spectrometry (HDX-MS). (A) Chiclet plots for NRAS T35S, Q61 and T35S/Q61R mutant proteins bound to GMPPNP, with NRAS wt GMPPNP taken as baseline. Numerical column values are time points given in minutes. White background indicates lacking data for the corresponding peptide comparisons due to mutation. Regions discussed in the text are labeled accordingly. (B) Coloring scale for chiclet plots: green = deprotection, blue = protection. (C) NRAS Q61R vs. wt comparison from (a) structurally mapped onto the crystal structure of KRAS Q61 GMPPNP, PBD 6XGU. Arg61 is shown in magenta carbons. (D) Chiclet plot for KRAS G12D GMPPNP vs. KRAS wt GMPPNP. All values used to make these graphs are found in the supplementary HDX data file (ESI†). | ||
In contrast to T35S, the oncogenic Q61R mutant, seen in 31P NMR to fully adopt the active “state-2”, was shown to have regions with decreased deuterium incorporation for the mutant NRAS compared to the wild-type in similar regions of the protein: However, it leads to protection from deuterium incorporation in the P-loop region and nucleotide pocket compared to NRAS wt, albeit only from the 60-minute time point onwards. At the earlier timepoints, mutation leads to protection from deuterium incorporation of switch-1 and also switch-2 as observed for peptides covering the backbone outside of position 61, e.g. 69–79. The protection of switch-1 observed in the HDX experiment is intriguing as it indicates a stabilizing interaction of Arg61 (located on switch-2) with partner residues on switch-1, including the backbone carbonyl in Thr35 as previously observed in our molecular dynamics simulations and in the cocrystal structure of KRAS Q61R GMPPNP in complex with RAF1 RBD-CRD (6XGU).62 In line with the observations by NMR, the T35S mutation appears to override the effects to backbone dynamics of the Q61R mutation in the T35S/Q61R double mutant, and leads to a de-protection of NRAS to a similar level as seen with the single T35S mutation. In light of the putative direct interaction of Arg61 with the Thr35 backbone carbonyl, this makes sense as Ser35 is not expected to retain the same relative position in the more flexible switch-1 that it induces.
For these mutants, the conclusions by the two independent methods, 31P NMR and HDX-MS, are much in line: Mutations that lead to a higher population of the inactive state-1 (such as T35S) are associated with localized increased backbone flexibility and subsequent increased deuterium incorporation, whereas mutations that lead to a higher population of the active state-2 (such as Q61R) are associated with localized backbone rigidification, as evidenced by protection from deuterium incorporation. We attribute the change from wildtype backbone dynamics to a fixated or released switch-1 which induces (de)protection of switch-2 and the P-loop and, as secondary effect, reduces or increases nucleotide affinity.
When comparing by HDX-MS the KRAS G12D mutant with the wildtype protein, we see increased flexibility in the G12D mutant around α1 and P-loop. Again, this is in line with the observations from 31P NMR, although the extent to which these regions saw increases in deuterium incorporation in HDX-MS was surprisingly large, given the relatively small shift seen in 31P NMR from active state-1 to inactive state-2 (70/30 in wildtype to 50/50 in G12D).
Elucidation of the mechanism of action of a KRAS G12D inhibitor
An understanding of the conformational dynamics of KRAS, NRAS and its oncogenic mutants is helpful to characterize or design RAS inhibitors as novel anti-tumor agents. Several KRAS inhibitors have recently been disclosed. Covalent KRAS G12C inhibitors, such as sotorasib,11 adagrasib10 and JDQ443,12,13 are on the market or in clinical trials. These covalent inhibitors have a strong preference for the GDP state as their acrylamide moieties occupy the same position as the terminal phosphate in GTP would. They are thus not amenable for 31P NMR analysis, since the gamma-phosphate which is most sensitive to the switch 1 dynamics is not present in GDP. An exception is the covalent KRAS G12C inhibitor BBO-8956, which was recently disclosed by Sharma et al.34 This inhibitor binds the GTP and GDP states of KRAS G12C with almost equal affinity and was thus investigated by 31P NMR. While the authors claim stabilization of state-1 or a state-1-like state (that can however bind Raf1 RBD) by BBO-8956, the 31P NMR spectra are complex and allow several interpretations.Recently, Mirati Therapeutics have disclosed their preclinical KRAS G12D inhibitor, MRTX1133.14 Like the G12C inhibitors, this compound binds to the switch-2 pocket of KRAS. While the covalent G12C inhibitors are thought to work by sequestering KRAS in the GDP-bound state and preventing its exchange to GTP, the mechanism of action of MRTX1133 is less clear. Due to the higher binding affinity of MRTX1133 to GDP-bound KRAS, it probably works like the G12C inhibitors and stabilizes KRAS in the GDP-bound form. However, an additional mechanism of action might be that MRTX1133 prevents the protein–protein interactions of GTP-bound KRAS with effectors by an allosteric mechanism. Note that the binding sites for MRTX1133 and the RAS-binding domain (RBD) or cysteine-rich domain (CRD) interaction domains of effector proteins are non-overlapping and there would be no steric clash between the two ligands even if both were simultaneously bound.
MRTX1133 also binds to GMPPNP-bound KRAS, albeit weaker than to GDP-bound KRAS. Therefore, we could investigate by 31P NMR its impact on KRAS G12D conformational dynamics. Fig. 6A shows the 31P NMR spectra of GMPPNP-bound KRAS G12D in absence (top) and presence (bottom) of MRTX1133. Clearly, MRTX1133 brings the dynamic equilibrium back to one single state, but whether this state is the inactive state-1 or the active state-2 is not clear from this experiment alone, due to small chemical shift changes of the 31P peaks. We therefore performed 1H,13C-HMQC experiments to see whether RAF1-RBD is able to bind GMPPNP-loaded KRAS G12D when bound to MRTX1133. Fig. 6B shows that RBD binds to KRAS G12D (chemical shift changes between blue and black spectrum) and that MRTX1133 binds to KRAS G12D (chemical shift changes between green and black spectrum). Adding both RBD and MRTX1133 to KRAS G12D could result in two outcomes: The ternary mixture could be identical to either of the two binary mixtures, indicating displacement of RBD or MRTX1133 and the formation of the stronger complex. Or the ternary mixture could result in yet another spectrum, which would indicate simultaneous binding of RBD and MRTX1133 and the formation of a ternary complex. Fig. 6B shows that the 1H,13C-HMQC of KRAS G12D in presence of both RBD and MRTX1133 (red spectrum) is almost identical to the 1H,13C-HMQC of KRAS G12D in presence of only MRTX1133 (green spectrum). This indicates that in the presence of both ligands, only MRTX1133 is bound and prevents binding of RBD, meaning that MRTX1133 binds with higher affinity and displaces RBD. This shows that the single state seen in 31P experiments (Fig. 6A) with MRTX1133 is indeed not the active state-2, and that RBD binding is prevented by MRTX1133. Based on a recent publication by Kim et al.,70 who detected weak binding of RBD to the KRAS:MRTX1133 complex by biolayer interferometry (BLI), we tested higher concentrations of RBD and saw quenching of KRAS signals at 100 μM RBD. This indicates weak interactions at these high concentrations, which we did not further characterize, since they are probably not relevant under physiological conditions.
 | ||
| Fig. 6 Characterization of MRTX1133 binding to KRAS G12D. (A) 31P NMR spectra of KRAS G12D:GMPPNP (top) and KRAS G12D:GMPPNP in complex with MRTX1133 (bottom). (B) 1H,13C-HMQC NMR spectra of GMPPNP-bound KRAS G12D (black), KRAS G12D + RBD (blue), KRAS G12D + MRTX1133 (green) and KRAS G12D + RBD + MRTX1133 (red). The concentrations were: KRAS G12D, 20 μM; MRTX1133, 40 μM; RBD, 60 μM. Note that RBD does not bind to the KRAS G12D/MRTX1133 complex at these concentrations. (C) Structural details on the allosteric relay of MRTX1133 binding from the switch-2 pocket to switch-1. Without MRTX1133 bound (PDB: 6QUU), the switch-2 loop folds such that Ala59's β-methyl points upwards. MRTX1133 binding leads to opening up of switch-2, formation of Asp12 and Gly60 ligand contacts, and rotation of Ala59 β-methyl such that switch-1 is disrupted with Thr35 rotating towards solvent, leading to an open conformation (PDB: 7T47). (D) HDX-MS chiclet plot for KRAS G12D:GMPPNP bound to compound 25 vs. KRAS G12D:GMPPNP alone. All values used to make these graphs are found in the supplementary HDX data file (ESI†). The coloring scheme is the same as in Fig. 4. Numerical column values are time points given in minutes. Compound 25 leads to strong protection across the protein. (E) HDX data structurally mapped onto the crystal structure of KRAS G12D GPPCP bound to MRTX1133 (PDB 7T47). D12 is shown in blue carbons. | ||
The displacement of RBD proves an additional mechanism of action of MRTX1133 by allosterically interfering with effector binding. Fig. 6C shows the molecular mechanism of this allostery: Binding of MRTX1133 in the switch-2 pocket substantially remodels switch-2 and pushes out the loop at the start of switch-2 (Ala59 and Gly60) in order to enable the formation of a salt bridge interaction between the rigidified piperazine and Asp12 in addition to a charge-assisted hydrogen bond to the Gly60 backbone carbonyl. This displaced loop, specifically the displaced Ala59 through its β-methyl group, in turn then pushes Thr35 and switch-1 from its closed, active conformation (state-2) to an open, inactive conformation (state-1). With this open, inactive conformation, RBD cannot bind to the KRAS G12D: MRTX1133 complex. Ala59 can be thus considered an allosteric node that signals between the switch-2 pocket and switch-1 and relays information between the two binding sites. It is intriguing to note that the allosteric network comprises Thr35 which has been shown by 31P NMR previously and in this work to be an essential residue for KRAS switch-1 dynamics. Taken together, MRTX is an allosteric modulator which binds to the switch-2 pocket and induces conformational changes that prevent binding of RBD.
We then investigated a close structural analog of MRTX1133, compound 25 recently described by Wang et al.14 by HDX-MS. As can be gleaned from Fig. 6D and E (and supplemental HDX data file, ESI†), when compared with the unbound protein, compound 25 induces significant protection from deuterium incorporation across a majority of the protein. Not surprisingly, we observed the greatest decreases in deuterium incorporation in regions that are in direct contact with the ligand, e.g. on peptides from helix α3 containing key residues for binding of that chemotype such as His95 and Tyr96, and on the P-loop containing the mutant residue Asp12 that is involved in a strong salt-bridge type interaction with the bridged piperazine that is conserved in Compound 25 and MRTX1133. In addition, the compound stabilizes switch-2, albeit to a somewhat lesser extent than α3 which is likely due to the very flexible nature of this loop. Interestingly, we do not observe an effect on switch-1 but intriguingly, as already observed for Q61R, the compound seems to have a stabilizing effect on parts of the nucleotide pocket, as observed through the increased protection for sequence stretch 115–133/134. This could be due to the doubly positive charge of the Mirati chemotype changing the electrostatic environment in the nucleotide pocket, making it more conducive to binding of the triple negatively charged GMPPNP (and presumably GTP).
Discussion and conclusions
Using multiple biophysical and computational techniques, we have characterized clinically relevant mutant forms of KRAS and NRAS with respect to their conformational equilibria in the GTP-state, and we have interpreted these equilibria on a structural basis. We find by 31P NMR that some mutations, such as G12D, G12C or G12V, have subtle but significant impact on the conformation of switch-1, and surprisingly tend to shift the populations towards an inactive state. Other mutations, such as Q61R, exert strong effects on this conformational equilibrium and fully shift it towards the active state. We corroborate these findings by three independent methods, NMR relaxation dispersion experiments, molecular dynamics simulations and HDX-MS which show that the inactive state-1 is associated with localized protein flexibility, whereas the active “state-2” is associated with reduced flexibility.Our studies have further elucidated an additional mechanism of action of the clinical KRAS G12D inhibitor MRTX1133. By binding tightly to the GDP-state of KRAS G12D, this inhibitor is thought to stabilize the GDP-bound state of KRAS and prevent its exchange to the GTP-bound state. However, MRTX1133 also binds KRAS G12D in its GTP state, and our experiments show that it allosterically modulates the conformation of switch-1 and shifts it to the fully inactive state, thereby preventing effector binding. Similar observations have previously been made for Zn-cyclen71 and for cmpd 2,72,73 a weak KRAS inhibitor.
Our results are generally in agreement with the recently published analysis of Sharma et al.34 which reported conformational equilibria of KRAS and its oncogenic mutants, both bound to GTP and GMPPNP. Our studies mainly investigate GMPPNP-bound KRAS, but add complementary information from multiple biophysical and computational techniques, such as NMR relaxation dispersion, HDX-MS, and molecular dynamics simulations, to give a holistic view of conformational states and dynamics in KRAS and its oncogenic mutants. Of particular interest is the elucidation of the mechanism of action (MoA) of MRTX1133, a clinical KRAS G12D inhibitor. The 31P NMR data, combined with 1H–13C-HMQC binding studies with effector proteins and crystal structures, unambiguously show that MRTX1133 de-populates the active state of KRAS G12D and results in conformational changes that prevent binding of effector proteins at physiological concentrations. In contrast, the recent MoA characterization of BBO-8956, an early-stage covalent KRAS G12C inhibitor, leads to less clear conclusions since RAF1-RBD can still bind to the KRAS G12C-BBO-8956 complex, and 31P NMR spectra are complex and allow several interpretations.
Our data on KRAS and NRAS dynamics, together with recently published data from Sharma et al.,34 extend our dynamical understanding of RAS family proteins to the therapeutically relevant KRAS and NRAS isoforms. They help to clarify some of the open questions in the literature, such as whether the KRAS G12D mutation stabilizes the active state-2 of KRAS.74 Taken together, our results complement the ongoing attempts to provide a better understanding of KRAS structure and dynamics, which ultimately will facilitate the development of more safe and efficient drugs.
Author contributions
Conceptualization and supervision: K. S. B., R. W., W. J., L. E. K. and J. R. E.; data curation: T. E. W.; investigation: E. R., C. H., C. J. D. and F. G.; methodology: D. E.; writing: W. J., with help and contributions from all co-authors.Data availability
The data supporting this article have been included as part of the ESI.† All HDX methods used and data generated or analyzed for this study are included in this manuscript and its ESI.† HDX MS source data have been deposited to the ProteomeXchange Consortium via the PRIDE75 partner repository with dataset identifier PXD054924.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Catherine Zimmermann, Marco Meyerhofer, Patrizia Fontana, Wassim Abdul Rahman, Julia Klopp, Sebastien Rieffel and Aurélie Winterhalter for their excellent protein expression and purification support. We also thank Mitsu Ikura and Ha-Neul Kim for very helpful discussions.References
- D. K. Simanshu, D. V. Nissley and F. McCormick, Cell, 2017, 170, 17–33 CrossRef PubMed.
- I. A. Prior, F. E. Hood and J. L. Hartley, Cancer Res., 2020, 80, 2969–2974 CrossRef PubMed.
- A. M. Waters and C. J. Der, Cold Spring Harbor Perspect. Med., 2018, 8, a031435 CrossRef PubMed.
- I. A. Prior, P. D. Lewis and C. Mattos, Cancer Res., 2012, 72, 2457–2467 CrossRef PubMed.
- I. R. Vetter and A. Wittinghofer, Science, 2001, 294, 1299–1304 CrossRef PubMed.
- J. L. Bos, Cancer Res., 1989, 49, 4682–4689 CAS.
- A. D. Cox, S. W. Fesik, A. C. Kimmelman, J. Luo and C. J. Der, Nat. Rev. Drug Discovery, 2014, 13, 828–851 CrossRef CAS PubMed.
- C. B. Marshall, F. KleinJan, T. Gebregiworgis, K. Y. Lee, Z. H. Fang, B. J. Eves, N. D. F. Liu, G. M. C. Gasmi-Seabrook, M. Enomoto and M. Ikura, J. Biomol. NMR, 2020, 74, 531–554 CrossRef CAS PubMed.
- T. Maurer, L. S. Garrenton, A. Oha, K. Pitts, D. J. Anderson, N. J. Skelton, B. P. Fauber, B. Pan, S. Malek, D. Stokoe, M. J. C. Ludlam, K. K. Bowman, J. S. Wu, A. M. Giannetti, M. A. Starovasnik, I. Mellman, P. K. Jackson, J. Rudolph, W. R. Wang and G. Fang, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5299–5304 CrossRef CAS PubMed.
- J. B. Fell, J. P. Fischer, B. R. Baer, J. F. Blake, K. Bouhana, D. M. Briere, K. D. Brown, L. E. Burgess, A. C. Burns, M. R. Burkard, H. Chiang, M. J. Chicarelli, A. W. Cook, J. J. Gaudino, J. Hallin, L. Hanson, D. P. Hartley, E. J. Hicken, G. P. Hingorani, R. J. Hinklin, M. J. Mejia, P. Olson, J. N. Otten, S. P. Rhodes, M. E. Rodriguez, P. Savechenkov, D. J. Smith, N. Sudhakar, F. X. Sullivan, T. P. Tang, G. P. Vigers, L. Wollenberg, J. G. Christensen and M. A. Marx, J. Med. Chem., 2020, 63, 6679–6693 CrossRef CAS PubMed.
- B. A. Lanman, J. R. Allen, J. G. Allen, A. K. Amegadzie, K. S. Ashton, S. K. Booker, J. J. Chen, N. Chen, M. J. Frohn, G. Goodman, D. J. Kopecky, L. Liu, P. Lopez, J. D. Low, V. Ma, A. E. Minatti, T. T. Nguyen, N. Nishimura, A. J. Pickrell, A. B. Reed, Y. Shin, A. C. Siegmund, N. A. Tamayo, C. M. Tegley, M. C. Walton, H. L. Wang, R. P. Wurz, M. Xue, K. C. Yang, P. Achanta, M. D. Bartberger, J. Canon, L. S. Hollis, J. D. McCarter, C. Mohr, K. Rex, A. Y. Saiki, T. San Miguel, L. P. Volak, K. H. Wang, D. A. Whittington, S. G. Zech, J. R. Lipford and V. J. Cee, J. Med. Chem., 2020, 63, 52–65 CrossRef CAS PubMed.
- E. Lorthiois, M. Gerspacher, K. S. Beyer, A. Vaupel, C. Leblanc, R. Stringer, A. Weiss, R. Wilcken, D. A. Guthy, A. Lingel, C. Bomio-Confaglia, R. Machauer, P. Rigollier, J. Ottl, D. Arz, P. Bernet, G. Desjonqueres, S. Dussauge, M. Kazic-Legueux, M. A. Lozac'h, C. Mura, M. Sorge, M. Todorov, N. Warin, F. Zink, H. Voshol, F. J. Zecri, R. C. Sedrani, N. Ostermann, S. M. Brachmann and S. Cotesta, J. Med. Chem., 2022, 65, 16173–16203 CrossRef CAS PubMed.
- A. Weiss, E. Lorthiois, L. Barys, K. S. Beyer, C. Bomio-Confaglia, H. Burks, X. Y. Chen, X. M. Cui, R. de Kanter, L. Dharmarajan, C. Fedele, M. Gerspacher, D. A. Guthy, V. Head, A. Jaeger, E. J. Núñez, J. D. Kearns, C. Leblanc, S. M. Maira, J. Murphy, H. Oakman, N. Ostermann, J. Ottl, P. Rigollier, D. Roman, C. Schnell, R. Sedrani, T. Shimizu, R. Stringer, A. Vaupel, H. Voshol, P. Wessels, T. Widmer, R. Wilcken, K. Xu, F. Zecri, A. F. Farago, S. Cotesta and S. M. Brachmann, Cancer Discovery, 2022, 12, 1500–1517 CrossRef PubMed.
- X. L. Wang, S. Allen, J. F. Blake, V. Bowcut, D. M. Briere, A. Calinisan, J. R. Dahlke, J. B. Fell, J. P. Fischer, R. J. Gunn, J. Hallin, J. Laguer, J. D. Lawson, J. Medwid, B. Newhouse, P. Nguyen, J. M. O’Leary, P. Olson, S. Pajk, L. Rahbaek, M. Rodriguez, C. R. Smith, T. P. Tang, N. C. Thomas, D. Vanderpool, G. P. Vigers, J. G. Christensen and M. A. Marx, J. Med. Chem., 2022, 65, 3123–3133 CrossRef CAS PubMed.
- C. J. Schulze, K. J. Seamon, Y. Zhao, Y. C. Yang, J. Cregg, D. Kim, A. Tomlinson, T. J. Choy, Z. Wang, B. Sang, Y. Pourfarjam, J. Lucas, A. Cuevas-Navarro, C. Ayala-Santos, A. Vides, C. Li, A. Marquez, M. Zhong, V. Vemulapalli, C. Weller, A. Gould, D. M. Whalen, A. Salvador, A. Milin, M. Saldajeno-Concar, N. Dinglasan, A. Chen, J. Evans, J. E. Knox, E. S. Koltun, M. Singh, R. Nichols, D. Wildes, A. L. Gill, J. A. M. Smith and P. Lito, Science, 2023, 381, 794–799 CrossRef CAS.
- Y. Lee, R. Lazim, S. J. Y. Macalino and S. Choi, Curr. Opin. Struct. Biol., 2019, 55, 147–153 CrossRef.
- T. Pantsar, Comput. Struct. Biotechnol. J., 2020, 18, 189–198 CrossRef PubMed.
- Throughout the text, the term “GTP-state” Is used to uefer to a state where GTP or a GTP analog (GMPPNP, GppCp or GTPgS) Is bound to RAS. In contrast, the terms “GTP-Bound” or “GMPPNP-Bound” refer to this particular nucleotide analog bound to RAS.
- M. Geyer, T. Schweins, C. Herrmann, T. Prisner, A. Wittinghofer and H. R. Kalbitzer, Biochemistry, 1996, 35, 10308–10320 CrossRef PubMed.
- M. Spoerner, C. Herrmann, I. R. Vetter, H. R. Kalbitzer and A. Wittinghofer, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 4944–4949 CrossRef PubMed.
- M. Spoerner, C. Hozsa, J. A. Poetzl, K. Reiss, P. Ganser, M. Geyer and H. R. Kalbitzer, J. Biol. Chem., 2010, 285, 39768–39778 Search PubMed.
- M. Spoerner, A. Wittinghofer and H. R. Kalbitzer, FEBS Lett., 2004, 578, 305–310 CrossRef PubMed.
- M. Araki, F. Shima, Y. Yoshikawa, S. Muraoka, Y. Ijiri, Y. Nagahara, T. Shirono, T. Kataoka and A. Tamura, J. Biol. Chem., 2011, 286, 39644–39653 CrossRef PubMed.
- F. A. Chao, A. H. Chan, S. Dharmaiah, C. D. Schwieters, T. H. Tran, T. Taylor, N. Ramakrishnan, D. Esposito, D. V. Nissley, F. McCormick, D. K. Simanshu and G. Cornilescu, Commun. Biol., 2023, 6, 594 CrossRef.
- J. L. Liao, F. Shima, M. Araki, M. Ye, S. Muraoka, T. Sugimoto, M. Kawamura, N. Yamamoto, A. Tamura and T. Kataoka, Biochem. Biophys. Res. Commun., 2008, 369, 327–332 Search PubMed.
- C. O'Connor and E. L. Kovrigin, Biochemistry, 2008, 47, 10244–10246 CrossRef PubMed.
- M. I. Parker, J. E. Meyer, E. A. Golemis and R. L. Dunbrack, Cancer Res., 2022, 82, 2485–2498 CrossRef PubMed.
- F. Shima, Y. Ijiri, S. Muraoka, J. L. Liao, M. Ye, M. Araki, K. Matsumoto, N. Yamamoto, T. Sugimoto, Y. Yoshikawa, T. Kumasaka, M. Yamamoto, A. Tamura and T. Kataoka, J. Biol. Chem., 2010, 285, 22696–22705 CrossRef CAS PubMed.
- M. Ye, F. Shima, S. Muraoka, J. L. Liao, H. Okamoto, M. Yamamoto, A. Tamura, N. Yagi, T. Ueki and T. Kataoka, J. Biol. Chem., 2005, 280, 31267–31275 CrossRef CAS PubMed.
- A. L. Hansen, X. Xiang, C. Yuan, L. Bruschweiler-Li and R. Brüschweiler, Nat. Struct. Mol. Biol., 2023, 30, 1446–1455 CrossRef CAS PubMed.
- G. Pálfy, D. K. Menyhárd, H. Ákontz-Kiss, I. Vida, G. Batta, O. Tőke and A. Perczel, Chem. - Eur. J., 2022, 28, e202201449 CrossRef PubMed.
- J.-S. Hu and A. G. Redfield, Biochemistry, 1997, 36, 5045–5052 CrossRef CAS PubMed.
- D. K. Menyhárd, G. Pálfy, Z. Orgován, I. Vida, G. M. Keserű and A. Perczel, Chem. Sci., 2020, 11, 9272–9289 RSC.
- A. K. Sharma, J. Pei, Y. Yang, M. Dyba, B. Smith, D. Rabara, E. K. Larsen, F. C. Lightstone, D. Esposito, A. G. Stephen, B. Wang, P. J. Beltran, E. Wallace, D. V. Nissley, F. McCormick and A. E. Maciag, J. Biol. Chem., 2024, 300, 105650–105658 CrossRef CAS.
- D. F. Hansen, P. Vallurupalli and L. E. Kay, J. Phys. Chem. B, 2008, 112, 5898–5904 CrossRef CAS PubMed.
- B. Jiang, B. Yu, X. Zhang, M. Liu and D. Yang, J. Magn. Reson., 2015, 257, 1–7 CrossRef CAS PubMed.
- G. N. B. Yip and E. R. P. Zuiderweg, J. Magn. Reson., 2004, 171, 25–36 CrossRef CAS.
- A. Cruz-Migoni, P. Canning, C. E. Quevedo, C. J. R. Bataille, N. Bery, A. Miller, A. J. Russell, S. E. V. Phillips, S. B. Carr and T. H. Rabbitts, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 2545–2550 CrossRef CAS.
- C. W. Johnson, D. Reid, J. A. Parker, S. Salter, R. Knihtila, P. Kuzmic and C. Mattos, J. Biol. Chem., 2017, 292, 12981–12993 CrossRef CAS PubMed.
- Molecular Operating Environment (MOE), 2022.02 Chemical Computing Group ULC, Canada, 2024.
- D. A. Case, K. Belfon, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham, III, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, G. Giambasu, M. K. Gilson, H. Gohlke, A. W. Goetz,R. Harris, S. Izadi, S. A. Izmailov, K. Kasavajhala, A. Kovalenko, R. Krasny, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, V. Man, K. M. Merz, Y. Miao, O. Mikhailovskii, G. Monard, H. Nguyen, A. Onufriev, F. Pan, S. Pantano, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, N. R. Skrynnikov, J. Smith, J. Swails, R. C. Walker, J. Wang, L. Wilson, R. M. Wolf, X. Wu, Y. Xiong, Y. Xue, D. M. York and P. A. Kollman, ( 2020), AMBER 2020, University of California, San Francisco Search PubMed.
- K. L. Meagher, L. T. Redman and H. A. Carlson, J. Comput. Chem., 2003, 24, 1016–1025 CrossRef CAS PubMed.
- Z. Li, L. F. Song, P. Li and K. M. Merz, Jr., J. Chem. Theory Comput., 2020, 16, 4429–4442 CrossRef CAS PubMed.
- C. Tian, K. Kasavajhala, K. A. A. Belfon, L. Raguette, H. Huang, A. N. Migues, J. Bickel, Y. Wang, J. Pincay, Q. Wu and C. Simmerling, J. Chem. Theory Comput., 2020, 16, 528–552 CrossRef PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef.
- I. S. Joung and T. E. Cheatham, III, J. Phys. Chem. B, 2008, 112, 9020–9041 CrossRef PubMed.
- R. W. Pastor, B. R. Brooks and A. Szabo, Mol. Phys., 1988, 65, 1409–1419 CrossRef.
- R. Salomon-Ferrer, A. W. Götz, D. Poole, S. Le Grand and R. C. Walker, J. Chem. Theory Comput., 2013, 9, 3878–3888 CrossRef PubMed.
- A. W. Götz, M. J. Williamson, D. Xu, D. Poole, S. Le Grand and R. C. Walker, J. Chem. Theory Comput., 2012, 8, 1542–1555 CrossRef PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef.
- M. P. Allen and D. J. Tildesley, Computer Simulation of Liquids, Oxford University Press, Inc., 2017 Search PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef.
- C. W. Hopkins, S. Le Grand, R. C. Walker and A. E. Roitberg, J. Chem. Theory Comput., 2015, 11, 1864–1874 CrossRef PubMed.
- D. R. Roe and T. E. Cheatham, III, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef PubMed.
- J. D. Hunter, Comput. Sci. Eng., 2007, 9, 90–95 Search PubMed.
- X. Chen, H. Yao, H. Wang, Y. Mao, D. Liu and D. Long, Angew. Chem., Int. Ed., 2019, 58, 2730–2733 CrossRef PubMed.
- D. Long, C. B. Marshall, G. Bouvignies, M. T. Mazhab-Jafari, M. J. Smith, M. Ikura and L. E. Kay, Angew. Chem., – Int. Ed., 2013, 52, 10771–10774 CrossRef PubMed.
- M. T. Mazhab-Jafari, C. B. Marshall, M. Smith, G. M. C. Gasmi-Seabrook, V. Stambolic, R. Rottapel, B. G. Neel and M. Ikura, J. Biol. Chem., 2010, 285, 5132–5136 CrossRef PubMed.
- J. A. Parker and C. Mattos, Cold Spring Harbor Perspect. Med., 2018, 8, a031427 CrossRef PubMed.
- C. E. Burd, W. Liu, M. V. Huynh, M. A. Waqas, J. E. Gillahan, K. S. Clark, K. Fu, B. L. Martin, W. R. Jeck, G. P. Souroullas, D. B. Darr, D. C. Zedek, M. J. Miley, B. C. Baguley, S. L. Campbell and N. E. Sharpless, Cancer Discovery, 2014, 4, 1418–1429 CrossRef PubMed.
- B. M. Murphy, E. M. Terrell, V. R. Chirasani, T. J. Weiss, R. E. Lew, A. M. Holderbaum, A. Dhakal, V. Posada, M. Fort, M. S. Bodnar, L. M. Carey, M. Chen, C. J. Burd, V. Coppola, D. K. Morrison, S. L. Campbell and C. E. Burd, Nat. Commun., 2022, 13, 3153 CrossRef PubMed.
- T. H. Tran, A. H. Chan, L. C. Young, L. Bindu, C. Neale, S. Messing, S. Dharmaiah, T. Taylor, J. P. Denson, D. Esposito, D. V. Nissley, A. G. Stephen, F. McCormick and D. K. Simanshu, Nat. Commun., 2021, 12, 1176–1191 CrossRef PubMed.
- D. Kessler, A. Bergner, J. Böttcher, G. Fischer, S. Döbel, M. Hinkel, B. Müllauer, A. Weiss-Puxbaum and D. B. McConnell, Future Med. Chem., 2020, 12, 1911–1923 CrossRef PubMed.
- A. G. Palmer, C. D. Kroenke and J. Patrick Loria, in Methods in Enzymology, ed. T. L. James, V. Dötsch and U. Schmitz, Academic Press, 2001, vol. 339, pp. 204–238 Search PubMed.
- D. M. Korzhnev and L. E. Kay, Acc. Chem. Res., 2008, 41, 442–451 CrossRef PubMed.
- A. A. Gorfe, B. J. Grant and J. A. McCammon, Structure, 2008, 16, 885–896 CrossRef.
- S. Vatansever, B. Erman and Z. H. Gurni, Sci. Rep., 2019, 9, 11730–11742 CrossRef.
- B. A. Kochert, R. E. Iacob, T. E. Wales, A. Makriyannis and J. R. Engen, Methods Mol. Biol., 2018, 1764, 153–171 CrossRef.
- G. R. Masson, M. L. Jenkins and J. E. Burke, Expert Opin. Drug Discovery, 2017, 12, 981–994 CrossRef PubMed.
- H.-N. Kim, M. Ikura, C. B. Marshall, G. Seabrook, M. Enomoto, T. Villeneuve, W. Mo, N. F. Liu and L. Zheng. Understanding the Mechanism of Action of Novel KRAS Inhibitors. Poster at the 30th International Conference of Magnetic Resonance in Biological Systems, Seoul, South Korea, 2024.
- H. R. Kalbitzer and M. Spoerner, Enzymes, 2013, 33, 69–94 Search PubMed.
- Z. Fang, C. B. Marshall, T. Nishikawa, A. D. Gossert, J. M. Jansen, W. Jahnke and M. Ikura, Cell Chem. Biol., 2018, 25, 1327–1336 CrossRef PubMed . e1324.
- J. M. Jansen, C. Wartchow, W. Jahnke, S. S. Fong, T. Tsang, K. Pfister, T. Zavorotinskaya, D. Bussiere, J. M. Cheng, K. Crawford, Y. M. Dai, J. Dove, E. Fang, Y. Feng, J. M. Florent, J. Fuller, A. D. Gossert, M. Hekmat-Nejad, C. Henry, J. Klopp, W. P. Lenahan, A. Lingel, S. Ma, A. Meyer, Y. Mishina, J. Narberes, G. Pardee, S. Ramurthy, S. Rieffel, D. Stuart, S. Subramanian, L. Tandeske, S. Widger, A. Widmer, A. Winterhalter, I. Zaror and S. Hardy, PLoS One, 2017, 12, e0174706 CrossRef.
- J. A. Parker, A. Y. Volmar, S. Pavlopoulos and C. Mattos, Structure, 2018, 26, 810 CrossRef.
- Y. Perez-Riverol, J. Bai, C. Bandla, D. García-Seisdedos, S. Hewapathirana, S. Kamatchinathan, Deepti J. Kundu, A. Prakash, A. Frericks-Zipper, M. Eisenacher, M. Walzer, S. Wang, A. Brazma and Juan A. Vizcaíno, Nucleic Acids Res., 2022, 50, D543–D552 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cb00233d |
| ‡ Current address: Cell Signaling Technology, Beverly, MA, USA. |
| § Current address: Flare Therapeutics, Cambridge, MA, USA. |
| This journal is © The Royal Society of Chemistry 2025 |