
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproved synthesis of the unnatural base NaM, and evaluation of its orthogonality in in vitro transcription and translation†
Anthony V.
Le
 ab and
Matthew C. T.
Hartman
*ab
ab and
Matthew C. T.
Hartman
*ab
aVirginia Commonwealth University, Department of Chemistry, 1001 W Main St. Richmond, VA 23284, USA. E-mail: mchartman@vcu.edu
bVirginia Commonwealth University, Massey Cancer Center, 401 College St. Richmond, VA 23219, USA
First published on 11th September 2024
Abstract
Unnatural base pairs (UBP) promise to diversify cellular function through expansion of the genetic code. Some of the most successful UBPs are the hydrophobic base pairs 5SICS:NaM and TPT3:NaM developed by Romesberg. Much of the research on these UBPs has emphasized strategies to enable their efficient replication, transcription and translation in living organisms. These experiments have achieved spectacular success in certain cases; however, the complexity of working in vivo places strong constraints on the types of experiments that can be done to optimize and improve the system. Testing UBPs in vitro, on the other hand, offers advantages including minimization of scale, the ability to precisely control the concentration of reagents, and simpler purification of products. Here we investigate the orthogonality of NaM-containing base pairs in transcription and translation, looking at background readthrough of NaM codons by the native machinery. We also describe an improved synthesis of NaM triphosphate (NaM-TP) and a new assay for testing the purity of UBP containing RNAs.
Introduction
Unnatural base pairs (UBP) have emerged as a promising strategy for expanding the genetic codes of organisms.1–4 Early UBPs based on altering the donor–acceptor hydrogen bonding patterns of nucleobases2,3,5–7 met with challenges with incomplete orthogonality due to the formation of tautomeric forms.8 Benner has solved some of these issues through careful nucleobase modifications, developing two new base pairs (dS-dB, and dZ-dP).9Kool, Hirao, and Romesberg developed new UBPs that rely solely on hydrophobic and pi–pi stacking interactions rather than H-bonding.10–12 The most advanced of these UBPs contain the NaM-5SICS and NaM-TPT3 pairs. These pairs have been validated in replication, transcription, and translation in living E. coli and have been used for industrial preparation of proteins with non-canonical amino acids.13–20
Of the 96 possible codons containing a single NaM or TPT3 base, only a small subset has been tested. Certain base pairs are not efficiently retained in the plasmid DNA,21 although this can be corrected using the CRISPR/Cas9 system.4 Some rules have emerged for the codons tested, including preference for locating the UBP in the second position and the requirement for at least one G–C base pair in the other codon positions for efficient translation.21
Although the downstream integration of UBPs into organisms is an exciting goal for synthetic biology, the requirements of working in vivo also present many challenges that prevent fundamental experiments exploring the function of UBPs. In vivo, an unnatural base-containing tRNA or mRNA must be efficiently replicated, and transcribed before it is read by the ribosome. The precise concentrations of each system component is also difficult to control and optimize. In vivo studies may therefore miss important fundamental insights about the efficiency of UBPs and their true orthogonality.
In this paper we focus on the efficiency and orthogonality of the NaM base in transcription and translation in vitro, learning new important insights about the functionality of this unnatural base. We also report a new, high-yielding synthetic pathway for the synthesis of NaM-ribose and its triphosphates.
Results and discussion
Romesberg's strategy for the synthesis of the NaM nucleoside involved first alkylation of a protected ribolactone, followed by deoxygenation and protecting group removal (Scheme 1a).22 The overall reported yield of his approach was low, and in our hands, the initial alkylation step was difficult to control. We therefore considered an alternative involving a Weinreb amide (Scheme 1b).23 Our synthesis started by reacting commercially available ribolactone 1 with dimethylhydroxylamine giving Weinreb amide 2 (Scheme 2). We noticed that 2 is very sensitive to reversion to 1, so we protected the C-4 OH. After some optimization (Table S1, ESI†), we found that the TES protecting group, as opposed to other silyl groups, gave the right balance of stability, reactivity, and removability for the subsequent alkylation and deprotection steps. | ||
| Scheme 1 (a) Original synthetic strategy involving direct alkylation of ribolactone. (b) Our new approach involving a Weinreb amide precursor. | ||
 | ||
| Scheme 2 Synthesis of NaM via Weinreb amide intermediate. | ||
The subsequent aryl-ketone formation with 2-iodo-3-methoxynapthalene24 required some optimization (Table S2, ESI†), but the maximum yield of ketone 4 was achieved using turbo-Grignard conditions (Scheme 2). Recyclization was achieved by removal of the TES group under acidic conditions to form 5 as a mixture ratio of the linear and ring closed isomers 1.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.76:0.40. Removal of the hydroxyl group was achieved with Et3SiH and BF3·OEt2 leading to 6 as a mixture of inseparable isomers 7.1:1 (β:α). For the final debenzylation to give the NaM nucleoside 7, we found that the milder Lewis Acid BCl325 was superior to BBr3. The overall yield for the 6 steps was 55% in Scheme 2.
:0.76:0.40. Removal of the hydroxyl group was achieved with Et3SiH and BF3·OEt2 leading to 6 as a mixture of inseparable isomers 7.1:1 (β:α). For the final debenzylation to give the NaM nucleoside 7, we found that the milder Lewis Acid BCl325 was superior to BBr3. The overall yield for the 6 steps was 55% in Scheme 2.
In parallel, we also prepared the 5SICS nucleoside using Romesberg's reported method.22 To form the NaM and 5SICS triphosphates, we found Romesberg's phosphorylation strategy gave multiple products, so we decided to selectively protect the 2′ and 3′ OH groups with benzoyl groups using a tritylation/benzoylation/detritylation sequence (Scheme 3). Each reaction proceeded in high yield for both the 5SICS and NaM base giving compounds 10a/b. We reacted 10a/b with di-t-butyl N,N-diisopropylphosphoramidite followed by oxidation to give the protected monophosphates 11a/b. Deprotection under acidic conditions gave 12a/b. We found that 5SICS was unstable in TFA, so we opted to use HCl in 1,4 dioxane for the deprotection instead. 12a/b was then coupled with pyrophosphate using 1-methyl-3-benzenesulfonylimidazolium triflate to produce a clean unnatural base-TP product 13a/b.2613a/b was then debenzoylated with NH4OH to give the final NaM and 5SICS triphosphates 14a/b (Scheme 3) as their sodium salts with a total yield of 19/15% starting from 7/20. Although we generated lower yields than the reported single step phosphorylation method, this route avoids difficult separations of water-soluble phosphate isomers.27
 | ||
| Scheme 3 Unnatural nucleotide triphosphorylation strategy. | ||
With the 5SICS and NaM triphosphates in hand, we moved on to tests of their efficiency in in vitro transcription. Using phosphoramidite chemistry, we first prepared 16 ssDNAs for transcription via T7 RNA polymerase (Fig. 1a). Each contained a single NYN codon (Y = 5SICS) for transcription into mRNAs containing NaM. We included a native template containing the CAG leucine codon as a control. We first initiated our studies by performing in vitro transcriptions in the absence of NaM-TP and were surprised to find that a full-length product was formed for all templates, even though the expected truncated products were also observed (Fig. S1, ESI†). We then titrated in NaM-TP and found that the percentage of full-length products increased with increasing NaM-TP concentration and saturated above 1.25 mM of NaM-TP (Fig. 1b and c). We then performed in vitro transcription reactions for each of the 16 NYN templates with and without 2.5 mM NaM-TP (Fig. S2, ESI†) and computed the improvements in full-length bands with each template (Fig. 1d). In every template we observed significant readthrough of the d5SICS base in the absence of NaM-TP, although improvements in the ratio of full-length to truncated bands were observed for every template in the presence of NaM-TP at 2.5 mM thus confirming at least partial NaM-TP incorporation.
 | ||
| Fig. 1 Optimizing NaM incorporation in T7 transcription with d5SICS. (a) ssDNAs containing all possible NYN codons where Y = d5SICS and X = NaM. The T7 promoter primer and the expected products from the transcription reaction are shown. (b) 10% Urea PAGE gel containing in vitro transcription products with increasing concentrations of NaM-TP (0–5 mM) with a ssDNA template containing the CYT codon. Bands were visualized by SYBR-green II (c) Plot showing the % full-length calculated by measuring the relative intensity of the full-length bands vs. truncated bands in each lane in (b). (d) % Full-length stacked bar graph for transcriptions with all 16 ssDNA templates with and without NaM-TP computed from gel image Fig. S2 (ESI†). The ssDNA template codons are shown in Table S3 (ESI†). | ||
Although we were able to optimize our NaM-TP incorporation efficiency, it was unclear how pure our RNA products were since in all cases, we observed a full-length band in the absence of NaM-TP. Several strategies have been employed to assess UBP incorporation efficiency. Hirao created a biotinylated version of his y-base UBP and used a gel shift assay to monitor incorporation.28 Romesberg envisioned a similar approach that utilized an amine modified MMO2 which was biotinylated post-transcriptionally and then analyzed via gel.29 Both approaches require a separate synthesis to deliver the desired product and assume that the biotinylated UBP will incorporate with the same efficiency as the unmodified UBP. Another common strategy to investigate incorporation efficiency involves radiolabeling the NTP preceding the UBP during transcription, followed by 2D-TLC after complete RNase I digestion to the 3′ monophosphates to analyze the monomer distribution.30,31 While powerful, this assay is typically performed with short model templates, to prevent the UBP signal from being overshadowed by the canonical bases. With longer templates slight misincorporations can easily fall within the measurement error.
We used two approaches to monitor the purity of our RNAs. First, we initiated a series of reverse transcriptions using superscript II (SSII)19,32,33 (Fig. 2a) on full-length, gel-purified RNAs prepared with increasing concentrations of NaM-TP. We omitted any deoxy UBP triphosphates in these reactions and therefore expected that SSII would terminate at the NaM base if present. RT reactions were performed with a fluorescein-labeled primer for detection (Fig. 2a). Surprisingly, we did not observe any truncation products from reverse transcription. This is a different result compared to that reported by Eggert et al. who observed 23% truncation when reverse transcribing NaM-containing RNAs with SSII in the absence of any complementary deoxy unnatural NTP.32 Notably, reverse transcriptions of RNAs prepared in the presence of NaM-TP produced a higher band than those transcribed in the absence of NaM-TP, and this band increased as more NaM-TP was added (Fig. 2b). Above 2.5 mM we observed almost exclusively this higher band (Fig. 2b and c). The band transcribed in the absence of NaM-TP was the same length as the band produced by a control template containing CUG in place of the NaM (Fig. S3, ESI†), thus this higher band is unique to RNAs containing NaM. This band was present in all 16 templates containing NaM (Fig. S3, ESI†) and its intensity ranged from 65–95% depending on the template (Fig. 2d).
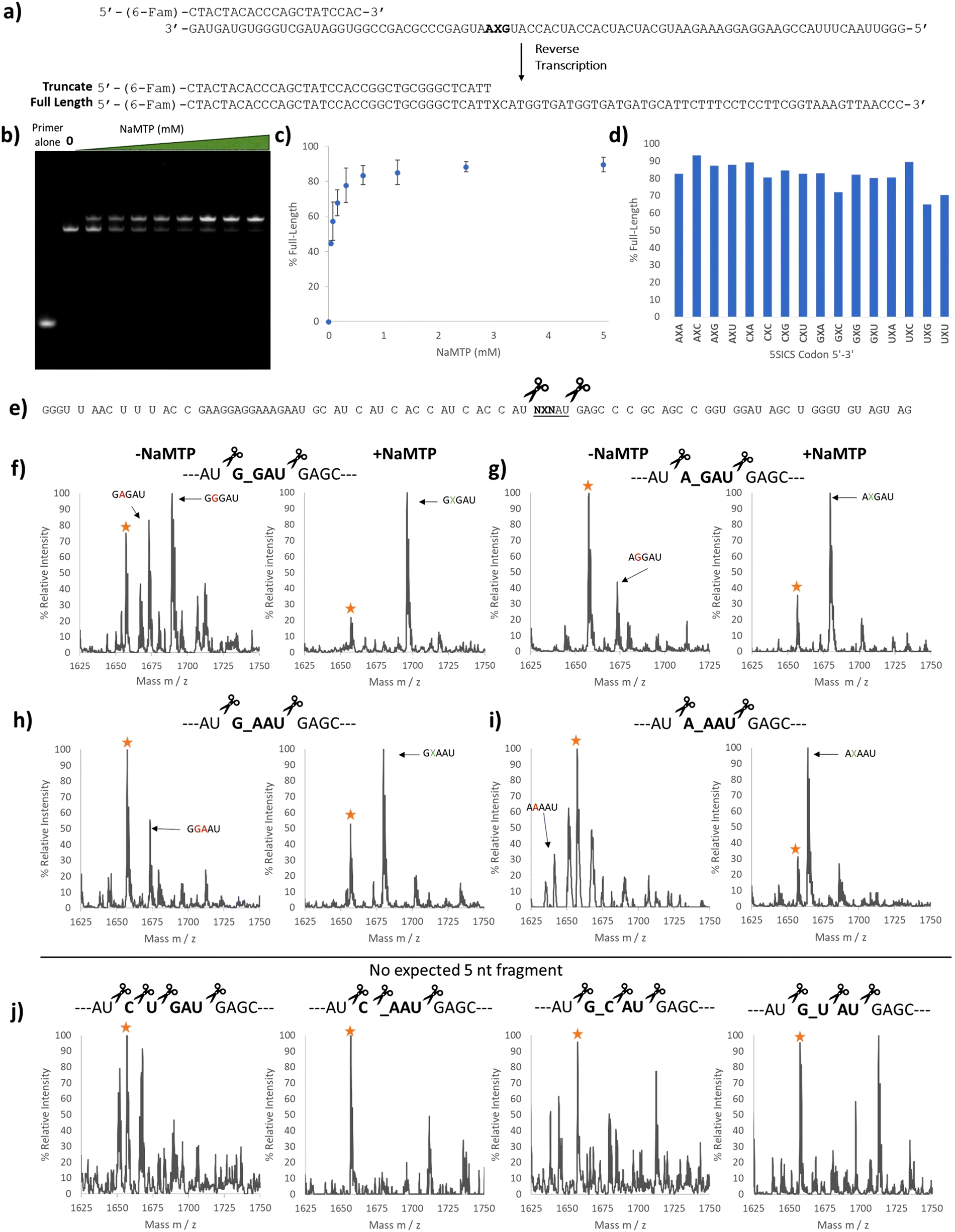 | ||
| Fig. 2 Purity validation of transcription products containing NaM. (a) Reverse transcription template containing NaM at the second position. The primer was labeled with 6-(FAM) and the expected full-length and truncated products are shown. (b) 10% Urea PAGE image with each lane containing SSII reverse transcription products using UBP mRNA containing the GXA codon prepared with increasing amounts of NaM-TP (0–5 mM) (Fig. 1b). (c) Plot showing the percentage of the higher band in each lane in (b). (d) Bar graph showing the percentage of the higher band for all 16 reverse transcribed templates. The RNA templates for the RT reactions were prepared with 2.5 mM NaM-TP. (e) Predicted RNase A digest products of the NaM-containing RNAs. RNase A cleaves on the 3′ end of C and U, so templates containing neither C nor U in the codon region would give a unique 5 nucleotide (nt) fragment. (f) MALDI-TOF MS of the RNase A digest products in the 5 nt region for the template encoding the GXG RNA transcribed without (left) and with (right) NaM-TP. Yellow star labeled as a background peak (g) MALDI-TOF MS of the RNase A digest products in the 5 nt region for the template encoding the AXG RNA transcribed with (Right) and without (Left) NaM-TP. Yellow star labeled as a background peak or potential AAGAU fragment. (h) MALDI-TOF MS of the RNase A digest products in the 5 nt region for the template encoding the GXA RNA transcribed with (Right) and without (Left) NaM-TP. Yellow star labeled as a background peak or potential GAAAU fragment. (i) MALDI-TOF MS of the RNase A digest products in the 5 nt region for the template encoding the AXA RNA transcribed with (Right) and without (Left) NaM-TP. Yellow star labeled as a background peak or potential AGAAU fragment. (j) MALDI-TOF MS of the RNase A digest products in the 5 nt region for the template mRNA # 01 CUG (Left), mRNA # 06 CXA (middle-left), mRNA # 11 GXC (middle-right) and mRNA # 13 GXC (right). Yellow star labeled as a background peak. Full mass spectrums are shown in Fig. S5–S16 (ESI†) and the expected and observed masses are summarized in Table S5 (ESI†). mRNA templates are shown in Table S4 (ESI†). | ||
To get a better idea of the identity of this higher band, we performed next-generation sequencing of the reverse transcribed products after PCR amplification (with only the standard dNTPs) of the template containing the GXA codon. Surprisingly, there was no indication that these longer sequences were present in the PCR products, although we could detect A, C, G, and T incorporation or a deletion in the PCR products (Fig. S4, ESI†) in the position where we would expect NAM to be. We therefore suspect that this higher band is a product that cannot be amplified from the cDNA by PCR using our primers.
To further confirm the purity of our mRNAs, we digested a subset of our transcripts using RNase A and looked for the incorporation of NaM in the expected fragments. RNase A cleaves RNAs at the 3′ side of C or U, and so we chose transcripts that were predicted to produce NaM-containing fragments within a unique mass range (the 5 nt region) (Fig. 2e). We compared the RNAs transcribed in the presence and absence of NaM-TP. The transcriptions performed in the absence of NaM-TP gave several peaks. In line with the sequencing results, we were able to identify G and A misincorporation (Fig. 2f–i left and Fig. S5–S16, ESI†). (We note here that C and U misincorporation would be digested by RNase A into smaller fragments indistinguishable from other fragments). In contrast, those mRNAs transcribed in the presence of NaM-TP (Fig. 2f–i, right, Fig. S5–S16, ESI†) all showed a major peak corresponding to NaM incorporation as well as an additional peak (labeled with a star). While this peak could correspond to a natural base misincorporation (G or A depending on the template), it was also present in four templates that would not be expected to give any fragments in this range: one natural control template and 3 UBP templates (Fig. 2j). We therefore suspect that this peak comes from another source (e.g. incomplete digestion). Taken together, these studies show that in vitro transcription using 2.5 mM NaM-TP delivers NaM-containing RNAs with minimal misincorporation of natural nucleotides, aligning our work with literature reports.27,34
With confidence in our NaM-mRNA purity, we moved to testing our NaM-mRNAs in in vitro translation. We first investigated the potential consequences of translating our mRNAs in the absence of any unnatural base-containing tRNAs. We wondered if the ribosome would be able to read through these codons, and if so, if there were any trends. We tested both wild-type (wt) and hyperaccurate (mS12) ribosomes, known to more readily reject near-cognate tRNAs.35,36 We used the recently developed affinity-clamp fluorescent translation assay for these studies in which each template has a C-terminal affinity-clamp sequence (encoding PQPVDSWV).37 In this assay peptides containing the C-terminal tag will bind to the cyPET-yPET-affinity clamp protein causing a decrease in the ratio of 527/475 fluorescence emission which correlates to yield (Fig. S17–S20, ESI†). Yields are shown in Fig. 3b alongside a control template with a native leucine codon (CUG). For most of the codons, very low yields were observed, further confirming the purity of our mRNA products, especially when observing the mS12 results. With wt ribosomes, there were exceptions. The CXA and GXC codons both showed >40% misdecoding yield; AXC and GXU showed misreading between 20 and 40%. For mS12 ribosomes the codons with appreciable yield tended to be the same, but misreading yields in all cases were significantly suppressed; no codons gave yields above 20% (Fig. 3b).
 | ||
| Fig. 3 Investigating the orthogonality of NaM codons. Translations were performed using wt or hyperaccurate (mS12) ribosomes. (a) Templates tested in translation. The C-terminus of each template encodes an affinity clamp peptide used to monitor yield. (b) In vitro translation yields in pmol calculated after 90 min at 37 oC. Bar graphs colored in blue are yields generated from using wt ribosomes and bar graphs colored in red are yields generated from using mS12 Ribosomes. The codon in the mRNA is shown below each data point. (c) Misdecoding events represented by a heat map. The yields were measured relative to the positive control mRNA containing the CUG codon and are shown in b. Each colored box in the heat map represents a detectable peak after His-tag purification. | ||
We also performed Ni-NTA capture of the translated peptides using the N-terminal His-tag and used MALDI to detect any full-length peptides formed (Fig. S21–S54, ESI†). Based on the amino acid incorporated, we inferred how the ribosome was able to interpret the UBP codon. These results are shown as heatmaps in Fig. 3c. Yields in the heatmaps are relative to the template containing a CUG leucine codon and a control translation reaction lacking mRNA.
There are global trends revealed by this dataset. First, most of the codons that are misread at detectable levels have at least one G or C (Fig. 3b) with AXU as an exception. This is in alignment with Romesberg's finding that successful UBP pairs contain at least 1 G–C pair. Second, there doesn’t seem to be a nucleotide bias in the misdecoding. Both the wild-type and mS12 ribosomes were able to misread the NaM base as either U, C, A or G depending on the context. Third, the hyperaccurate mS12 ribosomes were more efficient at discriminating against misdecoding by native AA-tRNAs than wild-type ribosomes. This is evidenced by the lower yields shown in Fig. 3b, many of which were at background levels. The difference in yields between hyperaccurate and wild-type ribosomes is further evidence of the purity of our NaM-containing mRNAs as templates containing standard nucleotides in place of NaM would have been expected to give the same results for both types of ribosomes.35 Finally, it is interesting to compare our results with Katoh and Suga's recent paper that investigated background readthrough with standard codons.38 Suga found that NUN and NCN codons are more easily mis-decoded that NAN and NGN codons. Our data shows that NXN codons overall appear to be misread at low frequencies in comparison with the native codons, which speaks to their relatively high level of orthogonality.
The CXA codon is notable for its particularly high background yield, due to misdecoding as glutamine. It is interesting that the tRNAGln that is the primary decoder of the CAA codon has a unique modified base, 5-carboxymethylaminomethyl-2-thiouridine, (cmnm5s2U) base in its anticodon.39,40 This particular base is not found in any other tRNAs in E. coli (although there are close analogs).40 The cmnm5s2U base is known to improve the geometry of the pair with A, preventing frameshifting.40 Perhaps this enhanced pairing also enables the ribosome to overcome the mismatched NaM:U base pair in the second codon position. It is also notable that next most readthrough codons (GXC, GXU, and AXC) in our data set were shown to have little background readthrough by Romesberg.21 This discrepancy suggests that the surrounding sequence context could influence UBP codon readthrough as it does for stop codon suppression.41 The fact that these same 3 codons are among the most efficient codons for in vivo incorporation with ncAAs is also intriguing and highlights that: (1) the ribosome has an innate ability to recognize these codons and (2) background readthrough can be overcome in the presence of a complementary unnatural base-containing tRNA. Finally, it is also interesting that some codons were read by multiple tRNAs. The GXC codon is prominent in this regard as we observed 4 different full length peptide products. This means that the ribosome was able to accept at least 4 different tRNAs in response to this codon (albeit at moderate to low yield).
We chose three NaM codons that have been shown by Romesberg to be highly efficient in translation, AXC, GXU, and GXC for further analysis.21In vivo work with these codons has utilized the pyrrolysyl tRNA (tRNAPyl); here we chose to use E. coli tRNALeu1 which natively has the CAG anticodon and reads the CUG codon.37,42,43E. coli LeuRS does not recognize the anticodon of the tRNA,44 making it amenable to substitution with an unnatural nucleotide. Using templates containing NaM (Table S4, ESI†), we created tRNAs containing the GYU, AYC, and GYC anticodons using 2.5 mM 5SICS triphosphate during transcription. All 3 tRNAs were able to be charged with Leucine and were detectable via MALDI-MS after undergoing the reductive amination/nuclease digestion assay (Fig. S55–S57, ESI†).45 AXC tRNA/GXU only showed misdecoding by native tRNAs. The GYU tRNA/AXC codon pairing gave some of the correct mass peak; however, it was impossible to distinguish this peak from the background isoleucine misdecoding (Fig. S58, compare with Fig. S25, ESI†) and so this codon was not pursued further. The GYC tRNA/GXC codon pairing showed the expected peptide product containing leucine as well as other misdecoding peaks when we tested at a concentration of 40 μM (Fig. S58–S60, ESI†).
We then titrated this tRNA into the translation mixture and observed clean MALDI-MS spectra at 100 μM (Fig. 4c), comparable to the native CUG codon experiment (Fig. 4b), highlighting the good orthogonality with this codon and that background readthrough can be overcome with sufficient complementary tRNA.
 | ||
Fig. 4 5SICS-tRNAGYC is able to read the GXC codon in vitro. (a) mRNAs used for testing of translation yield and their corresponding expected peptide sequences, where GXC codes for Leucine. (b) MALDI-MS spectrum of the translation products using mRNA 1 showing the expected peak corresponding to leucine incorporation. (c) MALDI-MS spectrum of the translation products using mRNA 2 at increasing concentrations of 5SICS-tRNAGYC. Peptide sequences: MH6![[V with combining low line]](https://www.rsc.org/images/entities/char_0056_0332.gif) MSPQPVDSWV labeled as a red circle, MH6 MSPQPVDSWV labeled as a red circle, MH6![[D with combining low line]](https://www.rsc.org/images/entities/char_0044_0332.gif) MSPQPVDSWV labeled as a yellow “X” and the desired product, MH6 MSPQPVDSWV labeled as a yellow “X” and the desired product, MH6![[L with combining low line]](https://www.rsc.org/images/entities/char_004c_0332.gif) MSPQPVDSWV, labeled as a green star. Masses for the labeled peaks are shown in Table S6 (ESI†). MSPQPVDSWV, labeled as a green star. Masses for the labeled peaks are shown in Table S6 (ESI†). | ||
Summary/conclusion
Here we report an improved synthesis of NaM-TP; we used the flexibility of in vitro transcription and translation systems to test its tolerance with the translation system. To monitor the purity of UBP products from transcription reactions we used a combination of reverse transcription and a newly developed RNase A assay. We then tested the ability of the translation apparatus to readthrough sixteen codons containing NaM (NXN) in the absence of complementary tRNAs. Codons CXA, AXC, GXC, GXU yielded >20% yield of mis-decoding using wt ribosomes; however, mS12 hyperaccurate ribosomes showed diminished misdecoding. Using MALDI-MS we were also able to infer how the ribosomes interpreted the NaM base during translation. Finally, we tested the fidelity and orthogonality of three UBP codon-anticodon in translation and found that mRNAGXC-tRNAGYC delivered high orthogonality at 100 μM of 5SICS-tRNAGYC. Taken together, our work shows that combining reconstituted in vitro translation and UBPs can lead to rapid assessment of UBP fidelity in transcription and translation. Our data shows that investigators performing UBP experiments with certain codons (in particular CXA and GXC) should be proceed with caution, realizing that they will likely be competing with background readthrough. It also points to the possibility of using hyperaccurate ribosomes as a strategy to minimize background readthrough of NaM codons. Down the line, it should also be possible to use this system alongside new UBP DNA synthesis strategies46 to quickly design and test the effects of engineered components for improved fidelity. A list of rigorously validated UBP codons and strategies maximize their incorporation will enable new advances in synthetic biology and the generation of peptide libraries via display technologies.Materials and methods
General
The RNAs were eluted by crushing and soaking in 2 mL dH2O at 80 °C for 30 min. The solution was then filtered through a membrane syringe filter and then extracted with butanol to a final volume of 100–500 μL of dH2O. The water layer was then separated and the RNA product was precipitated with 0.1 volumes of 3 M NaOAc (pH = 5.5) and 3 volumes of ethanol. The solution was then chilled at −20 °C for 30 min and then centrifuged. The precipitate was then filtered and washed with 70% Ethanol followed by ethanol to obtain the final RNA product.
The 10 × 10 gels were stained using SYBR green 1× (Thermo Fisher Scientific) in 1× TBE solution for 30 min. The gels were then imaged using the Bio-Rad ChemiDoc MP imaging system.
882 reads were analyzed. 59.3% of the reads consisted of the 5 outcomes described in Fig. S4 (ESI†).
:picolinic acid:citric acid diammonium (9:1:1) dissolved in MeCN:H2O (7:3).
000 × g. The pellet was washed with 500 μL of 70% Ethanol and then 500 μL of 100% ethanol to remove salts. Finally, the pellet was left to air dry and resuspended in NaOAc (12.5 μL, 100 mM pH = 5.0). Reductive amination was carried out by mixing 6.25 μL of the previous tRNA-aa preparation with 3.75 μL of dH2O, (4-formylphenoxypropyl) triphenylphosphonium bromide in MeOH (12.5 μL, 63 mM) and fresh NaBH3CN dissolved in 50 mM NaOAc pH = 5.0 (2.5 μL, 200 mM). The reaction was incubated at 37 °C on a tumbler for 2 hours before quenching with 0.1 volume of NH4OAc (4.4 M pH = 5.0). The reaction product was recovered through ethanol precipitation and the resulting pellet was resuspended in NH4OAc (2.25 μL, 200 mM pH = 5.0). 0.25 μL of Nuclease P1 (1 U μL−1 in 200 mM NH4OAc pH = 5.0) (Wako Cat#: 145-08221) was added and incubated at rt for 20 min. After incubation, the reaction mixture was quenched on ice, and 1 μL was mixed with 9 μL of MALDI matrix α-cyano-4-hydroxycinnamic acid (CHCA) 10 mg mL−1 in MeCN:1% TFA (1:1) and spotted onto the MALDI plate for further analysis.
:1) and 15 μL 0.2% TFA, centrifuging each time for 1 min at 1000 × g. The peptide eluted in TFA was loaded onto the tip. Each tip was washed 2× with 15 μL 0.2% TFA. The peptide was eluted with 4-chloro-α-cyanocinnamic acid (6.2 mg mL−1 in MeCN:0.2% TFA (7:3)), spotted in the MALDI-TOF plate and analyzed.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
Authors declare no conflicts of interest.Acknowledgements
The authors thank the NIH (R03CA259876 and R01GM143396) for funding of this work.References
- S. A. Benner and A. M. Sismour, Synthetic Biology, Nat. Rev. Genet., 2005, 6(7), 533–543 CrossRef CAS PubMed.
- I. Hirao, T. Mitsui, M. Kimoto and S. Yokoyama, An Efficient Unnatural Base Pair for PCR Amplification, J. Am. Chem. Soc., 2007, 129(50), 15549–15555 CrossRef CAS PubMed.
- J. D. Bain, C. Switzer, R. Chamberlin and S. A. Benner, Ribosome-Mediated Incorporation of a Non-Standard Amino Acid into a Peptide through Expansion of the Genetic Code, Nature, 1992, 356(6369), 537–539 CrossRef CAS PubMed.
- Y. Zhang, B. M. Lamb, A. W. Feldman, A. X. Zhou, T. Lavergne, L. Li and F. E. Romesberg, A Semisynthetic Organism Engineered for the Stable Expansion of the Genetic Alphabet, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(6), 1317–1322 CrossRef CAS PubMed.
- M. Ziemniak, J. Kowalska, M. Lukaszewicz, J. Zuberek, K. Wnek, E. Darzynkiewicz and J. Jemielity, Phosphate-Modified Analogues of M7GTP and M7Gppppm7G - Synthesis and Biochemical Properties, Bioorg. Med. Chem., 2015, 23(17), 5369–5381 CrossRef CAS PubMed.
- J. Horlacher, M. Hottiger, V. N. Podust, U. Hübscher and S. A. Benner, Recognition by Viral and Cellular DNA Polymerases of Nucleosides Bearing Bases with Nonstandard Hydrogen Bonding Patterns, Proc. Natl. Acad. Sci. U. S. A., 1995, 92(14), 6329–6333 CrossRef CAS PubMed.
- S. Moran, R. X.-F. Ren and E. T. Kool, A Thymidine Triphosphate Shape Analog Lacking Watson–Crick Pairing Ability Is Replicated with High SequenceSelectivity, Proc. Natl. Acad. Sci. U. S. A., 1997, 94(20), 10506–10511 CrossRef CAS PubMed.
- C. R. Geyer, T. R. Battersby and S. A. Benner, Nucleobase Pairing in Expanded Watson-Crick-like Genetic Information Systems, Structure, 2003, 11(12), 1485–1498 CrossRef CAS PubMed.
- S. Hoshika, N. A. Leal, M.-J. Kim, M.-S. Kim, N. B. Karalkar, H.-J. Kim, A. M. Bates, N. E. Watkins, H. A. SantaLucia, A. J. Meyer, S. DasGupta, J. A. Piccirilli, A. D. Ellington, J. SantaLucia, M. M. Georgiadis and S. A. Benner, Hachimoji DNA and RNA: A Genetic System with Eight Building Blocks, Science, 2019, 363(6429), 884–887 CrossRef CAS PubMed.
- A. M. Leconte, G. T. Hwang, S. Matsuda, P. Capek, Y. Hari and F. E. Romesberg, Discovery, Characterization, and Optimization of an Unnatural Base Pair for Expansion of the Genetic Alphabet, J. Am. Chem. Soc., 2008, 130(7), 2336–2343 CrossRef CAS PubMed.
- Y. Wu, A. K. Ogawa, M. Berger, D. L. McMinn, P. G. Schultz and F. E. Romesberg, Efforts toward Expansion of the Genetic Alphabet: Optimization of Interbase Hydrophobic Interactions, J. Am. Chem. Soc., 2000, 122(32), 7621–7632 CrossRef CAS.
- K. M. Guckian, T. R. Krugh and E. T. Kool, Solution Structure of a DNA Duplex Containing a Replicable Difluorotoluene–Adenine Pair, Nat. Struct. Biol., 1998, 5(11), 954–959 CrossRef CAS PubMed.
- J. Oh, J. Shin, I. C. Unarta, W. Wang, A. W. Feldman, R. J. Karadeema, L. Xu, J. Xu, J. Chong, R. Krishnamurthy, X. Huang, F. E. Romesberg and D. Wang, Transcriptional Processing of an Unnatural Base Pair by Eukaryotic RNA Polymerase II, Nat. Chem. Biol., 2021, 17(8), 906–914 CrossRef CAS PubMed.
- D. A. Malyshev, K. Dhami, T. Lavergne, T. Chen, N. Dai, J. M. Foster, I. R. Corrêa and F. E. Romesberg, A Semi-Synthetic Organism with an Expanded Genetic Alphabet, Nature, 2014, 509(7500), 385–388 CrossRef CAS PubMed.
- Y. J. Seo, D. A. Malyshev, T. Lavergne, P. Ordoukhanian and F. E. Romesberg, Site-Specific Labeling of DNA and RNA Using an Efficiently Replicated and Transcribed Class of Unnatural Base Pairs, J. Am. Chem. Soc., 2011, 133(49), 19878–19888 CrossRef CAS PubMed.
- E. L. Tae, Y. Wu, G. Xia, P. G. Schultz and F. E. Romesberg, Efforts toward Expansion of the Genetic Alphabet: Replication of DNA with Three Base Pairs, J. Am. Chem. Soc., 2001, 123(30), 7439–7440 CrossRef CAS PubMed.
- L. Li, M. Degardin, T. Lavergne, D. A. Malyshev, K. Dhami, P. Ordoukhanian and F. E. Romesberg, Natural-like Replication of an Unnatural Base Pair for the Expansion of the Genetic Alphabet and Biotechnology Applications, J. Am. Chem. Soc., 2014, 136(3), 826–829 CrossRef CAS PubMed.
- J. Oh, M. Kimoto, H. Xu, J. Chong, I. Hirao and D. Wang, Structural Basis of Transcription Recognition of a Hydrophobic Unnatural Base Pair by T7 RNA Polymerase, Nat. Commun., 2023, 14, 195 CrossRef CAS PubMed.
- E. S. Hoffmann, M. C. De Pascali, L. Neu, C. Domnick, A. Soldà and S. Kath-Schorr, Reverse Transcription as Key Step in RNA in Vitro Evolution with Unnatural Base Pairs. RSC, Chem. Biol., 2024, 5, 556–566 CAS.
- J. L. Ptacin, C. E. Caffaro, L. Ma, K. M. San Jose Gall, H. R. Aerni, N. V. Acuff, R. W. Herman, Y. Pavlova, M. J. Pena, D. B. Chen, L. K. Koriazova, L. K. Shawver, I. B. Joseph and M. E. Milla, An Engineered IL-2 Reprogrammed for Anti-Tumor Therapy Using a Semi-Synthetic Organism, Nat. Commun., 2021, 12, 4785 CrossRef CAS PubMed.
- E. C. Fischer, K. Hashimoto, Y. Zhang, A. W. Feldman, V. T. Dien, R. J. Karadeema, R. Adhikary, M. P. Ledbetter, R. Krishnamurthy and F. E. Romesberg, New Codons for Efficient Production of Unnatural Proteins in a Semisynthetic Organism, Nat. Chem. Biol., 2020, 16(5), 570–576 CrossRef CAS PubMed.
- Y. J. Seo, S. Matsuda and F. E. Romesberg, Transcription of an Expanded Genetic Alphabet, J. Am. Chem. Soc., 2009, 131(14), 5046–5047 CrossRef CAS PubMed.
- Y. Xie, T. Hu, Y. Zhang, D. Wei, W. Zheng, F. Zhu, G. Tian, H. A. Aisa and J. Shen, Weinreb Amide Approach to the Practical Synthesis of a Key Remdesivir Intermediate, J. Org. Chem., 2021, 86(7), 5065–5072 CrossRef CAS PubMed.
- R. R. Kadiyala, D. Tilly, E. Nagaradja, T. Roisnel, V. E. Matulis, O. A. Ivashkevich, Y. S. Halauko, F. Chevallier, P. C. Gros and F. Mongin, Computed CH Acidity of Biaryl Compounds and Their Deprotonative Metalation by Using a Mixed Lithium/Zinc-TMP Base, Chem. – Eur. J., 2013, 19(24), 7944–7960 CrossRef CAS PubMed.
- K. Okano, K. I. Okuyama, T. Fukuyama and H. Tokuyama, Mild Debenzylation of Aryl Benzyl Ether with BCl3 in the Presence of Pentamethylbenzene as a Non-Lewis-Basic Cation Scavenger, Synlett, 2008, 1977–1980 CAS.
- S. Mohamady, A. Desoky and S. D. Taylor, Sulfonyl Imidazolium Salts as Reagents for the Rapid and Efficient Synthesis of Nucleoside Polyphosphates and Their Conjugates, Org. Lett., 2012, 14(1), 402–405 CrossRef CAS PubMed.
- A. W. Feldman, V. T. Dien, R. J. Karadeema, E. C. Fischer, Y. You, B. A. Anderson, R. Krishnamurthy, J. S. Chen, L. Li and F. E. Romesberg, Optimization of Replication, Transcription, and Translation in a Semi-Synthetic Organism, J. Am. Chem. Soc., 2019, 141(27), 10644–10653 CrossRef CAS PubMed.
- K. Moriyama, M. Kimoto, T. Mitsui, S. Yokoyama and I. Hirao, Site-Specific Biotinylation of RNA Molecules by Transcription Using Unnatural Base Pairs, Nucleic Acids Res., 2005, 33(15), 1–8 CrossRef PubMed.
- Y. J. Seo, D. A. Malyshev, T. Lavergne, P. Ordoukhanian and F. E. Romesberg, Site-Specific Labeling of DNA and RNA Using an Efficiently Replicated and Transcribed Class of Unnatural Base Pairs, J. Am. Chem. Soc., 2011, 133(49), 19878–19888 CrossRef CAS PubMed.
- I. Hirao, T. Ohtsuki, T. Fujiwara, T. Mitsui, T. Yokogawa, T. Okuni, H. Nakayama, K. Takio, T. Yabuki, T. Kigawa, K. Kodama, T. Yokogawa, K. Nishikawa and S. Yokoyama, An Unnatural Base Pair for Incorporating Amino Acid Analogs into Proteins, Nat. Biotechnol., 2002, 20, 177–182 CrossRef CAS PubMed.
- Y. J. Seo, S. Matsuda and F. E. Romesberg, Transcription of an Expanded Genetic Alphabet, J. Am. Chem. Soc., 2009, 131(14), 5046–5047 CrossRef CAS PubMed.
- F. Eggert, K. Kurscheidt, E. Hoffmann and S. Kath-Schorr, Towards Reverse Transcription with an Expanded Genetic Alphabet, ChemBioChem, 2019, 20(13), 1642–1645 CrossRef CAS PubMed.
- A. X. Z. Zhou, X. Dong and F. E. Romesberg, Transcription and Reverse Transcription of an Expanded Genetic Alphabet in Vitro and in a Semisynthetic Organism, J. Am. Chem. Soc., 2020, 142(45), 19029–19032 CrossRef CAS PubMed.
- J. Oh, J. Shin, I. C. Unarta, W. Wang, A. W. Feldman, R. J. Karadeema, L. Xu, J. Xu, J. Chong, R. Krishnamurthy, X. Huang, F. E. Romesberg and D. Wang, Transcriptional Processing of an Unnatural Base Pair by Eukaryotic RNA Polymerase II, Nat. Chem. Biol., 2021, 17(8), 906–914 CrossRef CAS PubMed.
- B. Shakya, O. G. Joyner and M. C. T. Hartman, Hyperaccurate Ribosomes for Improved Genetic Code Reprogramming, ACS Synth. Biol., 2022, 11(6), 2193–2201 CrossRef CAS PubMed.
- C. A. L. McFeely, B. Shakya, C. A. Makovsky, A. K. Haney, T. Ashton Cropp and M. C. T. Hartman, Extensive Breaking of Genetic Code Degeneracy with Non-Canonical Amino Acids, Nat. Commun., 2023, 14(1), 5008 CrossRef CAS PubMed.
- G. N. Kerestesy, K. K. Dods, C. A. L. McFeely and M. C. T. Hartman, Continuous Fluorescence Assay for In Vitro Translation Compatible with Noncanonical Amino Acids, ACS Synth. Biol., 2024, 13(1), 119–128 CrossRef CAS PubMed.
- T. Katoh and H. Suga, A Comprehensive Analysis of Translational Misdecoding Pattern and Its Implication on Genetic Code Evolution, Nucleic Acids Res., 2023, 51(19), 10642–10652 CrossRef CAS PubMed.
- A. Cappannini, A. Ray, E. Purta, S. Mukherjee, P. Boccaletto, S. N. Moafinejad, A. Lechner, C. Barchet, B. P. Klaholz, F. Stefaniak and J. M. Bujnicki, MODOMICS: A Database of RNA Modifications and Related Information. 2023 Update, Nucleic Acids Res., 2024, 52(D1), D239–D244 CrossRef PubMed.
- M. E. Armengod, S. Meseguer, M. Villarroya, S. Prado, I. Moukadiri, R. Ruiz-Partida, M. J. Garzón, C. Navarro-González and A. Martínez-Zamora, Modification of the Wobble Uridine in Bacterial and Mitochondrial TRNAs Reading NNA/NNG Triplets of 2-Codon Boxes, RNA Biol., 2014, 11(12), 1495–1507 CrossRef PubMed.
- W. T. Pedersen and J. F. Curran, Effects of the Nucleotide 3′ to an Amber Codon on Ribosomal Selection Rates of Suppressor TRNA and Release Factor-1, J. Mol. Biol., 1991, 219(2), 231–241 CrossRef CAS PubMed.
- Z. Cui, V. Stein, Z. Tnimov, S. Mureev and K. Alexandrov, Semisynthetic TRNA Complement Mediates in Vitro Protein Synthesis, J. Am. Chem. Soc., 2015, 137(13), 4404–4413 CrossRef CAS PubMed.
- C. A. L. McFeely, K. K. Dods, S. S. Patel and M. C. T. Hartman, Expansion of the Genetic Code through Reassignment of Redundant Sense Codons Using Fully Modified TRNA, Nucleic Acids Res., 2022, 50(19), 11374–11386 CrossRef CAS PubMed.
- R. Giege, M. Sissler and C. Florentz, Universal Rules and Idiosyncratic Features in TRNA Identity, Nucleic Acids Res., 1998, 26(22), 5017–5035 CrossRef CAS PubMed.
- M. C. T. Hartman, K. Josephson and J. W. Szostak, Enzymatic Aminoacylation of TRNA with Unnatural Amino Acids, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(12), 4356–4361 CrossRef CAS PubMed.
- Y. Cao, J. Bai, J. Zou, Y. Du and T. Chen, One-Pot Enzymatic Preparation of Oligonucleotides with an Expanded Genetic Alphabet via Controlled Pause and Restart of Primer Extension: Making Unnatural Out of Natural, ACS Synth. Biol., 2023, 12(9), 2691–2706 CrossRef CAS PubMed.
- S. Carulli, C. D. Calvano, F. Palmisano and M. Pischetsrieder, MALDI-TOF MS Characterization of Glycation Products of Whey Proteins in a Glucose/Galactose Model System and Lactose-Free Milk, J. Agric. Food Chem., 2011, 59(5), 1793–1803 CrossRef CAS PubMed.
- J. Hoinka, R. Backofen and T. M. Przytycka, AptaSUITE: A Full-Featured Bioinformatics Framework for the Comprehensive Analysis of Aptamers from HT-SELEX Experiments, Mol. Ther. Nucleic Acid., 2018, 11, 515–517 CrossRef CAS PubMed.
- R. Matthiesen and F. Kirpekar, Identification of RNA Molecules by Specific Enzyme Digestion and Mass Spectrometry: Software for and Implementation of RNA Mass Mapping, Nucleic Acids Res., 2009, 37(6), e48 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cb00121d |
| This journal is © The Royal Society of Chemistry 2024 |