
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvanced piperazine-containing inhibitors target microbial β-glucuronidases linked to gut toxicity†
Amanda L.
Graboski‡
 a,
Joshua B.
Simpson‡
b,
Samuel J.
Pellock
b,
Naimee
Mehta
c,
Benjamin C.
Creekmore
b,
Yamuna
Ariyarathna
c,
Aadra P.
Bhatt
d,
Parth B.
Jariwala
b,
Josh J.
Sekela
b,
Mark E.
Kowalewski
e,
Natalie K.
Barker
f,
Angie L.
Mordant
f,
Valentina B.
Borlandelli
g,
Hermen
Overkleeft
g,
Laura E.
Herring
f,
Jian
Jin
h,
Lindsey
I. James
c and
Matthew R.
Redinbo
*be
a,
Joshua B.
Simpson‡
b,
Samuel J.
Pellock
b,
Naimee
Mehta
c,
Benjamin C.
Creekmore
b,
Yamuna
Ariyarathna
c,
Aadra P.
Bhatt
d,
Parth B.
Jariwala
b,
Josh J.
Sekela
b,
Mark E.
Kowalewski
e,
Natalie K.
Barker
f,
Angie L.
Mordant
f,
Valentina B.
Borlandelli
g,
Hermen
Overkleeft
g,
Laura E.
Herring
f,
Jian
Jin
h,
Lindsey
I. James
c and
Matthew R.
Redinbo
*be
aDepartment of Pharmacology, University of North Carolina, Chapel Hill, North Carolina, USA
bDepartment of Chemistry, University of North Carolina, Chapel Hill, North Carolina, USA. E-mail: redinbo@unc.edu
cCenter for Integrative Chemical Biology and Drug Discovery, Division of Chemical Biology and Medicinal Chemistry, UNC Eshelman School of Pharmacy, University of North Carolina, Chapel Hill, North Carolina, USA
dDivision of Gastroenterology and Hepatology, Department of Medicine, Center for Gastrointestinal Biology and Disease, and the Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
eDepartment of Biochemistry and Biophysics, University of North Carolina, Chapel Hill, North Carolina, USA
fUNC Proteomics Core Facility, Department of Pharmacology, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
gDepartment of Bio-organic Synthesis, Leiden Institute of Chemistry, Leiden University, Leiden, The Netherlands
hDepartment of Pharmacological Sciences, Icahn School of Medicine at Mount Sinai, New York, NY, USA
First published on 16th July 2024
Abstract
The gut microbiome plays critical roles in human homeostasis, disease progression, and pharmacological efficacy through diverse metabolic pathways. Gut bacterial β-glucuronidase (GUS) enzymes reverse host phase 2 metabolism, in turn releasing active hormones and drugs that can be reabsorbed into systemic circulation to affect homeostasis and promote toxic side effects. The FMN-binding and loop 1 gut microbial GUS proteins have been shown to drive drug and toxin reactivation. Here we report the structure–activity relationships of two selective piperazine-containing bacterial GUS inhibitors. We explore the potency and mechanism of action of novel compounds using purified GUS enzymes and co-crystal structures. Our results establish the importance of the piperazine nitrogen placement and nucleophilicity as well as the presence of a cyclohexyl moiety appended to the aromatic core. Using these insights, we synthesized an improved microbial GUS inhibitor, UNC10206581, that potently inhibits both the FMN-binding and loop 1 GUS enzymes in the human gut microbiome, does not inhibit bovine GUS, and is non-toxic within a relevant dosing range. Kinetic analyses demonstrate that UNC10206581 undergoes a slow-binding and substrate-dependent mechanism of inhibition similar to that of the parent scaffolds. Finally, we show that UNC10206581 displays potent activity within the physiologically relevant systems of microbial cultures and human fecal protein lysates examined by metagenomic and metaproteomic methods. Together, these results highlight the discovery of more effective bacterial GUS inhibitors for the alleviation of microbe-mediated homeostatic dysregulation and drug toxicities and potential therapeutic development.
Introduction
The human gut microbiome is a collection of trillions of microbes that inhabit the human gastrointestinal (GI) tract and play significant roles in the metabolism of dietary components, endobiotics, xenobiotics, as well as the regulation of host immunity.1–3 Decades of research harnessing ‘omics’ techniques have allowed us to better understand which bacterial taxa are abundant within the human gut microbiome and how they may contribute to homeostasis and disease. Gut microbial taxa have been linked to variations in xenobiotic and hormone metabolism with important pharmacological and homeostatic implications.1–4 Specific microbial enzymes can alter the efficacy of chemotherapeutics and immunotherapy that may influence treatment outcomes during and after the FDA-approval process.5–8 The remarkable variability in the composition of the gut microbiota and the enzymes they produce has contributed to our incomplete understanding of its role and significance in human health and disease.9–11One such microbial enzyme family that has been directly connected to host homeostasis and chemotherapeutic efficacy are gut bacterial β-glucuronidases (GUSs). These proteins hydrolyze the glucuronic acid (GlcA) appended to hydrophobic molecules by human uridine diphosphate glucuronosyl transferases (UGTs) within the liver and other metabolic tissues. This phase 2 metabolic process driven by UGTs improves solubility by attaching a GlcA to promote subsequent detoxification and excretion (Fig. S1a, ESI†). After conjugation, many glucuronidated metabolites are sent to the gut for elimination. However, while in the intestines they encounter gut microbiota expressing structurally diverse GUS enzymes that are capable of removing the inactivating glucuronide, releasing the original compound that can be further metabolized, act locally within the intestinal lumen, or be reabsorbed into systemic circulation (Fig. S1a, ESI†).12 Such reactivation and reabsorption processes have the potential to impact homeostasis and drug pharmacology.
The gut microbial GUS enzyme family is structurally and functionally diverse. Numerous distinct oligomerization states and active site-gating loops enable GUS enzymes to process highly distinct substrates ranging from small molecule glucuronide conjugates to large polysaccharides.13–15 Based upon these loop regions and other structural features, GUS enzymes have been classified into eight distinct functional categories.15,16 While human fecal metagenomics data have defined the genes that encode for the full scope of GUS structural classes, this information has not been found to correlate with the GUS activities present in donor samples. However, metaproteomic data from the same samples have identified and quantified GUS protein levels that do correlate with substrate-specific activities, and these results have shown that loop 1 and FMN GUSs are the most relevant for small molecule glucuronide processing (Fig. S1b, ESI†).17 These two structural classes of bacterial GUS preferentially reactivate a range of xenobiotic and hormone glucuronides. For example, loop 1 GUS enzymes efficiently process the glucuronide conjugates of the active moiety of the chemotherapeutic irinotecan (SN-38), the consumer products toxin triclosan, and the non-steroidal anti-inflammatory drug (NSAID) diclofenac, in turn promoting GI toxicity and gut epithelial cell damage (Fig. 1A).6,18–21 Similarly, FMN GUS enzymes preferentially reactivate the immunosuppressant mycophenolate (MPA-G), the chemotherapeutic regorafenib, and also process triclosan-glucuronide (Fig. 1A).17,19,22 Thus, the development of a small molecule GUS inhibitor that is specific for and potent against both loop 1 and FMN GUS enzymes has been of considerable interest.
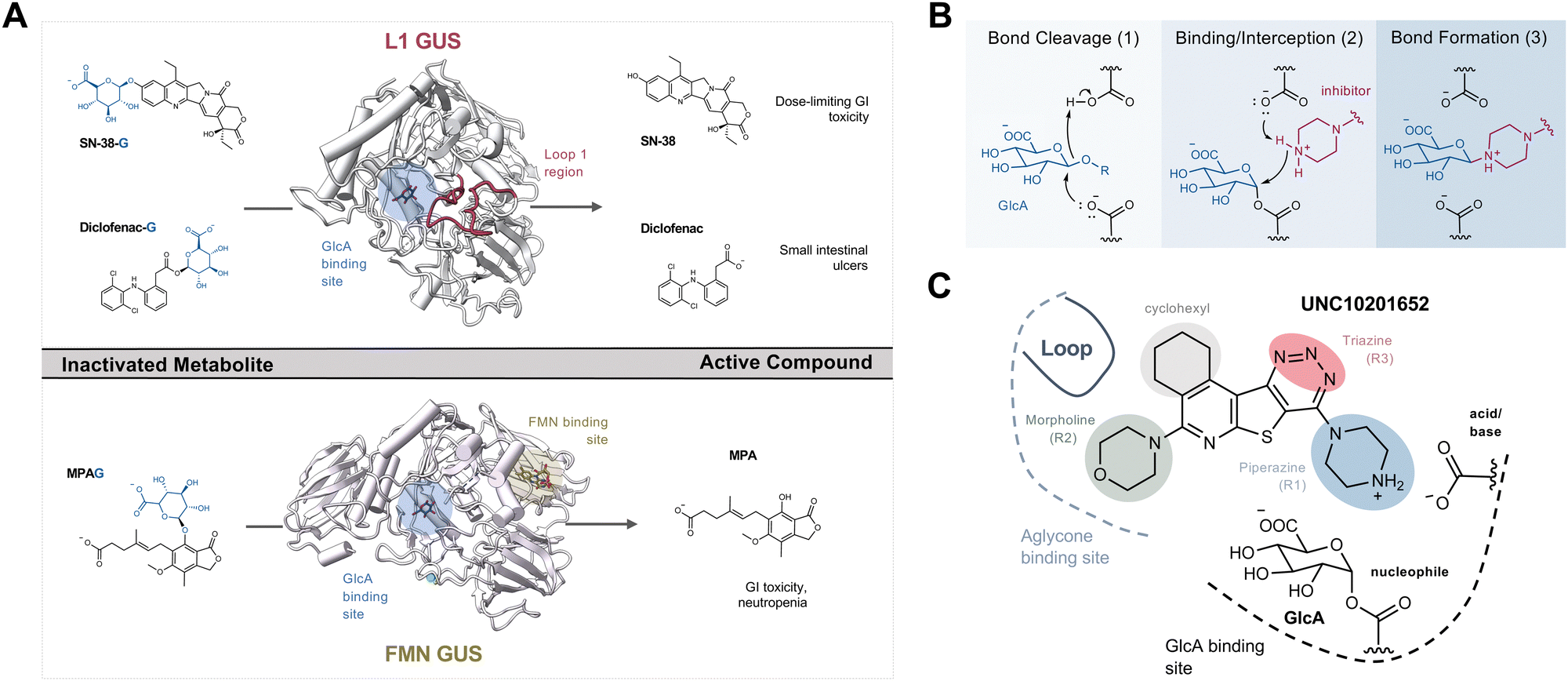 | ||
| Fig. 1 Metabolite reactivation by GUSs causes GI toxicity and extant inhibitors, their slow-binding mechanism, and binding pose. (A) Well-established examples of substrates that are reactivated in the gut by L1 and FMN GUS and cause GI toxicity as well as additional dose-limiting side effects. (B) Proposed mechanism of substrate-dependent catalytic interception by GUS inhibitors in the GlcA binding pocket. (C) Binding pose and highlighted moieties of UNC10201652 in the GlcA binding pocket of L1 GUS. Cp, Clostridium perfringens; FMN, flavin mononucleotide; GI, gastrointestinal; GlcA, glucuronic acid; L1, loop 1; MPA, active form of mycophenolate; MPAG, mycophenolate glucuronide; pNPG, 4-nitrophenyl-β-D-glucopyanoside; SN-38, active form of irinotecan; SN-38-G, irinotecan-glucuronide. | ||
The first generation of selective bacterial GUS inhibitors, (e.g., inhibitor 1, UNC10201652, and UNC4917) were identified via high throughput screening using the loop 1 GUS derived from E. coli and subsequent medicinal chemistry efforts.23,24 These inhibitors displayed selectivity for bacterial GUS and were non-toxic, setting the stage for non-antibiotic approaches to modulating the metabolic output of the gut microbiome. More recent efforts exploring UNC10201652 and UNC4917 defined a consistent mechanism of inhibition using kinetic and structural analysis, demonstrating a unique slow-binding and substrate-dependent mechanism via catalytic interception by the conserved piperazine moiety in each compound (Fig. 1B and C).23 Moreover, these GUS inhibitors have been shown to alleviate the GI toxic side effects of irinotecan in mice, which significantly enhances tumor regression by reducing dose-limiting intestinal damage.18
Here we discuss the structure–activity relationships (SAR) around UNC10201652 and UNC4917, which display the most promising non-specific loop 1 GUS inhibition, slow-binding kinetics, selectivity for microbial GUS enzymes, and efficacy in mouse models.18,23 We synthesized related analogs and measured their activities against distinct bacterial loop 1 GUSs that are present in the human gut microbiome. These studies validate that the piperazine is essential for potent bacterial GUS inhibition and show that any modification to this moiety reduces potency; however, modification to other regions of UNC10201652 and UNC4917 moderately improved potency and enabled improved pan-activity among bacterial GUS enzymes. Our most potent analogs efficiently inhibit GUS in E. coli cells without reducing cell viability while maintaining selectivity against closely related proteins. Of all analogs tested, UNC10206581 demonstrated the most favorable time-dependent properties and greatest efficacy against the loop 1 and FMN structural classes of GUS enzymes, which we show by proteomics are the most prevalent and abundant in human fecal samples. Finally, extant and novel crystal and co-crystal structures reveal key differences in GUS loop structures that dictate inhibitor potency and selectivity, further supporting the unique substrate-dependent slow-binding mechanism of these compounds. Together, our findings define the molecular features that enable potent inhibition of both the loop 1 and FMN gut microbial GUS enzymes and highlight several promising candidates for further tool compound and even therapeutic development.
Results
Piperazine moiety is essential for potent GUS inhibition
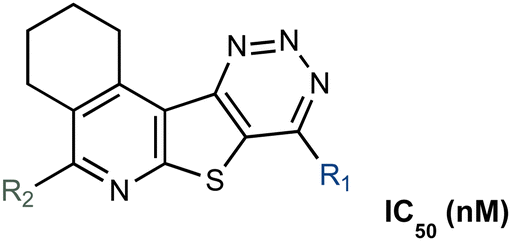
We embarked on an SAR campaign to better understand the structural requirements for inhibition and to determine what changes improve potency and/or alter selectivity against GUS enzymes. We modified the UNC10201652 (1) and UNC4917 (13) GUS inhibitor scaffolds at three key positions (R1, R2 and R3, Fig. 1C, Table 1 and Table S1, ESI†) in a systematic fashion. We assessed half-maximal inhibitory concentration (IC50) values for all synthesized analogs using a reporter substrate (p-nitrophenyl-β-D-glucuronide; pNPG) assay.14,23 Even within a structural class of GUS enzymes, we have observed that differences in residues immediately surrounding the active site and in neighboring loops contribute to differential substrate processing efficiencies and inhibitor binding. As we sought to understand the SAR of our new compounds in the context of extant co-crystal structures, we initially screened our inhibitors against a panel of loop 1 GUS enzymes. In particular, we assessed the inhibition of four loop 1 GUS enzymes representative of the human fecal microbiome that exhibit relatively low sequence identities: Escherichia coli GUS (Ec), Streptococcus agalactiae GUS (Sa), Clostridium perfringens GUS (Cp), and Eubacterium eligens GUS (Ee) (Fig. S2a, ESI†).23,25|
|
|||||||
|---|---|---|---|---|---|---|---|
| # | Name | R1 | R2 | Ec GUS | Ee GUS | Cp GUS | Sa GUS |
| Parent scaffold data is highlighted in bold. p-Nitrophenyl-β-D-glucuronide was used as a substrate. Cp, Clostridium perfringens, Ec, Escherichia coli; Ee, Eubacterium eligens GUS; Sa, Streptococcus agalactiae. | |||||||
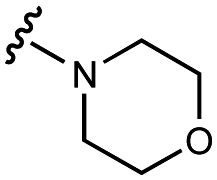
| 1 | UNC10201652 |
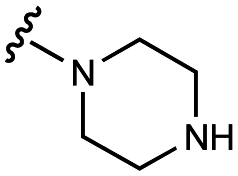
|
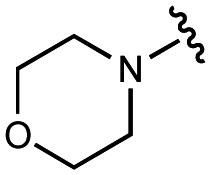
|
108 ± 8 | 127 ± 9 | 60 ± 10 | 600 ± 200 |
| 2 | UNC4510 |
|
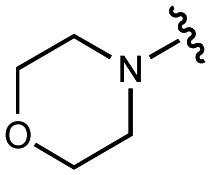
|
10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ± 2000 000 ± 2000 |
30000 ± 2000 |
5000 ± 1000 | 14000 ± 1000 |
| 3 | UNC4601 |
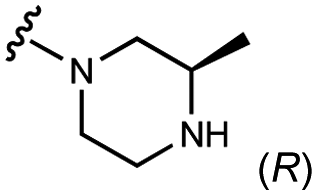
|
|
800 ± 100 | 820 ± 40 | 900 ± 100 | 2400 ± 400 |
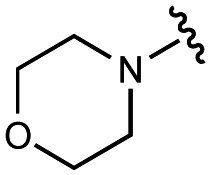
| 4 | UNC4684 |
|
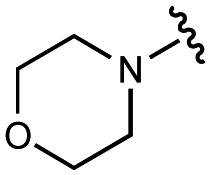
|
>100000 |
>100000 |
>100000 |
1900 ± 400 |
| 5 | UNC4511 |
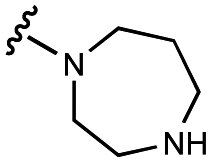
|
|
1200 ± 800 | 3800 ± 300 | 2000 ± 100 | 11200 ± 800 |
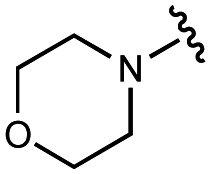
| 6 | UNC4540 |
|
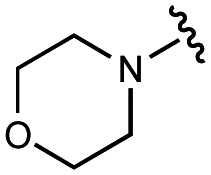
|
2600 ± 400 | 21000 ± 1000 |
3200 ± 600 | 11000 ± 300 |
| 7 | UNC4351 |
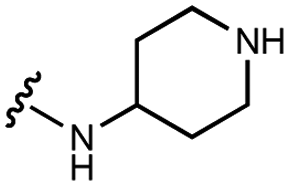
|
|
10200 ± 800 |
8000 ± 1000 | 6000 ± 1000 | 40000 ± 10000 |
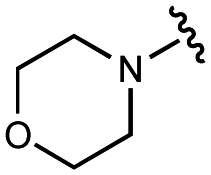
| 8 | UNC4365 |
|
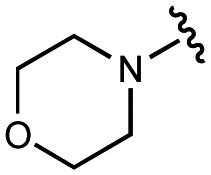
|
>100000 |
>100000 |
>100000 |
>100000 |
| 9 | UNC10201651 |
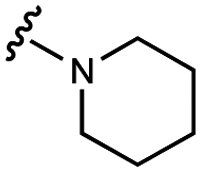
|
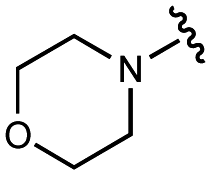
|
>100000 |
>100000 |
>100000 |
>100000 |
| 10 | UNC10206579 |
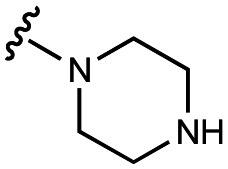
|
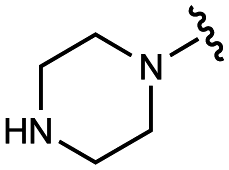
|
100 ± 20 | 123 ± 4 | 32 ± 2 | 300 ± 30 |
| 11 | UNC10206577 |
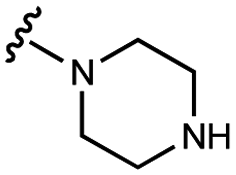
|
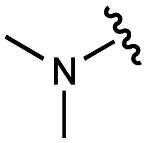
|
70 ± 10 | 118 ± 4 | 49 ± 2 | 700 ± 90 |
| 12 | UNC10206581 |
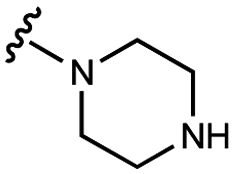
|
|
29 ± 6 | 88 ± 8 | 34 ± 2 | 770 ± 90 |
An extant co-crystal structure of the parent compound, UNC10201652 (1), bound to Ee GUS reveals GlcA within the active site as an N-linked adduct to the piperazine (Fig. 2A and B, PDB 8GEN). This covalent interaction between the substrate (GlcA) and the nucleophilic secondary amine of the piperazine emphasizes its importance for GUS inhibition. Thus, we first synthesized analogs with different cyclic amines and piperazine-like functional groups to better understand the limits of this covalent bond formation. We found that the addition of steric bulk to the piperazine ring proved to negatively impact GUS inhibition. When a methyl was added to the secondary amine (UNC4510, 2) or the C3 position of the piperazine (UNC4601, 3 and UNC4684, 4), significant reductions in inhibitor potency was observed across our panel of L1 GUSs (Table 1). Interestingly, the stereochemistry of the methyl significantly impacts the degree of inhibition, with the (R) enantiomer (3) retaining sub-micromolar potency against three of the four enzymes whereas the (S) enantiomer (4) displayed a complete loss of inhibitory potential against nearly all loop 1 GUS enzymes. This stereospecificity can potentially be explained by the proximity of the piperazine to one of the catalytic glutamates (E425 in Ee GUS), a key stabilizing contact. The enantiomers may position the methyl towards nearby E425, causing a steric clash, disrupting this critical interaction, and leading to reduced potency (Fig. 2B and Fig. S2b, ESI†).
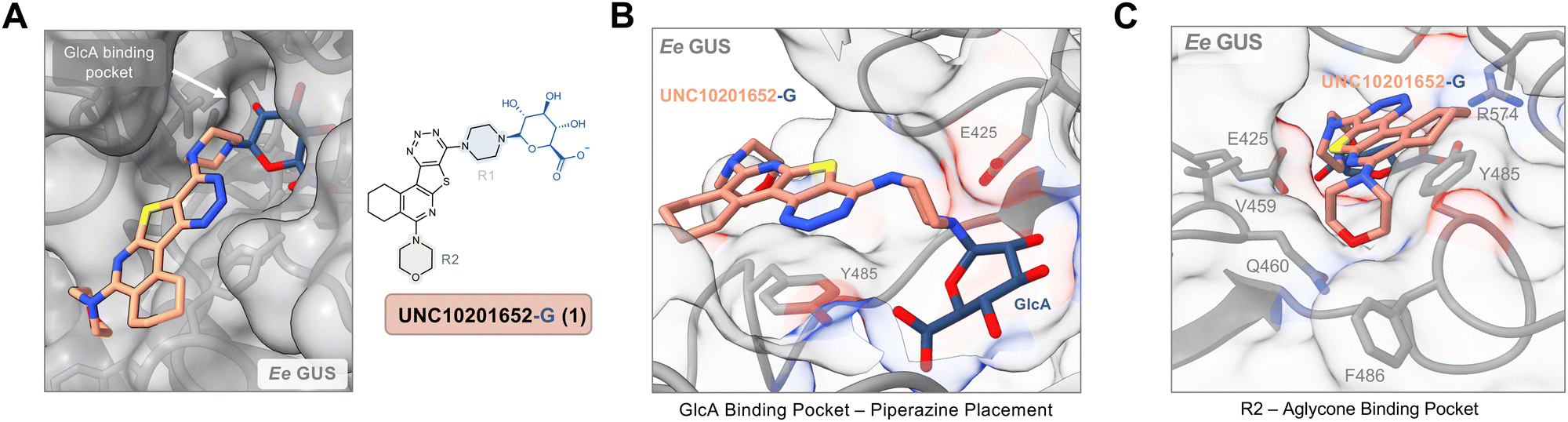 | ||
| Fig. 2 UNC10201652 co-crystal structures with L1 GUSs reveal key contacts and rationalize SAR. (A) Structure and binding pose of UNC10201652-G in the GlcA binding pocket of Ee GUS. The inhibitor is shown in peach while the glucuronic acid is rendered in blue (PDB 8GEN). (B) GlcA binding pocket highlighting the binding pose and interactions of the glucuronic acid and piperazine (R1) conjugate of UNC10201652-G (PDB 8GEN). (C) Shallow groove within the aglycone pocket region of Ee GUS highlighting the placement of the morpholine substituent (R2) of UNC10201652-G (PDB 8GEN). Ee, Eubacterium eligens; GlcA, glucuronic acid. | ||
Furthermore, altering the nitrogen position through expansion of the ring (UNC4511, 5), increasing distance from the core (UNC4351, 7), or replacing the piperazine with a more flexible ethyl amine (UNC4540, 6), significantly reduced potency across our panel of GUSs. This may be attributed to poor placement of the basic nitrogen within the active site leading to weaker interactions with the catalytic glutamate or a steric clash with GlcA (Fig. 2B). Lastly, when the nucleophilic nitrogen is replaced by oxygen or carbon (UNC4365, 8 and UNC10201651, 9, respectively), inhibitory activity is abolished. Taken together, inhibitor potency is reduced when the position of the nucleophilic nitrogen in UNC10201652 (1) is altered or removed (Table 1).
Aglycone binding pocket is tolerant of different functional groups
We next explored modifications of the morpholine group of UNC10201652 (1) (Table 1). Replacing the morpholine with a piperazine (UNC10206579, 10) moderately improved inhibition (2-fold) for two loop 1 GUS enzymes, Cp and Sa GUS. When the morpholine is reduced in size to a dimethyl amine (UNC10206577, 11) or monomethyl amine (UNC10206581, 12), slight improvements in potency against Ee, Ec, and Cp GUS were also observed (Table 1).We next shifted to explore modifications to the UNC4917 (13) scaffold while leaving the piperazine unchanged given its importance in UNC10201652 (1) (Table S1, ESI†). Similar to what was observed with analogs 10–12 in Table 1, we found that substitution at R2 with a morpholine (UNC4785, 17), phenyl (UNC4708, 19), and hydrogen (UNC4910, 23) were well tolerated and resulted in minimal changes in potency (Table S1, ESI†). The co-crystal structure of Ee GUS bound to UNC10201652 and aligned with other L1 GUSs reveals a shallow groove surrounding the R2 morpholine, no significant stabilizing contacts, and differences in residue sidechain size and polarity (Fig. 2C and Fig. S2c, ESI†). The difference in sidechain physiochemical properties and the shallow nature of the groove likely makes targeting this protein region difficult to improve potency towards L1 GUS enzymes. Moreover, the GUS loop 1 region is localized at a dimer interface in the quaternary structure and reaches into the neighboring aglycone binding pocket to form stabilizing interactions with substrates and inhibitors. The sequence of this loop region can contribute to enzyme–inhibitor interactions unique to specific GUS enzymes within the same structural class (Fig. S3a and b, ESI†). These results demonstrate that GUS enzymes from the same class can form differential contacts with similar inhibitor scaffolds leading to changes in potency.
UNC4917 analogs demonstrate conserved activity against loop 1 GUS enzymes
Lastly, we explored changes to R3 (Table S1, ESI†) and found that replacement of the R3 monomethyl amine of UNC4708 (19) with a tertiary dimethyl amine (UNC4707, 20), ethanol amine (UNC4847, 21), or hydrogen (UNC4600, 18) resulted in minimal changes in GUS inhibition. Additionally, the incorporation of the triazole of UNC10201652 (1) into this scaffold results in minimal changes in inhibition (UNC4764, 14 and UNC4830, 15), further supporting the conclusion that a functional group at R3 is not required.Taken together, the analogs described in Table 1 and Table S1 (ESI†) reveal that the position and nucleophilicity of the piperazine amine is critical to potent inhibition of gut microbial GUS enzymes. In contrast, there is considerably more tolerance for modifications at other positions, as changes to R2 and R3 resulted in only modest impacts on GUS inhibition. Finally, UNC4917 (13) and related analogs generally demonstrated more potent inhibition of Ec GUS and Cp GUS over Ee GUS and Sa GUS, suggesting that loss of the cyclohexyl ring in UNC10201652 (1) may alter the selectivity profile of these compounds.
Lead inhibitors selectively inhibit bacterial GUS enzymes without reducing cell viability
We selected three compounds from our SAR studies, UNC10206579 (10), UNC10206581 (12), and UNC4707 (20), along with the parent compound (UNC10201652, 1) to assess in cell-based experiments. These molecules were chosen due to their improvements in potency, pan-inhibition against our panel of loop 1 GUSs, and structural diversity. We cultured E. coli K12 MG1655, a GUS-expressing strain, and performed cell-based assays to examine GUS inhibition (Fig. 3A). Strong target engagement was observed in culture, recapitulating the IC50 values of these compounds against recombinant Ec GUS, with UNC4707 (20) and UNC10206581 (12) displaying the greatest in-cell GUS inhibition at 7 and 6 nM, respectively. To confirm that inhibition observed in E. coli was due to selective targeting of GUS and not cellular toxicity, we performed microbial growth kinetic assays in the presence of 10 μM of our parent compound (1) and lead analogs (10, 12, 20) (Fig. 3B). None of the compounds tested reduced or impacted microbial growth kinetics, indicating strong tolerability and selective targeting of GUS enzymes in cells.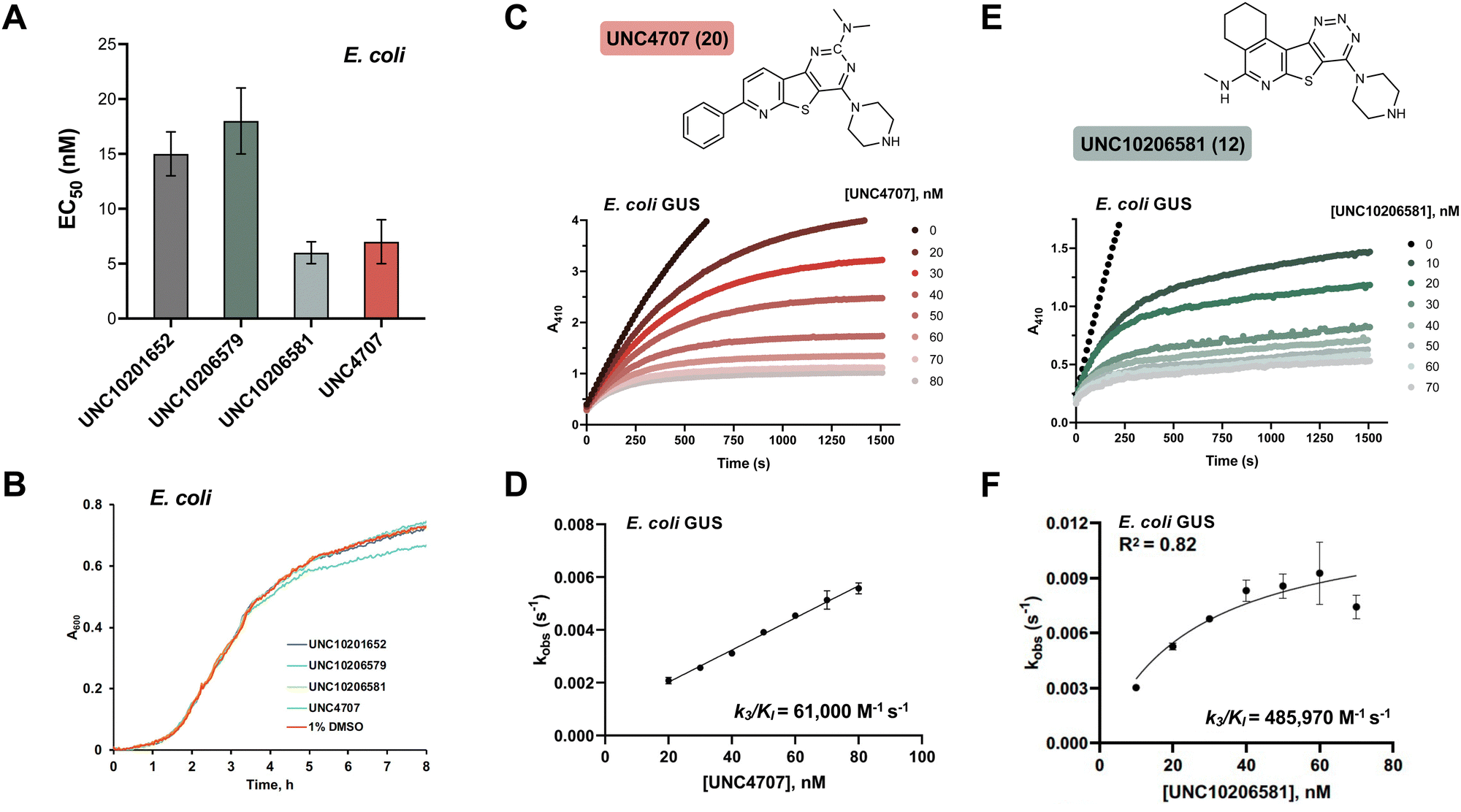 | ||
| Fig. 3 Top 3 analogs inhibit GUS in microbial culture without displaying toxicity and kinetic assays identify UNC10206581 as the most potent slow-binding inhibitor. (A) EC50 assays using pNPG as a substrate reveal successful GUS inhibition in E. coli culture for the top 3 most potent and non-specific analogs of the SAR campaign. Error bars represent mean ± SEM from n = 3 independent experiments. (B) Growth kinetics assay in E. coli culture show no toxicity of GUS inhibitors at a concentration of 10 μM. (C) Non-linear inhibitory progress curves of UNC4707 at various concentrations using pNPG as a substrate. (D) Secondary kobsvs. [UNC4707] plots against Ec GUS confirms a substrate-dependent and one-step, slow-binding mechanism of inhibition. Error bars represent mean ± SEM from n = 3 independent experiments. (E) Non-linear inhibitory progress curves of UNC10206581 at various concentrations using pNPG as a substrate. (F) Secondary kobsvs. [UNC10206581] plots against Ec GUS confirms a substrate-dependent and one-step, slow-binding mechanism of inhibition. Error bars represent mean ± SEM from n = 3 independent experiments. pNPG, p-nitrophenyl-β-D-glucuronide; E. coli, Escherichia coli. PNPG, 4-nitrophenyl-β-D-glucopyanoside; SAR, structure–activity relationship. | ||
Humans express a GUS ortholog involved in lysosomal processing and mutations to this gene cause Sly Syndrome (Mucopolysaccharidosis type 7), a lysosomal storage disease that impacts development, often causing skeletal abnormalities and intellectual disabilities.26 To gauge selectivity for microbial GUS over a mammalian ortholog, we screened our lead inhibitors against purified bovine GUS. Neither the parent compound nor lead inhibitors (UNC10296579, 10; UNC10206581, 12; and UNC4707, 20) reduced the activity of bovine GUS at a concentration of 10 μM (Fig. S4a, ESI†). Moreover, all compounds showed limited inhibition of E. coli β-galactosidase (β-gal), a closely related glycoside hydrolase (Fig. S4b, ESI†). While nearly 25% inhibition of Ec β-gal was observed at 10 μM inhibitor concentrations, 10 μM is greater than 500-times the concentration that inhibition was observed in cultures of E. coli (6–18 nM, Fig. 3A). Therefore, lead inhibitors do not reduce the activity of a eukaryotic GUS ortholog and have limited effects on a related microbial glycoside hydrolase enzyme.
Cellular toxicity, cellular inhibition, and selectivity data indicate that UNC10206581 (12) and UNC4707 (20) are promising inhibitors for further evaluation. However, as IC50 and classical potency assessments do not account for time-dependent and covalent mechanisms of inhibition, we next sought to more thoroughly assess enzyme binding affinity. Kobsvs. [inhibitor] plots were generated from the non-linear inhibitory progress curves to extrapolate kinetic parameters such as k3/KI and to assess curve fit, enabling us to better identify lead candidates with favorable time-dependent inhibition. As expected, both compounds produced non-linear progress curves, indicating a slow-binding mechanism as previously observed for the parent scaffolds of these piperazine-containing analogs (Fig. 3C and E).23k3/KI values derived from secondary plots of kobsvs. [inhibitor] reveal UNC10206581 (12) as notably more potent, with a value of 485970 M−1 s−1, approximately 8-fold greater than UNC4707 (20) (Fig. 3D and F). The greater k3/KI while maintaining a comparable IC50 value suggests that the initial binding interaction of both inhibitors with GUS is similar; however, formation of the GlcA-inhibitor conjugate is significantly faster for UNC10206581 (12). As potent inhibition is achieved once the substrate-dependent and slow-binding mechanism ensues, these kinetic studies reveal UNC10206581 (12) is a more promising inhibitor.
We next sought to examine the molecular details of bacterial GUS inhibition by UNC10206581 (12). We resolved a co-crystal structure of UNC10206581-GlcA bound to Ee GUS (Table S2, PDB 8UGT, ESI†), which reveals several analogous contacts to those observed with the parent scaffold, UNC10201652 (1). Indeed, the piperazine forms a conjugate with GlcA and is stabilized by the catalytic residue E425, π–π stacking between the UNC10206581 aromatic core and Y485, and the R2 amine is seated in a shallow groove within the aglycone binding site (Fig. 4A). However, when comparing this structure to that of Ee GUS and UNC10201652-GlcA, there is a slight difference in 3-dimensional positioning of the aromatic core and piperazine moiety, likely driven by the accommodation of UNC10201652s (1) morpholine moiety (Fig. 4A).
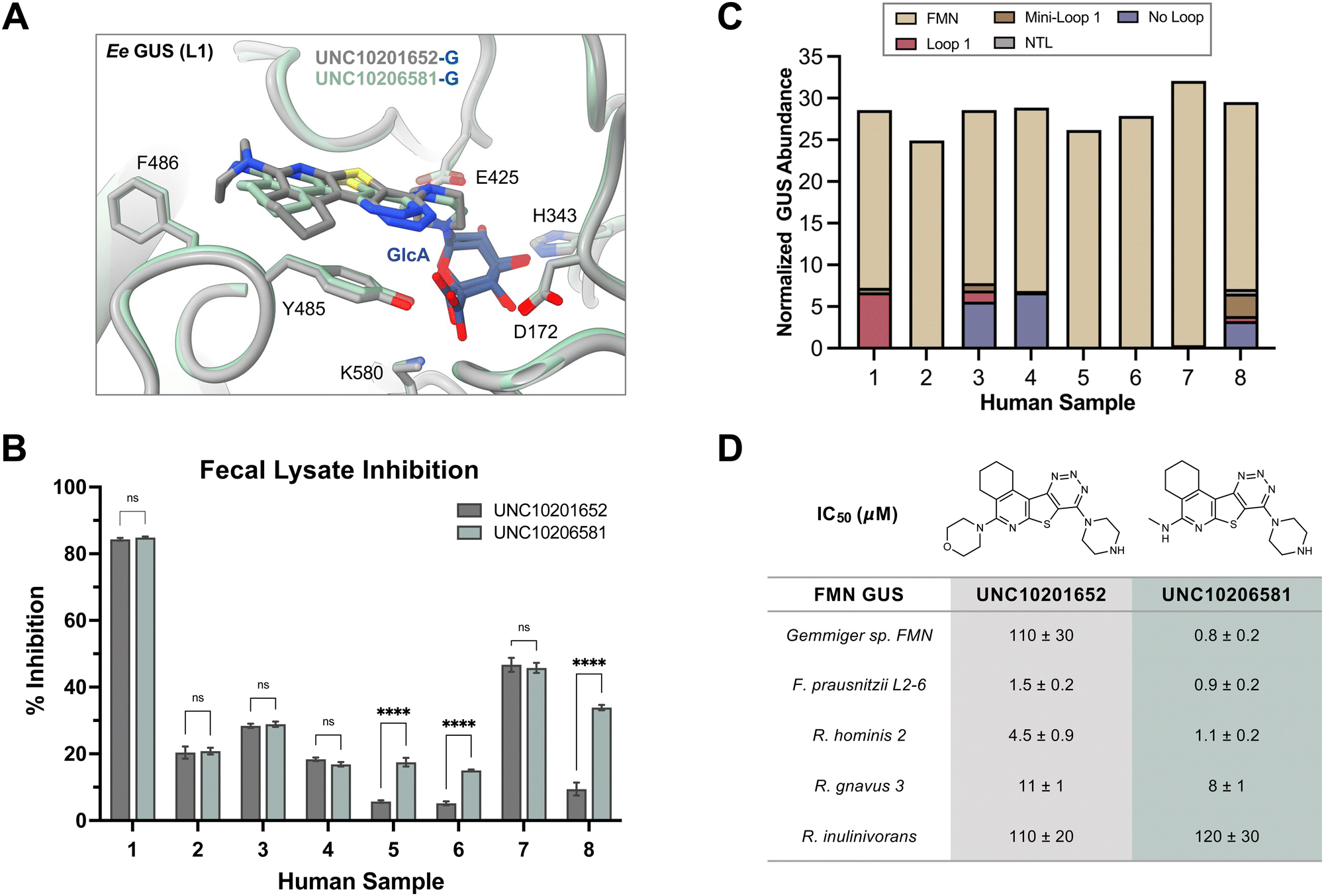 | ||
| Fig. 4 UNC10206581 is the most potent GUS inhibitor within human fecal lysates and against FMN GUSs. (A) Aligned co-crystal structures of Ee GUS with UNC10201652-G and UNC10206581-G reveals very similar binding pose and key stabilizing contacts within the GlcA and aglycone binding sites. A slight displacement in the aromatic core and positioning of R2 is observed to provide favorable seating of the parent scaffold morpholine. (PDB 8GEN and 8UGT). (B) Inhibition data for UNC10201652 and UNC10206581 in 8 human fecal samples reveals UNC10206581 as the superior GUS inhibitor in physiologically relevant samples. 4MU-G was used as the substrate. Two-way ANOVA with multiple comparison (mean ± SEM), n = 3, ****p < 0.0001, ns = not significant. (C) Abundance and loop class distribution of GUS enzymes found in 8 human fecal samples using probe-enabled proteomics reveal FMN GUSs as the most abundant across this cohort. (D) Inhibitory data of the parent scaffold and UNC10206581 against a panel of FMN GUSs reveals activity of both compounds against this loop class. 4MU-G, 4-methylumbelliferyl-β-D-glucuronide; FMN, flavin mononucleotide; GlcA, glucuronic acid; L1, loop 1. Fp2, Faecalibacterium prausnitzii L2-6; Rg3, Ruminococcus gnavus; Rh3, Ruminococcus hominis; Ri, Ruminococcus inulinivorans; GemFMN, Gemmiger sp. | ||
UNC10206581 inhibits GUS enzymes in fecal samples
To assess efficacy in a more physiologically relevant context, we tested GUS inhibition by our initial (1) and lead (12) compounds in human fecal extracts using in fimo reaction profiling.27 Eight fecal samples from healthy individuals were processed into protein lysates and percent inhibition assays were performed using 4-methylumbelliferyl-β-D-glucuronide (4MU-G) as a substrate and a concentration of 10 μM for both UNC10201652 (1) and UNC10206581 (12) (Fig. 4B). 4MU-G was used as the substrate in fecal extracts rather than PNPG because the fluorescence-based readout allows for clean signal detection within complex samples. Both PNPG and 4MU-G are pan-substrates that are processed by GUSs from all structural classes and are often used interchangeably to monitor total GUS activity. UNC10206581 (12) potently inhibited GUS activity in fecal lysates with statistically significant improvements in inhibition over compound 1 in three samples. Complete inhibition was not observed for any of the human fecal lysates; however, this result was anticipated as all structural classes of GUS can process 4-MUG and fecal lysates contain multiple GUSs from various structural classes.To better understand the distribution of bacterial GUSs present within each of the eight samples, probe-enabled proteomics was employed to identify and quantitate GUS abundance, as previously described.17,21 Proteins in five of the eight structural classes were identified, with FMN GUS showing the broadest presence and highest abundance across the cohort (Fig. 4C). No loop and loop 1 GUSs were the next most abundant but only FMN GUS was detected in all samples. Taken together, our findings indicate that UNC10206581 (12) is a potent and broad-acting inhibitor of bacterial GUS in physiologically relevant samples primarily containing FMN and loop 1 GUS enzymes.
UNC10206581 inhibits FMN GUS enzymes
Because we found that FMN GUS proteins were abundant in the human fecal samples inhibited by our compounds, we sought to better understand inhibitory activity against the FMN GUS loop class. We selected five FMN GUSs that were the most prevalent across our eight fecal samples and representative of diverse taxa: Gemmiger sp. (GemFMN), Faecalibacterium prausnitzii L2-6 (Fp2), Ruminococcus hominis (Rh2), Ruminococcus gnavus (Rg3), and Ruminococcus inulinivorans (Ri). These proteins were expressed recombinantly in E. coli, purified, and then used in the same inhibitory GUS assays as the L1 GUSs. UNC10206581 (12) displayed similar potency against the FMN GUSs compared to UNC10201652 (1) with the exception of GemFMN GUS, which was inhibited by UNC10206581 (12) 135-fold more potently than UNC10201652 (1) (Fig. 4D). A wide range in inhibitory activity was observed for the FNM GUS panel, from a relatively weak IC50 of 120 μM for Ri to high nanomolar inhibition against Fp (Fig. 4D).To rationalize these differences in inhibitor potency across FMN GUS, we compared existing structures (Rh2, Rg3, Fp2) with AlphaFold models of GemFMN and Ri GUS aligned to the Ee GUS UNC10206581-GlcA co-crystal structure. We found that nearly all binding site contacts were conserved for both FMN and loop 1 GUSs, although Ri GUS has a leucine (L431) in place of the glycine conserved for all other proteins (Fig. S5a, ESI†). The Ri GUS L431 is positioned in the R2-accomodating groove of the aglycone binding site and thus is expected to generate a steric clash with both UNC10201652 (1) and UNC10206581 (12) (Fig. S5a, ESI†). This feature may contribute to the reduction in potency of both compounds for Ri GUS compared to all other FMN GUSs assayed (Fig. 4D).
Further analysis of our proteomic data revealed that GemFMN was the most prevalent FMN GUS across our cohort. It was detected in 7 of 8 samples and was the most abundant GUS in 5 of 8 samples. The second most abundant GUS across the cohort was another FMN GUS originating from Roseburia spp. AM16-25. While this enzyme was only 68% identical to the R. hominis 2 GUS examined in our recombinant panel (Fig. 4D), the AlphaFold structure of AM16-25 GUS is remarkably similar to Rh2 GUS (RMSD of 0.4 Å across Cα positions; Fig. S5b, ESI†). These results suggest the proteomic Roseburia protein is functionally comparable to the Rh2 GUS examined in our in vitro panel and may be inhibited by UNC10206581.
In the context of our human cohort, we observed GemFMN to be the only FMN GUS present within donors 5 and 6. Donor 8 contained both GemFMN GUS and the Roseburia AM16-25 GUS, with the abundance of GemFMN ∼2.5-fold higher than that of AM16-25. These findings provide a rationale for the improved GUS inhibition by UNC10206581 (12) in fecal lysates from donors 5, 6, and 8. Additionally, these results indicate that our in vitro explorations in Fig. 4D are representative of the GUS proteins present in these human samples. In summary, FMN GUS are subject to potent inhibition by compounds like UNC10206581 (12), suggesting potential as a therapeutic adjuvant to address microbiome-related disruptions in homeostasis and drug-induced toxicities.
SiteMap analysis of GlcA pockets reveal minimal optimization opportunities
Because UNC10206581 (12) displayed improved potency toward some but not all L1 and FMN GUSs, we sought to better understand the binding pocket properties across protein isoforms to inform further inhibitor optimization. We leveraged the wealth of GUS structures available to perform SiteMap analysis using FMN, L1, and human GUS structures to quantitate the surface area and physiochemical properties of the active site (Fig. S6A, ESI†). These analyses help demonstrate the considerable variability present in these enzymes. FMN GUSs have small pockets, averaging 853 Å3 between the three resolved structures (Rg3, Rh2, Fp2), not extending much further beyond the space occupied by UNC10206581 (Fig. S6B, ESI†). Thus, there appears to be limited opportunities to improve potency because expansion off our most potent compound will lead into solvent. While L1 GUSs have larger binding pocket surfaces (average 1829 Å3) with more opportunities for stabilizing interactions, there are large sequence and physiochemical property variabilities in these regions, making it difficult to maintain or improve potency across all enzyme isoforms. Additionally, the active site gating loops are often dynamic and not resolved in crystal structures, limiting our visualization into these regions and ability to specifically engage them. Taken together, these structural and functional analyses indicate that further functionalization of the R2 and R3 positions would not be expected to improve potency for all FMN and L1 GUS enzymes.Conclusion
Metabolic pathways catalyzed by the gut microbiota can transform drugs and dietary compounds, and subsequently impact metabolite availability and potentially host physiology. While numerous microbes have been identified as disease-associated, disruptive of homeostatic functions, and influential in pharmacological efficacy, in many cases the specific proteins involved are not known. Gut microbial GUS represent a bona fide example of a family of microbial enzymes that can disrupt homeostatic metabolism and pharmacological efficacy.6,17–19,28 Despite their established roles in host–microbial interactions, a comprehensive scope of activities for the hundreds of GUS orthologs present in the human gut microbiome and their differential roles in homeostasis and disease has not been defined.Two structural classes of GUS, loop 1 and FMN, are the most efficient at metabolizing small molecule glucuronide-conjugates, like those of hormones and xenobiotics.14,15,17,19,22 Loop 1 GUSs have been well-characterized as driving the reactivation of irinotecan glucuronide (SN-38-G) and non-steroidal anti-inflammatory drug (NSAID) glucuronides, both of which are inactivated via glucuronidation by host UGTs. The subsequent reactivation of such drugs by microbial GUS leads to severe GI toxicity, small intestinal ulcers, poor surgical outcomes, and reduced drug efficacy.6,18,20 Recent studies have also shown that another structural class, the FMN GUS enzymes, play a significant role in reactivating the glucuronides of mycophenolate (MPA), triclosan, and regorafenib,17,19,22 leading to similar GI toxicities. While both loop 1 and FMN GUS enzymes generally work well with small molecule glucuronides, distinct substrate preferences as well as microbial compositional differences may influence disease and treatment outcomes with specific drugs and toxins. Therefore, the development of a small molecule GUS inhibitor that is specific for and potent against both loop 1 and FMN GUS enzymes has been of considerable interest.
To date, potent and non-toxic GUS inhibitors have been generated that are selective for microbial GUS over human GUS. The majority of these GUS inhibitors are specific for loop 1 GUS over other structural classes, and co-administration of these compounds with irinotecan, the clinical prodrug of SN-38, alleviated GI toxicity and significantly improved antitumor efficacy.6,18 Here, we sought to improve the potency of these inhibitors against loop 1 GUS and to examine their potential to inhibit other gut microbial GUS structural classes.
We subjected two known GUS inhibitors, UNC10201652 (1) and UNC4917 (13), to a focused medicinal chemistry campaign to define critical pharmacophores and better understand structure–activity relationships. As expected, altering the piperazine of these scaffolds causes moderate to significant reductions in potency, as the piperazine secondary amine must be correctly positioned for covalent reaction with the substrate (Table 1).23 Additionally, inclusion of the cyclohexyl ring in UNC10201652 (1) and related analogs both improved activity against our panel of loop 1 GUSs and potently inhibited a greater portion of the loop 1 panel compared to analogs of UNC4917 (13). Lastly, alteration of moieties at the R2 and R3 positions displayed in Table 1 and Table S1 (ESI†) reveal little to no changes in potency, indicating that functionalization at these positions does not improve ligand efficiency.
Cellular studies reveal that UNC4707 (20) and UNC10206581 (12) are efficacious, selective for microbial GUS enzymes, and non-toxic. While the IC50 and EC50 values of the lead compounds UNC4707 (20) and UNC10206581 (12) may be comparable, indicating sufficient permeability into microbes, the k3/KI reveal disparate slow-binding and covalent properties. Similar to the parent scaffolds,23 both compounds produced non-linear progress curves that are indicative of a slow-binding mechanism (Fig. 3C and E). These results corroborate our conserved mechanism of catalytic interception and formation of a piperazine-GlcA conjugate (Fig. 1C). Plots demonstrating kobsvs. [inhibitor] from the non-linear progress curves of UNC10206581 (12) and UNC4707 (20) reveal ∼8-fold disparity in k3/KI values between the compounds. Thus, UNC10206581 (12) has more favorable time-dependent inhibition properties (Fig. 3D and F). While the initial binding event between the inhibitors and GUSs are similarly favorable, the rate of formation of the GlcA conjugate varies significantly between these two compounds and the improved formation of the inhibitor-GlcA conjugate with UNC10206581 (12) results in more potent inhibition.
Previous studies that explored GUS inhibition have attributed the selectivity of inhibitors such as UNC10201652 (1) for bacterial over human GUS to the presence of the loop 1 region.6 Here we show this potent inhibition extends to FMN GUS enzymes, which do not contain a loop 1 or 2 region. FMN GUSs contain a C-terminal domain (CTD) that is modeled to be placed adjacent to the GlcA binding site (Fig. S5c, ESI†). It is likely this domain behaves similarly to the loop 1 region by closing over the active site to improve substrate specificity as well as inhibitor stabilization. This may explain why our piperazine-containing GUS inhibitors display activity for FMN and loop 1 GUS yet maintain selectivity against bovine GUS which lacks these bacterial loops and CTD.
The variability in L1 region and CTD also likely play a large role in the differences observed in inhibitor potency. Crystal structures of L1 GUSs show remarkable conservation in the GlcA and aglycone binding sites, with key protein–ligand interactions consistent across protein isoforms. However, the L1 regions are often too dynamic to be resolved within crystal structures. As such, we have an incomplete understanding of the specific interactions made between L1 residues and inhibitors. Furthermore, the amino acid sequences and physiochemical properties of L1 regions vary greatly between protein isoforms and may explain the differences in potency we have observed between Ec, Ee, Cp, and Sa GUS (Table 1 and Fig. S3, ESI†). Our medicinal chemistry efforts have been successful in significantly improving potency against Ec and Ee GUS, moderately improving potency for Cp GUS, but we have not improved potency for Sa GUS. Until these L1–inhibitor interactions and protein regions are better understood, we appear to be limited in our ability to improve potency against all L1 GUS isoforms, specifically Sa GUS, with the current chemical series.
When tested in human fecal lysates, UNC10206581 (12) displayed improved GUS inhibition in 3 of 8 samples compared to the parent compound (Fig. 4B). Though complete GUS inhibition was not observed, this can be explained by the presence of gut microbial GUS enzymes of structural classes beyond loop 1 and FMN. Lysates from all eight samples efficiently process a pan-GUS reporter substrate but many are not inhibited by these loop 1- and FMN-targeting inhibitors at a concentration of 10 μM. Indeed, our IC50 data of UNC10201652 (1) and UNC10206581 (12) reveal a wide range in inhibitor potency across diverse FMN GUS enzymes ranging from high nanomolar to 120 μM (Fig. 4C). However, we show that inhibition by UNC10206581 (12) extends to FMN GUS and exhibits the most efficient substrate-dependent slow-binding inhibition kinetics, indicating that (12) is a promising candidate for therapeutic use.
Bacterial GUS enzymes within a particular structural class can drive the reactivation of specific metabolites that may be implicated in altered homeostasis or drug-induced toxicity.17,18 For each independent metabolite and disease state, inhibitor efficacy may vary due to the vast structural and functional differences in GUSs, even those within the same structural class. Here, we show for the first time that both loop 1 and FMN GUSs can be potently inhibited by the same slow-binding and substrate-dependent inhibitor. While the implications of GUS inhibition within human fecal lysates warrants more extensive investigation, we show that UNC10206581 (12) demonstrates significant potential as a candidate to inhibit the GUS enzymes driving the gut toxicity associated with several xenobiotics.
Gut microbial GUSs can disrupt homeostasis and alter drug efficacy by reversing host phase 2 metabolism, releasing various small-molecule drugs and hormones into the gut and bloodstream to cause undesirable side effects. In this study, we investigate the SAR of extant piperazine-containing inhibitors developed to specifically target bacterial GUS enzymes. We establish that piperazine nucleophilicity and increased core hydrophobicity contribute to more potent GUS inhibition. We develop UNC10206581 (12), which selectively targets microbial GUS enzymes of both the loop 1 and FMN GUS structural classes via a slow-binding mechanism similar to the parent scaffolds. Furthermore, UNC10206581 (12) potently inhibits bacterial GUS enzymes in microbial cultures and human fecal protein lysates, suggesting potential as a therapeutic adjuvant to address microbial-induced disruptions in homeostasis and drug-related toxicities.
Materials & methods
Protein expression and purification
All proteins were purified as previously described.15,16 In brief, proteins were expressed recombinantly as N-terminal His-tag fusions in E. coli (BL21-G) cells and purified by both Ni-affinity chromatography and size exclusion chromatography. All proteins displayed at least 95% purity by SDS-PAGE and were snap frozen in liquid nitrogen and stored at −80 °C.In vitro GUS inhibition assay
p-Nitrophenyl-β-D-glucuronide (pNPG) was purchased as a solid (Sigma Aldrich) and resuspended in water to a concentration of 50 mM. Inhibitors were synthesized and suspended in 100% DMSO at various concentrations above 20 mM. Each inhibitor was then diluted in ddH2O and an equivalent % DMSO for all final concentrations. Assays were conducted in Costar half-area 96-well assay plates at a total volume of 50 μL. The reaction consisted of 10 μL assay buffer (125 mM HEPES, 125 mM NaCl, pH 6.5), 5 μL of purified GUS enzyme (15 nM of Ec, Sa, Cp, Ee, Rh2, Rg3, FPL2-6, Gemmiger FMN, and 60 nM final of Ri; see Results for species names), 5 μL inhibitor (at various concentrations), and 30 μL pNPG (900 μM final). After the addition of inhibitor, the reaction was incubated for 5 minutes at 37 °C, then initiated by the addition of pNPG and incubated at 37 °C for 1 hour. The reaction was quenched with 50 μL sodium carbonate (0.2 M). Analysis of the reaction mixture was performed using a CLARIOstar Plus Microplate Reader (BMG Lab Tech), measuring the absorbance of PNP at 410 nm. Resultant data were converted to percent inhibition and fit with a 4-parameter logistic function in Graphpad Prism for determination of the IC50 values.29Slow-binding continuous kinetic assay
The same procedure as outlined for the in vitro GUS inhibition assay was followed for reaction volumes and concentrations. Product formation was monitored continuously at 410 nm in a BMG lab tech PHERAstar plate reader. Progress curves were truncated such that only data where the uninhibited reaction was linear were utilized to eliminate any nonlinear artifacts from substrate depletion. The resultant progress curves were fit by nonlinear regression analysis in MATLAB with the following equation:vi is the initial velocity of the reaction, vs is the steady-state velocity, kobs is the first-order rate constant for the transition from vi to vs, t is time, and A0 is the initial absorbance. The two-step slow-binding is described by the general kinetic scheme below:
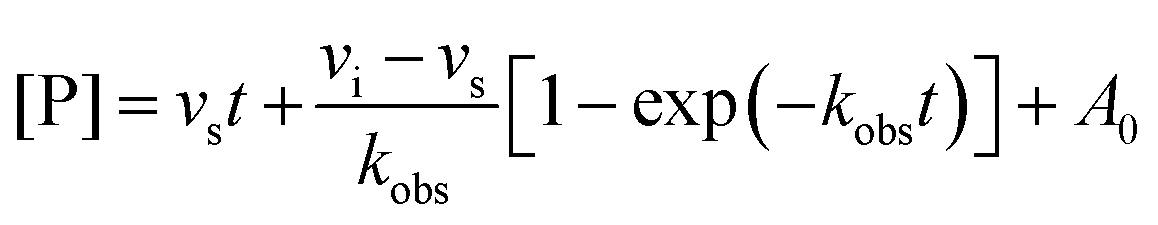
E is enzyme, and I is inhibitor. As the kobsversus [I] plots generated were linear, we assumed that the initial isomerization was kinetically insignificant (i.e., [I] ≪ KI) and used a one-step kinetic scheme to fit the linear data of kobsversus inhibitor concentration:
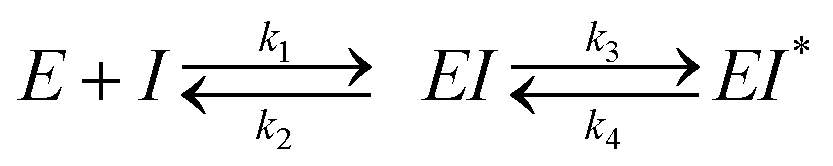
[I] is the concentration of inhibitor and KI is the equilibrium that describes the initial binding complex (k1/k2).
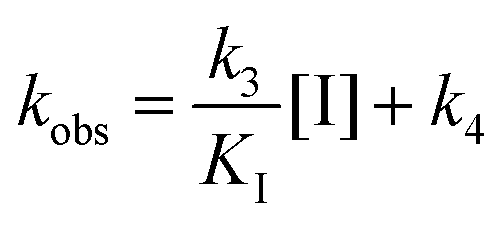
Protein crystallography
Crystals of Ee GUS bound to UNC10206581-GlcA were produced via the sitting drop vapor diffusion method using an Orxy4 robot (Douglas Instruments). Ee GUS (11.5 mg mL−1) was preincubated with 10-fold molar excess UNC10206581 for one hour, then pNPG was added to the solution and the mixture was cooled on ice before adding into the crystalline solution. Crystals were formed at 20 °C by incubating ligand bound Ee GUS in 0.075 M HEPES, pH 7.5, 15% (w/v) PEG 1000, and 25% (v/v) glycerol. The mother liquor also acted as a cryoprotectant for the crystals thus protein crystals were removed directly from the crystallization drop and quickly flash cooled (<1 minute) in liquid nitrogen. Diffraction data were collected at 100 K at GM-CA 23-ID-D (Advanced Photon Source, Argonne National Laboratory). CCP4i (v7.1.015) was used to scale the raw data, and Phenix (v.1.17) was used to refine the model to the statistics shown in Table S1 (ESI†). The final structural coordinates were deposed in the PDB under the accession code 8UGT.Cell-based inhibitory assay
E. coli K-12 MG1655 cells were grown overnight in 10 mL of Lysogeny broth (LB). The following morning, a 100 μL aliquot was subcultured into 5 mL of fresh LB. Cells were grown until an OD of 0.6 was achieved and then used for the assay. Reactions were carried out in Costar 96-well black clear bottom plates at a volume of 90 μL of cells (premixed with 700 μM pNPG) and 10 μL of various concentrations of inhibitor. This reaction was incubated for 24 h at 37 °C with a low evaporation lid before being quenched by the addition of 50 μL of 0.2 M sodium carbonate. Absorbance values were measured at 410 nm in a BMG lab tech PHERAstar plate reader. Percent inhibition and EC50 values were determined as described previously for the in vitro IC50 assay.23Bacterial cell toxicity assay
Bacterial cell toxicity was assessed as described previously.23 In brief, overnight cultures of WT E. coli MG1655 K-12 were grown and sub-cultured the following morning. Sub-cultures were subject to 10 μM of various inhibitors and growth was monitored over the course of 8 hours to assess inhibitor toxicity.β-Galactosidase selectivity assays
Initial enzyme substrate preferences were assessed using reporter substrate conjugates of the two sugars. For each enzyme, preferences were determined using the fluorescent β-glucuronidase substrate 4-methylumbelliferyl-β-D-glucuronide (4MU-G, Cayman) and the fluorescent β-galactosidase substrate 4-methylumbelliferyl-β-D-galactoside (Cayman) as described previously.30Bovine GUS selectivity assay
Bovine liver GUS was obtained from Sigma-Aldrich as a lyophilized powder and dissolved in storage buffer (10 mM sodium acetate and 10 mM sodium chloride, pH 5.0) and stored at 4 °C. Final assay wells contained 5 μL of bovine liver GUS (0.132 mg mL−1), 10 μL of assay buffer (25 mM sodium chloride, 25 sodium acetate, pH 5), 5 μL of inhibitor (10 μM final), and 30 μL of pNPG. Buffer, inhibitor, and protein was added to the plate then assays were initiated by the addition of pNPG and incubated for 1 hour at 37 °C. Reactions were quenched using 25 μL of 0.2 M sodium carbonate, and absorbance at 410 nm was measured in a BMG lab tech PHERAstar plate reader. Percent inhibition was calculated as described for the in vitro IC50 assay.Fecal sample collection
Fecal samples were collected from healthy volunteers at the University of North Carolina at Chapel Hill (IRB#17-1528) or purchased from healthy donors supplied by a commercial vendor, BioIVT. Fecal samples were collected using a toilet specimen collection kit (Fisher Scientific) and were stored at −80 °C until further use.Preparation of human fecal extract
Approximately 5–10 g of thawed fecal material collected from each donor was resuspended in 25 mL cold extraction buffer (25 mM HEPES pH 6.5, 25 mM NaCl, one Roche Complete EDTA-free protease inhibitor tablet in 50 mL buffer) and 500 mg autoclaved garnet beads then vortexed. Samples were centrifuged at 300 × g for 5 min at 4 °C and supernatant was collected. 25 mL cold extraction buffer was added to the centrifuged pellet, which was again vortexed and centrifuged. Both supernatants were combined and centrifuged at 300 × g for 5 min at 4 °C two additional times to further remove insoluble fiber. The supernatant was then sonicated twice on a Fischer Scientific Sonic Dismembrator Model 500 with 0.5 second pulses for 1.5 min and the lysate was mixed by inversion between each sonication. Lysate was then centrifuged at 17000 × g for 20 min at 4 °C to remove insoluble debris then decanted. The lysate was then concentrated with Amicon Ultra 15 mL 30 kDa centrifugal filters and exchanged with fresh extraction buffer three times to remove metabolites. After buffer exchanging, the total protein concentration of the final fecal lysate for each sample was measured with a Bradford assay using purified Escherichia coli β-glucuronidase as a reference standard. Complex protein lysates were aliquoted at 500 μL then flash frozen in liquid nitrogen and stored at −80 °C until later use in proteomics and fecal lysate assays.
Preliminary metagenomics analysis
Raw metagenomics genes were trimmed, filtered, and annotated, then assembled into gene and protein sequences using Metagenomics Analysis Toolkit (MOCAT2 v2.0.1).31Identification and characterization of GUS sequences
Metagenomic amino acid sequences were each aligned pairwise to 17 representative GUS enzymes with reported crystal structures using Protein–Protein BLAST (BLASTP v2.5.0+).32 Candidate sequences with ≥25% identity to any representative GUS enzyme were then assessed for the presence of 7 conserved residues.15 Sequences that both met the identity threshold and contained all 7 conserved residues were accepted as putative GUS enzymes. Accepted sequences were filtered for redundancies at a sequence identity threshold of 100% using CD-HIT (v4.8.1), and the output was used to form a representative set of GUS sequences for downstream analysis.33 Accepted sequences were aligned to representative sequences from each loop class in a Multiple Sequence Alignment (MSA) using Clustal Omega (v1.2.4), and GUS class was assigned according to parameters reported previously and shown in Fig. S3 (ESI†).15,16,30 The resulting GUS classes were further screened for “No loop” class enzymes which conserve both a c-terminal domain with previously reported FMN-binding GUS (PDB: 6MVF, 6MVG, 6MVH).16,25 Resulting sequences were assigned the class “FMN”. Taxonomy was assigned to representative GUS sequences by mapping queries to the UHGP using Diamond (v2.0.15.153) as reported previously, and resulting taxonomic identifiers were used to rename these sequences.15,34,35 Raw metagenomics data with key results have been uploaded to Zenodo and are accessible at the following https://doi.org/10.5281/zenodo.11110310.Activity-based probe (ABP)
Cyclophellitol-based probe JJB397 was synthesized and purified as previously described to form a biotin-linked covalent inhibitor of GUS enzymes.36Metaproteomics
General proteomics workflow was adapted from our previously reported GUS-targeted activity based proteomic profiling pipeline.19,21 Briefly, 3.5 mg purified fecal extract was incubated with 10 μM biotin-activity-based probe complex in 500 μL extraction buffer with 1% DMSO (final) for 1 h at 37 °C. 125 μl 10% sodium dodecyl sulfate (SDS) was added to quench the reaction, then samples were heated to 95 °C for 5 min. Samples were then cooled on ice and washed with extraction buffer containing 0.05% SDS three times by centrifugation for 5 min at 13000 × g in 1.5 mL 10 K cutoff spin concentrators (Amicon). After centrifugation, the total volume was normalized to 1 mL using extraction buffer + 0.05% SDS. 15 μL streptavidin sepharose beads (GE) were added to the protein mixture, and samples were then incubated at room temperature for 1 h. Afterwards, beads were washed 3 times with 300 μL extraction buffer with 0.1% SDS, three times with 300 μL extraction buffer alone, and finally three times with 300 μL 50 mM NH4HCO3. Samples were centrifuged at 400 × g for 2 min at 4 °C between washes, and the supernatant decanted. Beads were then resuspended in 100 μL 50 mM NH4HCO3 and stored at −20 °C, then subjected to subsequent LC-MS/MS analysis exactly as described previously.19,21 Raw metaproteomic data with key results have been uploaded to Zenodo and are accessible at the following https://doi.org/10.5281/zenodo.11110310.
Metaproteomics data analysis
Raw data were processed as described previously, with the following modifications.17 Data were processed using Metalab (v1.1.148) with MaxQuant (v1.6.2) to identify peptides and protein groups. A sample-specific database was derived from cohort metagenomic sequences then combined with the UniProtKB/Swiss-Prot human sequence database (containing 26122 entries).37 Processed data were searched against this combined database with the following search parameters enabled: static carbamidomethyl cysteine modification, specific trypsin digestion with up to two missed cleavages, variable protein N-terminal acetylation and methionine oxidation, and match between runs. A false discovery rate (FDR) of 1% was used for filtering protein identifications at the unique peptide level, and potential contaminants and decoys were removed. For each protein, peptide peak areas were extracted then summed and the protein intensities were used for relative quantitation. Best-match protein headers were mapped back to their corresponding amino acid sequences from the sample metagenomes, and GUS enzymes were identified as described in “Identification and Characterization of GUS Sequences” above. Proteomic intensities for GUS enzymes were log2-transformed to reach the normalized GUS abundance values shown in Fig. 4C.
Fecal lysate inhibitor assays
Reaction mixtures contained 5 μL fecal extract (0.1 mg mL−1 final), 10 μL 4MU-G (100 μM final), 5 μL inhibitor (10 μM final), and 30 μL assay buffer (25 mM HEPES, 25 mM NaCl, pH 6.5). Reactions were incubated with inhibitor for five minutes, initiated by the addition of 4MU-G, then quenched after 1 hour with 50 μL sodium carbonate (0.2 M). Analysis of the reaction mixture was performed using a CLARIOstar Plus Microplate Reader (BMG Lab Tech), measuring the fluorescence of 4MU at excitation 350 nm per emission 450 nm. The absorbance data collected was converted to percent inhibition as described previously.21SiteMap binding pocket analysis
Protein monomers were first prepared in PyMOL by isolating a single chain, removing waters, and overlaying a ligand into the active site, if not already present. The prepared monomers were next loaded into Schrodinger's Maestro and prepared using the default settings for protein preparation. Once prepared, structures were subject to SiteMap task using the “evaluate a single binding site region” function and centering on the bound ligand. Default settings were then used to execute the SiteMap function.Author contributions
S. J. P. and M. R. R.: conceptualization; J. B. S.: data curation; J. B. S., S. J. P., and J. J. S.: formal analysis; A. L. G., J. B. S., S. J. P., N. M., Y. A., P. B. J., M. E. K., B. C. C., N. K. B., and L. E. H.: investigation; A. L. G.: writing – original draft; A. L. G., J. B. S., L. I. J., and M. R. R.: writing – review and editing; A. L. G., J. B. S., and S. J. P.: visualization; J. J., L. I. J. and M. R. R.: supervision; J. B. S., S. J. P., N. M., Y. A., and A. P. B.: methodology; M. R. R.: funding acquisition; M. R. R.: project administration.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for E. eligens GUS bound to UNC10206581-G has been deposited at the PDB under accession number 8UGT and will become available upon publication. Raw metagenomics and proteomics data with key results have been uploaded to Zenodo and are accessible at the following https://doi.org/10.5281/zenodo.11110310.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NIH grants GM135218, GM137286, and GM152079 (M. R. R.), and by NSF DGE-1650116 (J. B. S.). A. P. B. was supported by a Career Development Award from the Crohn's and Colitis Foundation. We would like to thank Dr Jarod Waybright and Rebecca Johnson for assistance with the experimental chemistry. Fig. S1 (ESI†) was generated using BioRender.References
- J. Y. Lee, R. M. Tsolis and A. J. Bäumler, The microbiome and gut homeostasis, Science, 2022, 377(6601), eabp9960 CrossRef CAS PubMed.
- G. A. Cresci and E. Bawden, The Gut Microbiome: What we do and don’t know, Nutr. Clin. Pract., 2015, 30(6), 734–746 CrossRef CAS PubMed.
- M. J. Bull and N. T. Plummer, Part 1: The human gut microbiome in health and disease, Integr. Med., 2014, 13(6), 17–22 Search PubMed.
- R. K. Weersma, A. Zhernakova and J. Fu, Interaction between drugs and the gut microbiome, Gut, 2020, 69(8), 1510–1519 CrossRef CAS PubMed.
- A. Saqr, B. Carlson, C. Staley, A. Rashidi, M. Al-Kofahi and T. Kaiser, et al., Reduced Enterohepatic Recirculation of Mycophenolate and Lower Blood Concentrations Are Associated with the Stool Bacterial Microbiome after Hematopoietic Cell Transplantation, Transplant. Cell. Ther., 2022, 28(7), 372 Search PubMed.
- B. D. Wallace, H. Wang, K. T. Lane, J. E. Scott, J. Orans and J. S. Koo, et al., Alleviating Cancer Drug Toxicity by Inhibiting a Bacterial Enzyme, Science, 2010, 330(6005), 831–835 CrossRef CAS PubMed.
- D. Chrysostomou, L. A. Roberts, J. R. Marchesi and J. M. Kinross, Gut Microbiota Modulation of Efficacy and Toxicity of Cancer Chemotherapy and Immunotherapy, Gastroenterology, 2023, 164(2), 198–213 CrossRef CAS PubMed.
- K. L. Flannigan, M. R. Taylor, S. K. Pereira, J. Rodriguez-Arguello, A. W. Moffat and L. Alston, et al., An intact microbiota is required for the gastrointestinal toxicity of the immunosuppressant mycophenolate mofetil, J. Heart Lung Transplant., 2018, 37(9), 1047–1059 CrossRef PubMed.
- L. M. Olsson, F. Boulund, S. Nilsson, M. T. Khan, A. Gummesson and L. Fagerberg, et al., Dynamics of the normal gut microbiota: A longitudinal one-year population study in Sweden, Cell Host Microbe, 2022, 30(5), 726–739 CrossRef CAS PubMed.
- A. B. Hall, A. C. Tolonen and R. J. Xavier, Human genetic variation and the gut microbiome in disease, Nat. Rev. Genet., 2017, 18(11), 690–699 CrossRef CAS PubMed.
- G. L. Davidson, A. C. Cooke, C. N. Johnson and J. L. Quinn, The gut microbiome as a driver of individual variation in cognition and functional behaviour, Philos. Trans. R. Soc., B, 2018, 373(1756), 1–12 CrossRef PubMed.
- C. D. Klaassen and J. Y. Cui, Mechanisms of how the intestinal microbiota alters the effects of drugs and bile acids, Drug Metab. Dispos., 2015, 43(10), 1505–1521 CrossRef CAS PubMed.
- P. Dashnyam, R. Mudududdla, T. J. Hsieh, T. C. Lin, H. Y. Lin and P. Y. Chen, et al., β-Glucuronidases of opportunistic bacteria are the major contributors to xenobiotic-induced toxicity in the gut, Sci. Rep., 2018, 8(1), 1–12 CAS.
- K. A. Biernat, S. J. Pellock, A. P. Bhatt, M. M. Bivins, W. G. Walton and B. N. T. Tran, et al., Structure, function, and inhibition of drug reactivating human gut microbial β-glucuronidases, Sci. Rep., 2019, 9(1), 1–15 CrossRef CAS PubMed.
- R. M. Pollet, E. H. D’Agostino, W. G. Walton, Y. Xu, M. S. Little and K. A. Biernat, et al., An Atlas of β-Glucuronidases in the Human Intestinal Microbiome, Structure, 2017, 25(7), 967–977 CrossRef CAS PubMed.
- S. J. Pellock, W. G. Walton, S. M. Ervin, D. Torres-Rivera, B. C. Creekmore and G. Bergan, et al., Discovery and Characterization of FMN-Binding β-Glucuronidases in the Human Gut Microbiome, J. Mol. Biol., 2019, 431(5), 970–980 CrossRef CAS PubMed.
- J. B. Simpson, J. J. Sekela, A. L. Graboski, V. B. Borlandelli, M. M. Bivins and N. K. Barker, et al., Metagenomics combined with activity-based proteomics point to gut bacterial enzymes that reactivate mycophenolate, Gut Microbes, 2022, 14(1), 1–20 CrossRef PubMed.
- A. P. Bhatt, S. J. Pellock, K. A. Biernat, W. G. Walton, B. D. Wallace and B. C. Creekmore, et al., Targeted inhibition of gut bacterial β-glucuronidase activity enhances anticancer drug efficacy, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(13), 7374–7381 CrossRef CAS PubMed.
- J. Zhang, M. E. Walker, K. Z. Sanidad, H. Zhang, Y. Liang and E. Zhao, et al., Microbial enzymes induce colitis by reactivating triclosan in the mouse gastrointestinal tract, Nat Commun., 2022, 13(1), 1–14 Search PubMed.
- S. T. K. Yauw, M. Arron, R. M. L. M. Lomme, P. van den Broek, R. Greupink and A. P. Bhatt, et al., Microbial Glucuronidase Inhibition Reduces Severity of Diclofenac-Induced Anastomotic Leak in Rats, Surg. Infect., 2018, 19(4), 417–423 CrossRef PubMed.
- P. B. Jariwala, S. J. Pellock, D. Goldfarb, E. W. Cloer, M. Artola, J. B. Simpson, A. P. Bhatt, W. G. Walton, L. R. Roberts, M. B. Major, G. J. Davies, H. S. Overkleeft and M. R. Redinbo, Discovering the Microbial Enzymes Driving Drug Toxicity with Activity-Based Protein Profiling Parth, ACS Chem. Biol., 2020, 17(15), 217–225 CrossRef PubMed.
- S. M. Ervin, R. P. Hanley, L. Lim, W. G. Walton, K. H. Pearce and A. P. Bhatt, et al., Targeting Regorafenib-Induced Toxicity through Inhibition of Gut Microbial β-Glucuronidases, ACS Chem. Biol., 2019, 14(12), 2737–2744 CrossRef CAS PubMed.
- S. J. Pellock, B. C. Creekmore, W. G. Walton, N. Mehta, K. A. Biernat and A. P. Cesmat, et al., Gut Microbial β-Glucuronidase Inhibition via Catalytic Cycle Interception, ACS Cent. Sci., 2018, 4(7), 868–879 CrossRef CAS PubMed.
- S. Ahmad, M. A. Hughes, K. T. Lane, M. R. Redinbo, L. A. Yeh and J. E. Scott, A high throughput assay for discovery of bacterial β-glucuronidase inhibitors, Curr. Chem. Genomics, 2011, 5(1), 13–20 CrossRef CAS PubMed.
- M. E. Walker, J. B. Simpson and M. R. Redinbo, A structural metagenomics pipeline for examining the gut microbiome, Curr. Opin. Struct. Biol., 2022, 75 Search PubMed.
- A. M. Montaño, N. Lock-Hock, R. D. Steiner, B. H. Graham, M. Szlago and R. Greenstein, et al., Clinical course of sly syndrome (mucopolysaccharidosis type VII), J. Med. Genet., 2016, 53(6), 403–418 CrossRef PubMed.
- A. P. Bhatt, L. Grillo and M. R. Redinbo, In Fimo: A Term Proposed for Excrement Examined Experimentally, Gastroenterology, 2019, 156(5), 1232 CrossRef PubMed.
- S. M. Ervin, H. Li, L. Lim, L. R. Roberts, X. Liang and S. Mani, et al., Gut microbial β-glucuronidases reactivate estrogens as components of the estrobolome that reactivate estrogens, J. Biol. Chem., 2019, 294(49), 18586–18599 CrossRef CAS PubMed.
- J. F. Morrison and C. T. Walsh, The behavior and significance of slow-binding enzyme inhibitors, Adv. Enzymol. Relat. Areas Mol. Biol., 1988, 61, 201–301 CAS.
- S. J. Pellock, W. G. Walton, K. A. Biernat, D. Torres-Rivera, B. C. Creekmore and Y. Xu, et al., Three structurally and functionally distinct -glucuronidases from the human gut microbe Bacteroides uniformis, J. Biol. Chem., 2018, 293(48), 18559–18573 CrossRef CAS PubMed.
- J. R. Kultima, S. Sunagawa, J. Li, W. Chen, H. Chen and D. R. Mende, et al., MOCAT: A Metagenomics Assembly and Gene Prediction Toolkit, PLoS One, 2012, 7(10), 1–6 CrossRef PubMed.
- C. Camacho, G. Coulouris, V. Avagyan, N. Ma, J. Papadopoulos and K. Bealer, et al., BLAST +: Architecture and applications, BMC Bioinf., 2009, 10, 1–9 CrossRef PubMed.
- W. Li and A. Godzik, Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences, Bioinformatics, 2006, 22(13), 1658–1659 CrossRef CAS PubMed.
- G. Yu, D. K. Smith, H. Zhu, Y. Guan and T. T. Y. Lam, Ggtree: an R Package for Visualization and Annotation of Phylogenetic Trees With Their Covariates and Other Associated Data, Methods Ecol. Evol., 2017, 8(1), 28–36 CrossRef.
- C. Kanz, P. Aldebert, N. Althorpe, W. Baker, A. Baldwin and K. Bates, et al., The EMBL nucleotide sequence database, Nucleic Acids Res., 2005, 33(1), 29–33 Search PubMed.
- L. Wu, J. Jiang, Y. Jin, W. W. Kallemeijn, C. L. Kuo and M. Artola, et al., Activity-based probes for functional interrogation of retaining β-glucuronidases, Nat. Chem. Biol., 2017, 13(8), 867–873 CrossRef CAS PubMed.
- J. Li, H. Jia, X. Cai, H. Zhong, Q. Feng and S. Sunagawa, et al., An integrated catalog of reference genes in the human gut microbiome, Nat. Biotechnol., 2014, 32(8), 834–841 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cb00058g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |