
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbohydrate-active enzyme (CAZyme) discovery and engineering via (Ultra)high-throughput screening
Jacob F.
Wardman
 ab and
Stephen G.
Withers
*abc
ab and
Stephen G.
Withers
*abc
aDepartment of Biochemistry and Molecular Biology, University of British Columbia, Vancouver, BC V6T 1Z3, Canada. E-mail: withers@chem.ubc.ca
bMichael Smith Laboratories, University of British Columbia, Vancouver, BC V6T 1Z4, Canada
cDepartment of Chemistry, University of British Columbia, Vancouver, BC V6T 1Z1, Canada
First published on 23rd May 2024
Abstract
Carbohydrate-active enzymes (CAZymes) constitute a diverse set of enzymes that catalyze the assembly, degradation, and modification of carbohydrates. These enzymes have been fashioned into potent, selective catalysts by millennia of evolution, and yet are also highly adaptable and readily evolved in the laboratory. To identify and engineer CAZymes for different purposes, (ultra)high-throughput screening campaigns have been frequently utilized with great success. This review provides an overview of the different approaches taken in screening for CAZymes and how mechanistic understandings of CAZymes can enable new approaches to screening. Within, we also cover how cutting-edge techniques such as microfluidics, advances in computational approaches and synthetic biology, as well as novel assay designs are leading the field towards more informative and effective screening approaches.
Introduction
Carbohydrates are among the most stable biological polymers. Indeed, intact cellulose and chitin can be found in fossils dating back 45 million years.1,2 At the same time, the combinations and permutations of carbohydrates far exceed the complexity that can be achieved with other polymers such as nucleic acids or polypeptides.3 The surface of every cell is coated in carbohydrates, and it is this complexity that primes these carbohydrates – especially in the form of conjugates with lipids or proteins – for vital roles in cellular signaling.4 To degrade, synthesize, or otherwise manipulate carbohydrates, Nature has developed and utilized a suite of enzymes known as carbohydrate-active enzymes (or CAZymes). CAZymes are a remarkable class of enzymes that carry out diverse reactions with often exquisite selectivity and large rate enhancements. As an example, glycoside hydrolases (GHs), the primary enzymes involved in carbohydrate degradation, commonly act with specificity both for the types of sugar(s), and the stereo- and regiochemistry of the linkage; while also offering up to 1017-fold rate enhancements compared to background hydrolysis.5CAZymes have found a number of applications beyond what Nature has developed them for. At an industrial level, CAZymes can play roles in biofuel production,6 textile processing,7 and the production of prebiotics8 and other designer food sugars.9 Applications in biomedical science include the re-modelling of blood and organ antigens,10–13 development of longer lasting biopharmaceuticals,14,15 and the manipulation and analysis of protein glycosylation patterns to provide more potent biopharmaceuticals.16,17 In addition, CAZymes often provide useful fundamental research tools for deciphering the roles of specific carbohydrate and glycoconjugate structures in biological systems (often called the glyco-code).4,18,19
The central role of carbohydrates in cellular structure, signaling and metabolism at all levels of life has led to an incredible diversity of enzymes for glycan biosynthesis and degradation. Consequently, Nature can often provide us with ready-made enzymatic solutions for our specific needs since, in many instances, organisms have spent millennia evolving enzymes to carry out the exact reactions that a researcher is interested in. Such enzymatic diversity can be accessed through creation of synthetic gene libraries or through methods such as functional metagenomics (for which random fragments of bacterial DNA are cloned into laboratory strains of bacteria for expression and screening).20 However, enzymes obtained from Nature may not always have the exact desired activity or specificity. Directed evolution – for which the Nobel Prize in chemistry was awarded in 2018 – has offered a useful way of modifying these enzymatic activities for different purposes.21 In principle: the process of directed evolution subjects genes to accelerated evolution through mutagenesis and subsequent screening/selection for the transformations or properties of interest to the researcher.
In order to find an enzyme for a desired purpose it is typically necessary to screen many different clones containing sequence variants. High-throughput screening is thus essential in CAZyme discovery and engineering campaigns. While differing in the source material, the screens applied in directed evolution, interrogation of synthetic gene libraries, and functional metagenomics are similar in their design and often interchangeable. This review will cover both recent advances and general principles in the field of (ultra)high-throughput screening for CAZymes. In so doing it will highlight both the scientific principles behind the approaches and the creativity of the authors in the development of these methods. Additionally, we will highlight underexplored reaction spaces and anticipated next steps in the field.
CAZyme fundamentals – a primer
CAZymes are classified within the CAZy database according to their primary reaction type.22 The largest and best studied of these are the glycoside hydrolases (GHs), which hydrolytically cleave glycosidic linkages (Fig. 1a–c). Also included are polysaccharide lyases (PLs) which degrade uronic acid-containing polysaccharides via a non-hydrolytic elimination mechanism (Fig. 1d). The enzymes primarily responsible for carbohydrate synthesis are the glycosyltransferases (GTs) which generally synthesize glycosides through transfer from activated nucleotide sugars or lipid phosphate sugars (Fig. 1e). Finally, the classification includes carbohydrate esterases (CEs) which hydrolyze ester and amide linkages on carbohydrates (Fig. 1f), and auxiliary enzymes (AAs) which catalyze diverse oxidative chemistries involved in the degradation of carbohydrates and associated structures such as lignin (Fig. 1g).23 Carbohydrate binding modules (CBMs) are also classified within the CAZy database but do not themselves have catalytic activity. Rather they improve the activity of CAZymes against their substrates by improving binding, especially for polymeric substrates.24 The general reactions carried out by these CAZymes and select mechanisms for these CAZymes are shown in Fig. 1. Where appropriate, the mechanisms will be discussed within this review as they are necessary to understand certain screening methodologies. For more detailed explanations of these enzymatic mechanisms and others not included here, interested readers are directed to other recent reviews.18,25,26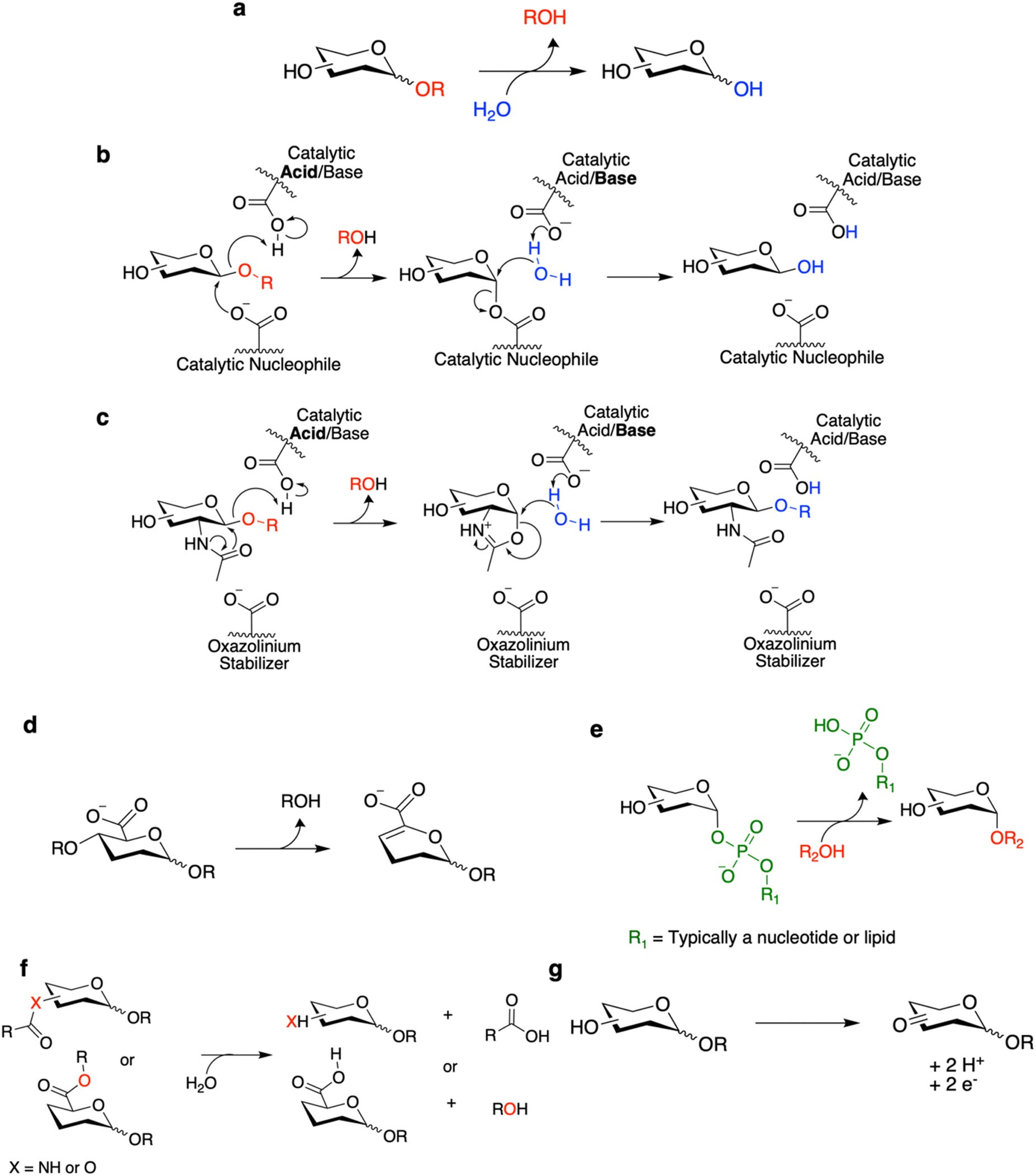 | ||
| Fig. 1 General reactions and mechanisms of CAZymes. (a) General reaction catalyzed by GHs. (b) The Koshland retaining GH mechanism which is generally followed by many GHs. (c) A variation upon the retaining GH mechanism utilized by certain hexosaminidases. General reactions catalyzed by (d) PLs, (e) GTs, (f) CEs, and (g) AAs. Note that there are variations upon these mechanisms depending upon the enzyme of interest. As well, for simplicity only a single stereochemical configuration is generally shown. In many cases, the initial stereochemistry of the reactants and the ultimate stereochemical outcome will differ depending upon the enzyme and the reaction catalyzed. | ||
Outside of these primary classifications within the CAZy database are enzymes such as transglycosylases (TGs) and glycoside phosphorylases (GPs). TGs are mechanistically similar to GHs and classified within GH families. Rather than hydrolysis of glycosidic linkages, TGs preferentially catalyze the transfer of sugars to different non-water acceptors.27 TGs have been of great interest for carbohydrate synthesis as they do not require the same costly sugar donors as GTs since they can often use relatively inexpensive feedstocks such as sucrose.9,27,28 GPs catalyze the reversible phosphorolysis of carbohydrates and are found in several GH and GT families.29 The reversibility of this reaction is of synthetic utility as one can drive glycan synthesis through use of an excess of relatively accessible sugar-1-phosphates and/or removal of phosphate or glycan polymer product.30
While GHs are useful for a number of purposes – principally glycan degradation – the engineering of GHs as biosynthetic catalysts bestows additional utility. Towards this end, classes of redesigned enzymes such as glycosynthases31 and (thio)glycoligases32 have been developed (Fig. 2a and b). Both of these classes of enzymes were developed by the Withers laboratory based upon a mechanistic understanding of GHs in order to create mutants that are crippled hydrolytically but can perform glycan synthesis with the appropriate substrates.31,32 Use of these engineered enzymes may be preferred over GTs – the natural enzymes for glycan synthesis – since these mutant GHs are typically easily expressed and do not need expensive nucleotide/lipid-linked donor sugars for glycan biosynthesis. While this may also be true of TGs, an advantage of these engineered enzymes is that they do not degrade their reaction products – and can thus offer improved yields.28 Classically, glycosynthases and (thio)glycoligases can be developed from GHs through mutagenesis of the catalytic nucleophile and the catalytic acid/base residues respectively.31,32
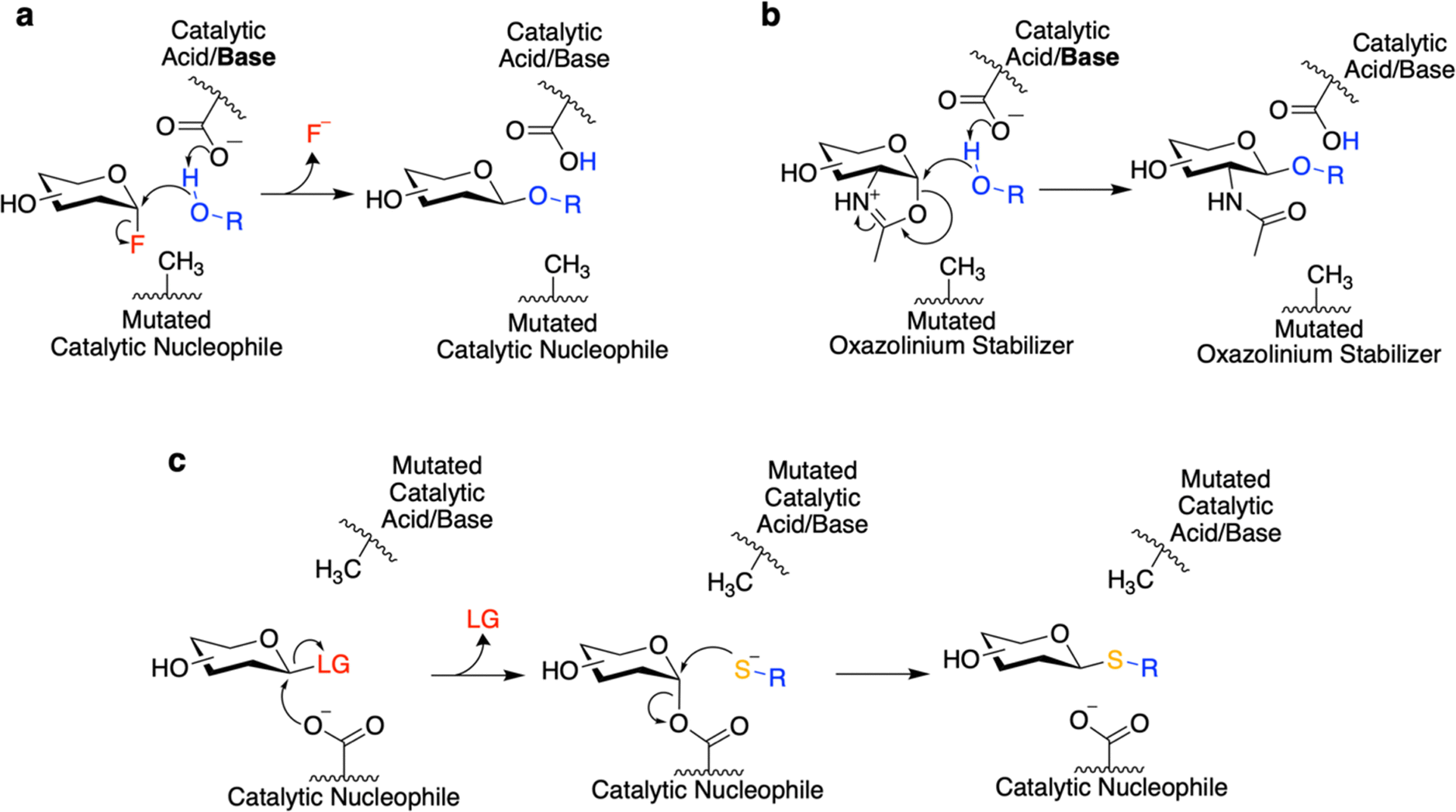 | ||
| Fig. 2 Mechanisms of glycosynthases and thioglycoligases. (a) Mechanism utilized by the classical glycosynthases derived from a retaining GH. (b) Mechanism for a glycosynthase derived from an N-acetylhexosaminidase which uses neighboring group participation. (c) Mechanism of a thioglycoligase. Note that LG denotes a good leaving group. | ||
Glycosynthases accomplish glycoside bond synthesis by using a glycosyl fluoride (1-fluoro sugar) of the opposite anomeric configuration to that of their native substrate (Fig. 2a).28,31 This activated sugar mimics the glycosyl-enzyme covalent intermediate found in the mechanism for retaining GHs (Fig. 1b), setting the enzyme up to accept different nucleophiles and form new glycosidic linkages.28 The glycosynthase concept has found widespread use for variants of Koshland GHs that carry out their reaction through a neighbouring group participation mechanism involving an oxazoline intermediate (Fig. 1c and 2b). Mutants of such enzymes lacking the oxazoline-stabilising residue can carry out glycosyl transfer using an oxazoline-bearing glycan donor, but are unable to hydrolyse the product so formed.28 These oxazoline donors can be readily prepared from glycans bearing an N-acetyl sugar at the reducing end using a mild and selective chemical reagent.33 And so, in this way, complex glycan donors – often derived from N-glycans that have been synthesised or isolated from natural sources – can be prepared and used in the synthesis of a variety of products, most notably glycoproteins.28
Thioglycoligases are mutant retaining glycosidases missing their acid/base residue. When reacted with a glycan donor containing a leaving group such as DNP or F that does not need acid catalysis, they rapidly form the glycosyl-enzyme intermediate (Fig. 2c).32,34–37 However, in the absence of the catalytic base, these enzymes are unable to efficiently turn over the covalent intermediate through hydrolysis. However introduction of a good nucleophile – such as a thiol – that does not need base catalysis results in ready turnover and formation of new thioglycosidic bonds.32,34–37 A further variation on this theme is that of the O-glycoligases, which are again mutants at the acid/base position.38,39 To date only mutants of α-glycosidases have been shown to react usefully with oxygen nucleophiles, thus to function as O-glycoligases. This is thought to be due to the higher reactivity of the β-glycosyl-enzyme intermediate than its alpha counterpart.38 Further combining these approaches, thioglycosynthases have also been developed through mutation of both catalytic residues.40 However, their application has been relatively limited due to their low reactivity. While this review covers efforts to evolve and improve these enzymes, interested readers are directed to other recent reviews for a more thorough interrogation of the transformations which can be catalyzed by these enzymes.18,28
An overview of different modes of screening
Microtitre plate-based screening is the most common and widespread method for high-throughput screening of CAZymes. Most glycosidases can be screened for using commonly available substrates whereby the release of a chromophore from the anomeric position produces a change in absorbance and/or fluorescence.41–44 Such substrates have been used extensively in functional metagenomic screening and directed evolution and form the bedrock for most GH screens. In the absence of available chromogenic glycoside substrates, variations upon this have also measured reducing end formation upon glycoside cleavage. This is commonly carried out with detection systems such as the DNS assay, allowing use of natural carbohydrates or glycoproteins as substrates.45–47 One of the great benefits of plate-based screening is that many different methods of detection and sample manipulation are possible. While too numerous to list all examples, we have highlighted some interesting versions based upon this basic concept throughout this review.In the absence of a colorimetric screening method, HPLC offers a near universal (albeit time intensive) detection method for carbohydrate modification. Desmet and colleagues have shown the power of this technique for monitoring the effects of modification of TGs in the formation of rare sugars from sucrose.9,48 These products are inherently difficult to screen for as the detection method must be able to distinguish the different stereo- and regioisomers which can be formed.9,48
Agar plate assays provide an accessible method for semi-quantitative screening. Many researchers will be familiar with the use of compounds such as X-Gal to detect β-galactosidase activity as part of blue-white screening. However, this approach can be extended for other CAZymes. As a nice example, an entirely new family of sialidases (GH156) was discovered through functional metagenomic screening of a hot spring library using X-Neu5Ac-containing agar plates.49 Other options exist such as halo assays with indicators such as Congo Red to detect polysaccharide degradation,50,51 or through incorporation of dyed carbohydrates into the media (often carbohydrates with conjugated azo dyes).52–54 An advantage of such screens is that they enable use of a near native substrate. While many approaches are purely qualitative, more quantitative screens have also been carried out using these methods. For example, transglycosylation activities have been engineered using screens based upon a comparison of reaction velocities in the presence/absence of a suitable acceptor.55 While these agar plate screens tend to use a large amount of material, they do enable screening of many variants relatively easily.
With the availability and expertise of suitable core facilities in many institutes, FACS offers an accessible method for ultrahigh-throughput screening of many enzymatic activities. The throughputs of FACS are typically on the order of >103 s−1 enabling ready screening of very large libraries.56 Further benefits of FACS includes an ability to easily multiplex reactions, limited specialized equipment needed to conduct the screen, and quantitative results. A typical FACS screen will involve either production/trapping of a fluorescent signal within the cell or at its cell surface.56 While this does make it amenable to many purposes, it does complicate certain screens as it can require efficient uptake of substrates or surface display and capture of enzymes/substrates, which can be non-trivial.
Of considerable recent interest in the CAZyme engineering space has been the use of microfluidic technologies, of which droplet-based microfluidics has been the most widely adopted.57 In most studies, aqueous reaction mixtures containing single clones are encapsulated within an oil medium that acts in the same manner as the walls of a microtiter plate (Fig. 3a).58 The assay is then allowed to proceed in the droplet with subsequent sorting of hits based upon formation of a fluorescent signal (Fig. 3a). A number of other methods (e.g. absorbance, fluorescence anisotropy, mass spectrometry, etc.) to measure activity within droplets have also been developed.57 As well, technologies such as pico-injection and controlled droplet merging allow for further manipulation of droplets and the performance of complex reactions and screens (Fig. 3b).57 Thus, at least part of the promise of droplet microfluidics is that the plethora of assays developed for plate-based screening can be readily adapted for microfluidics and thus conducted at much higher throughputs and with substantially less substrate.59 A particular advantage of droplet screening is that entry of the substrate into the cell is not necessarily required if the cell can be lysed within the droplet. As an example of the assay complexity possible and the manipulations that can be performed with droplet microfluidics, in work by Holstein et al., a cytotoxic protease was evolved via a multistep process (Fig. 3b). This involved encapsulation of single DNA molecules into droplets, followed by rolling circle amplification and in vitro transcription and translation steps, and then pico-injection of the substrate with subsequent sorting by FADS (Fig. 3b).60 As a key point for screen adaptation, in general, it seems that charged substrates and products are required for effective retention within droplets.61,62 While the exact mechanism of fluorophore exchange is not known, certain commonly used hydrophobic, un-charged fluorophores (e.g. hydroxycoumarin and resorufin derivatives) can diffuse into the oil or between droplets under certain conditions – thus leading to loss of genotype-phenotype linkage.61,62 Besides changes to the fluorophore itself, optimization of screening conditions (e.g. pH, surfactant concentration, inclusion of other additives) can also improve fluorophore retention.57,59
 | ||
| Fig. 3 A selection of droplet microfluidic workflows used to screen for enzymatic activities. (a) Typical workflow employed for droplet microfluidic screening. Note that FADS requires specialized equipment. (b) Droplets can be readily manipulated as demonstrated in the workflow employed by Holstein et al. to screen for improved protease variants.60 (c) A common workflow for generation of double emulsion droplets for subsequent sorting on widely available FACS instruments.59,66 | ||
It should be noted that, while microfluidic screens require more technical expertise and set-up compared to FACS assays, they also offer considerable flexibility and open the door to entirely new ways of screening and characterisation. For example, in a recent study, Markin et al. were able to carry out the near simultaneous high-throughput kinetic characterization (kcat, KM, Ki) of thousands of enzyme variants with different substrates (albeit this was achieved using a complex microfluidic chip rather than droplets for compartmentalization).63 Even absorbance-based assays, which cannot be done by FACS but are widely used in plate-based screens, are possible with microfluidics.64,65
While microfluidics does require some specialized expertise, strides towards democratization of the methodology and more widespread use have been made with the development of double-emulsion (water/oil/water) droplet systems (Fig. 3c).59,66 These double emulsions are relatively straightforward to produce and can be sorted on widely available conventional FACS instruments – thus making droplet microfluidics available to those without experience in droplet sorter design or engineering.66 A nice example of this being used for CAZyme discovery is the work by Tauzin et al. wherein they applied the approach to the discovery of new β-GalNAcases.59 There are many further interesting examples of the applications of droplet microfluidics which are beyond the scope of this review. Interested readers are thus directed to the excellent recent review by Gantz et al. on the use of microfluidics for enzyme discovery and engineering.57
Using mechanistic differences to identify enzymes
Nasseri and colleagues offered up an inventive method for the selective identification of GHs that do not follow the canonical Koshland mechanisms.67 This strategy relies on the fact that Koshland GHs do not catalyze the cleavage of non-activated thioglycosidic bonds.25,67 In fact, in Nature, carbon-sulfur bonds are generally cleaved enzymatically by elimination mechanisms.68 Indeed, this is true also for the cleavage of thioglycosides which is nicely carried out by glycosidases in GH4, GH88, GH105, and GH109, which use elimination mechanisms, as well as by PLs.25 By use of a thioglycoside substrate in which the chromogen is linked to the 1-thiosugar via a self-immolative linker, the authors were able to selectively identify unsaturated glucuronidases (of GH88) in a functional metagenomic screen.67 Particularly impressive is the fact that this campaign was successful even in the presence of high background activity from the E. coli host glucuronidase, which does indeed follow the canonical Koshland mechanism, thereby highlighting the selectivity of these reagents (Fig. 4a).67 More recently, this approach has been used to identify a surprisingly widespread, but heretofore un-appreciated, set of gene clusters that carry out the degradation of carbohydrates using a mechanism akin to that used by glycosidases of GH4 or GH109.69 However, intriguingly, each step of the mechanism is carried out by a different enzyme within these gene clusters rather than a single enzyme providing enzyme activities of unprecedentedly broad specificities.69 | ||
| Fig. 4 Applying mechanistic differences to screen for distinct enzyme activities. (a) Use of an unactivated thioglycoside substrate enables selective identification of non-Koshland GHs. In this screen, the released hemithioacetal undergoes a self-immolative reaction to produce a fluorescent signal. (b) GPs catalyze the reversible reaction between an oligosaccharide and the corresponding glycosyl phosphate. (c) GPs can be screened for production of free phosphate in the presence of a suitable glycosyl phosphate and glycan acceptor. In this assay, the released phosphate is detected via the Molybdenum Blue reaction. | ||
Other differences in enzyme mechanism can also be exploited in screening. Glycoside phosphorylases (GPs) differ from GHs in that inorganic phosphate acts as a nucleophile rather than H2O (Fig. 4b). Screens have been successfully applied for enhancements in the rate of substrate cleavage in the presence of phosphate in order to distinguish GPs from GHs.44,70 In a remarkable demonstration of the power of this technique, Franceus and colleagues were able to readily convert a GH to a GP via site-saturation libraries at “gatekeeping” residues with screens for rate enhancement upon addition of Pi.70
Owing to the near equal free energies of sugar-1-phosphates and oligosaccharides, reactions catalyzed by GPs are also readily reversible (Fig. 4b). This reversibility also allows for screening by measurement of the release of inorganic phosphate from sugar phosphate substrates using the molybdenum blue assay (Fig. 4c).71,72 This approach has been successfully applied to the directed evolution of a cellobiose phosphorylase to expand acceptor specificity,72 as well as in the screening of metagenomic libraries71 and synthetic gene libraries73 for GP activity. A key benefit of screening for reverse phosphorolysis is that the acceptor and donor pairs can be readily varied to rapidly probe substrate specificities.73 This approach enabled the discovery of the GlcNAc-β1,3-GlcNAc biopolymer – acholetin – from the activity of its corresponding GP from Acholeplasma laidlawii.73
PLs are a class of CAZyme that degrades uronic acid-containing substrates via a unique catalytic mechanism involving eliminative cleavage of the C4-oxygen bond to the sugar on the non-reducing side. However, they have been largely unexplored in protein engineering. PL activity can be assayed by monitoring the formation of the unsaturated uronic acid product via its absorbance at 235 nm (Fig. 1d).7 This method, while allowing use of natural substrates, suffers from low sensitivity, and is likely susceptible to interference from other assay components. Agar plate-based assays based upon halo formation in carbohydrate-containing media are also frequently used, though these are not specific for PLs.50,54,74 Nevertheless, combinations of these techniques have been used successfully. This is nicely demonstrated by Solbak et al.'s work in which they used azo-rhamnogalacturonan to detect pectinase and pectate lyase activity in an agar plate-based screen of a metagenomic library.54 They subsequently applied directed evolution to the best pectate lyase to enhance its thermostability and activity by monitoring activity on polygalacturonic acid after heat treatment. This process yielded variants with up to 16 °C increases in protein TM and 20 °C increases in temperature optima (from 50 °C to 70 °C).54 Thermostable PLs could be useful in applications such as textile processing where harsh, environmentally damaging, alkali solutions are otherwise used.54 Curiously, there has been limited application of artificial chromogenic substrates of PLs which might offer easier detection. Such substrates have been described by Rye et al. and were used in the determination of the catalytic mechanisms of these enzymes.75,76 Moreover, these substrates are selective for PLs over GHs as the chromophore is attached to the C4 hydroxyl rather than at the anomeric position.75,76
Coupled assays enable screening of (near)-native substrates
One of the principal benefits of coupled assays is that they enable the user to screen using substrates closer in structure to that of the natural substrate than to the simpler aryl glycosides. A fruitful line of work has been the use of oxidoreductases to screen for GH activity.77–80 For example, cleavage of cellulose with the appropriate enzymes might result in production of free glucose. This glucose can then be readily oxidized by glucose oxidase to produce H2O2 which can be detected by further reaction with horse radish peroxidase and a dye or by some other method of detection (Fig. 5a).77 The Schwaneberg laboratory has further elaborated upon this with a FACS-based screen79,81 wherein the production of H2O2 by glucose oxidase initiates localized polymerization of PEG-diacrylate and N-vinyl-2-pyrrolidone along with fluorescent acrylic monomers (e.g. Polyfluor 570) to form a fluorescent hydrogel around the cell (Fig. 5a).79,81 This can subsequently be sorted using a conventional cell sorter. In addition to cellulases, Lülsdorf et al. showed that the method can be further used to screen for CEs through use of glucose pentaacetate as a substrate.81 Alternatively, detection of H2O2 production can be used to directly screen for oxidases. Many campaigns have applied this approach to evolve galactose-6-oxidase such that it is active against other sugars such as glucose, mannose, GlcNAc, and Neu5Gc.82–84 Recently, it has been shown that this form of oxidase screening is also amenable to droplet microfluidics.85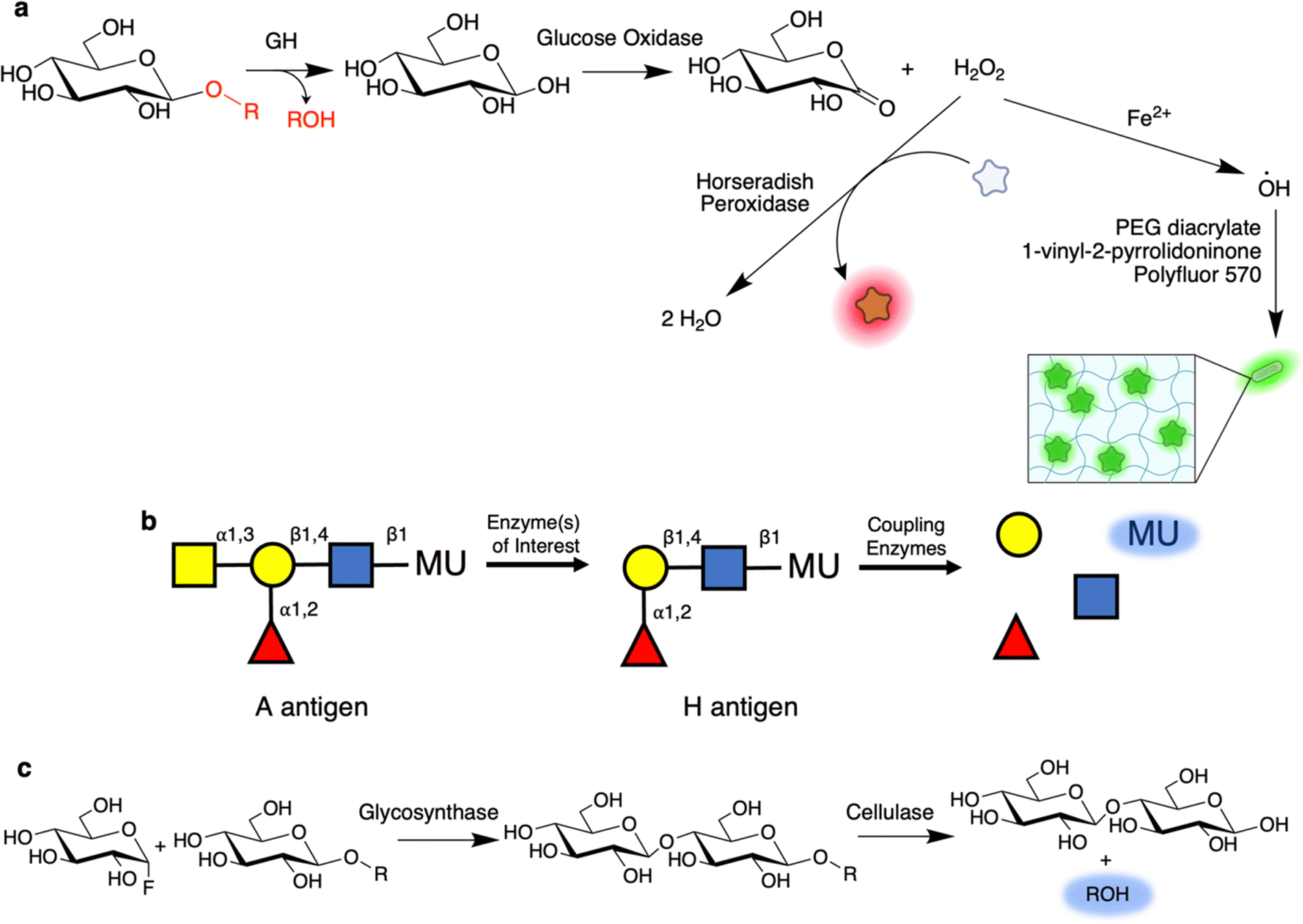 | ||
| Fig. 5 Coupled reactions enable high-throughput screening with highly relevant substrates. (a) Overview of a common coupled assay in which glucose is subsequently oxidized by glucose oxidase to form gluconolactone and H2O2. H2O2 can then be detected by use as a substrate for horseradish peroxidase along with a dye co-substrate. Alternatively, the produced H2O2 can also be used to initiate polymerization of a fluorescent hydrogel around the enzyme-producing cells for subsequent sorting.79,81 (b) Coupled assay used to screen for blood group converting enzymes. In this, once the initial GalNAc is removed, thereby converting the A antigen to the H antigen found in O-type blood, a series of coupling enzymes sequentially degrade the remaining trisaccharide to produce a fluorescent signal. (c) A coupled assay for use in screening for glycosynthases. As shown, a glucosynthase is used to produce a chromogenic cellobioside substrate. This cellobioside, but not the parent glucoside, can then be cleaved by an endo-acting cellulase to produce a fluorescent signal. | ||
More recent work by the Hollfelder laboratory has expanded this approach by using sugar dehydrogenases to enable measurement of the release of free sugars such as glucuronic acid, xylose, and galactose.78,86 This method uses the reduction of NAD(P)H by these enzymes during sugar oxidation to initiate an enzymatic cascade that results in reduction of a tetrazolium derivative (WST-1) to form a UV/Vis absorbent formazan.78 Ladeveze et al. show in their proof of concept that enzymes active on beechwood xylan, wheat arabinoxylan and xylobiose can be detected and that absorbance-activated droplet sorting can be used to sort active clones.78 Innovative work from the same laboratory offers further improvement through development of a highly sensitive fluorescence assay which operates under similar principles. In brief, the production of NAD(P)H by the sugar dehydrogenase is then coupled to the reduction of glutathione disulfide to glutathione by glutathione reductase.86 This free reduced glutathione can then release a quenched carboxy-coumarin fluorophore through nucleophilic cleavage of a sulfonic ester.86 The resulting fluorescent signal offers a much lower limit of detection compared to the absorbance based assay.86 This approach should prove useful in enzyme discovery campaigns via methods such as functional metagenomics where low enzyme expression levels are common.
Many GHs derive specificity from key interactions with both the components at the non-reducing end of the glycan and the reducing end (within their +1, +2, etc. subsites). Consequently, certain enzymes do not effectively recognize common chromogenic substrates, even if the enzymes are exo-acting.10,87 Such difficulties can be readily overcome through use of coupled assays with oligosaccharide substrates. An excellent example is in a screen utilized during the discovery of the blood group-cleaving GalNAc deacetylase and GH36 exo-α-GalNase by Rahfeld and colleagues (Fig. 5b).10 In this coupled assay, the terminal GalNAc is removed to expose a trisaccharide substrate which can then be degraded by added enzymes, in this case a fucosidase, galactosidase, and N-acetylhexosaminidase, to release the fluorophore. A similar approach had been previously applied by Kwan and colleagues for the directed evolution of a broad specificity blood group A cleaving enzyme from a Streptococcus pneumoniae GH98.11 Notably for the GH36 GalNase, this enzyme has almost 1000-fold lower activity against GalN-pNP as compared to a more natural substrate based on a blood group antigen tetrasaccharide.10
Coupled assays have also found use in screening for enzymes that assemble glycosides. In the general scheme introduced by Mayer and colleagues, the chromogenic substrate is cleaved by the coupling enzyme(s) only when the desired product – with both correct stereo- and regio-chemistry – is produced (Fig. 5c).88 This stands in contrast to indirect measurements such as enzymatic turnover or generalized product formation. This approach has been applied for the directed evolution of glycosynthases,89 and thioglycoligases.34 In both of these examples, the biosynthetic exo-β-glucosidase mutant produces a substrate for an endo-cellulase, resulting in ultimate fluorophore release (Fig. 5c).34,89 Interestingly, during the directed evolution of the Abg exo-β-glucosynthase by this approach, the identified mutations recapitulated interactions lost in creation of the glycosynthase mutant.89 It seems that by removing the catalytic nucleophile for creation of the glycosynthase, key interactions with the C2 hydroxyl (which are typically quite strong90) were also lost and only restored upon directed evolution.89 Excitingly, the best mutant from this screen turned over the glycosyl fluoride substrate with Glc-β-pNP as an acceptor with a kcat value over 1000-fold higher than the starting mutant and comparable to that for the native enzyme's hydrolysis of Glc-β-pNP.89,91 Such work highlights the ability of directed evolution campaigns to provide new understanding of how enzymes work.
Glycosyltransferase assays
While GHs are likely the best studied and engineered class of CAZymes, a number of assays for GTs have also been developed and implemented allowing high throughput engineering. In fact, some GT activities can be monitored using activated chromogenic substrates in a manner similar to those of many screens for GHs. In these assays, the GT of interest generates their nucleotide sugar donors in situ upon being presented with activated substrates of the opposite anomeric configuration to that of their nucleotide sugar donors (e.g. Neu5Ac-α-pNP and Neu5Ac-β-CMP) and a catalytic amount of the corresponding nucleotide (Fig. 6a).92 This in situ generation of nucleotide sugar donor results in the release of a small amount of chromophore. In the presence of a suitable acceptor, the required nucleotide can be regenerated, and so the enzyme can carry out repeated turnovers of the chromogenic substrate, thereby enabling direct detection of catalytic activity. Alternatively, GTs can also be used to glycosylate hydroxyl groups on fluorescent molecules (commonly 4-methylumbelliferone) such that the enzymatic modification results in loss of fluorescence.93,94 This latter approach has been applied to both GTs94 and O-glycoligases.95 These methodologies have been useful not only in screening the donor and acceptor specificity of GTs96,97 but also in directed evolution. In particular, OleD from Streptomyces antibioticus has been evolved to produce diverse nucleotide sugars using this method.98 As well, the approach has been further extended to GT evolution for the glycorandomization of natural products.93,94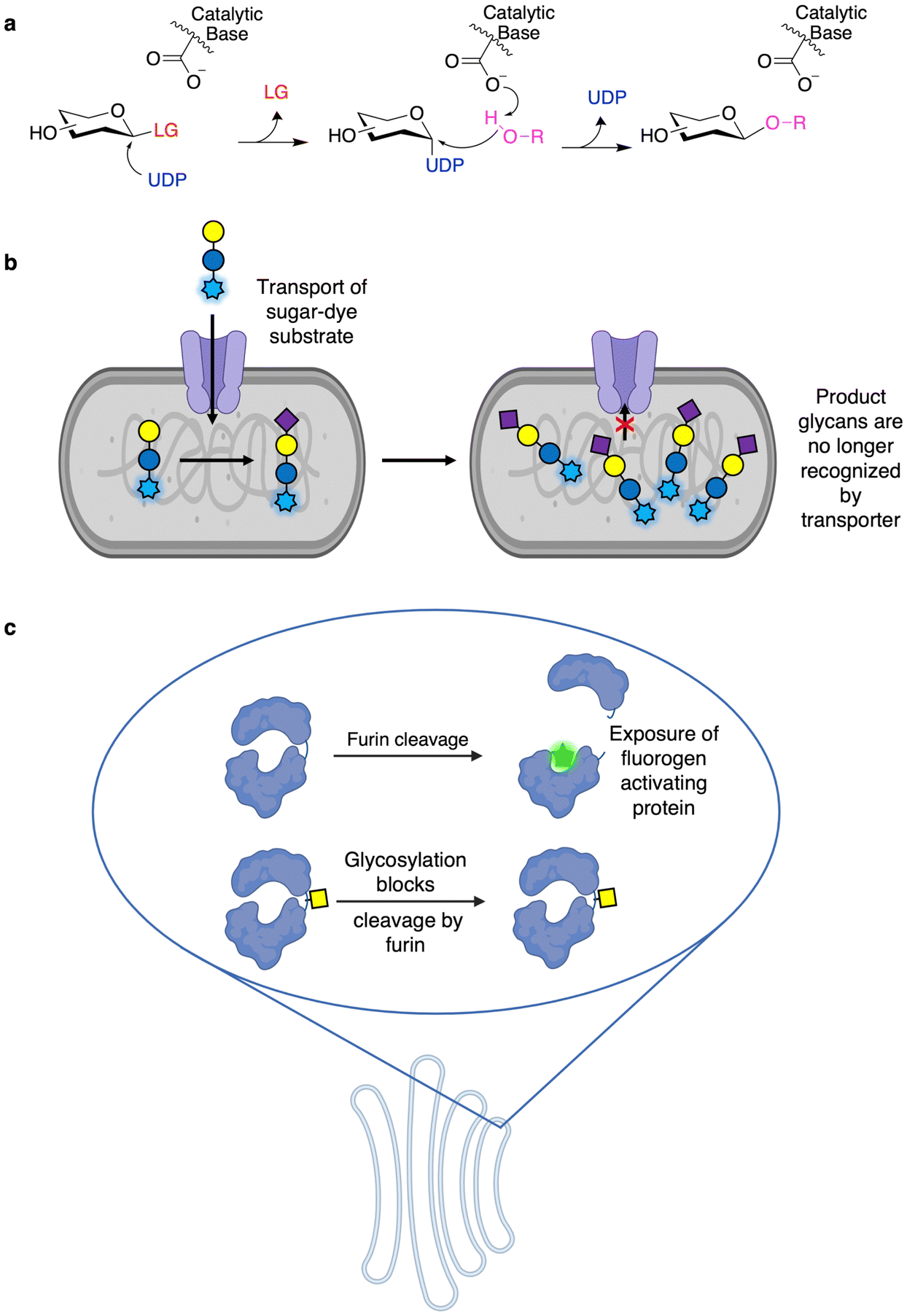 | ||
| Fig. 6 A selection of screens used for GTs. (a) Use of activated donors to screen for GT activities. In this assay, the GT catalyzes the formation of a nucleotide sugar when presented with a sufficiently activated glycan donor and a catalytic amount of nucleotide. This results in the release of a small amount of chromophore from the activated sugar. In the presence of a suitable glycan acceptor, the nucleotide can be regenerated multiple times resulting in readily detectable release of chromophore. (b) The general method for GT screening by FACS. In this method, fluorescently labeled sugars are taken up by a transporter-mediated mechanism. They are subsequently modified by the GT of interest resulting in a product that cannot be recognized by the transporter, hence the accumulation of a fluorescent product. This allows FACS sorting of cells containing active enzyme. (c), Use of mammalian processing pathways for GT activity screening. In this approach glycosylation of a furin cleavage site results in protection from furin cleavage within the Golgi apparatus. However, if no glycosylation occurs, the protein can be cleaved to expose a fluorogen-activating protein for subsequent labeling. | ||
Fundamental work on the screening of CAZymes by FACS includes the generalizable GT assay developed by Aharoni and colleagues.99 In this method, a glycoside conjugated to a fluorophore is transported into the cell via a sugar transporter (Fig. 6b). The fluorescent substrate is then glycosylated in the cell by the GT of interest and, once so modified, is no longer recognized by the transporter, resulting in accumulation of fluorescent substrate within cells containing active enzymes, which can then be sorted by FACS.8,99,100 This method has been applied to the directed evolution of sialyltransferases, galactosyltransferases, and fucosyltransferases.8,99,100 In an example of “you get what you screen for”, the initial iteration of this methodology resulted in the development of a hydrophobic binding pocket on the enzyme to which the dye moiety could bind, thereby improving reaction rates.99 This has been avoided in subsequent studies through the simultaneous use of two chemically distinct dyes such that the enzyme is not selected just through improved dye binding.100 While thus far this method has only been applied towards galactoside substrates (as transported by LacY), expression of different sugar transporters may further enhance the substrate scope of this method.
In mammalian cells, processing steps in the protein export pathway can be exploited for screening. In the approach developed by the Lindstedt laboratory, loss of O-glycosylation can be detected by furin cleavage of the aglycosylated protein within the trans Golgi network to reveal a fluorogen-activating protein (Fig. 6c).101,102 This has been used to probe compartmentalization of enzymes within the Golgi apparatus, the roles of different glycan biosynthesis genes, as well as in the identification of inhibitors of mucin-type O-glycosylation.101,102 While this method has not been used for directed evolution – and directed evolution is typically much more difficult in mammalian cells compared to bacteria or yeast103 – this does offer a useful method for engineering enzymes in a meaningful environment. This is especially the case since it offers access to enzymes which may be recalcitrant to recombinant expression in microbial systems.
Glycan binding proteins for product detection
Most glycoconjugates are extracellular. This simple fact enables convenient screening methods through use of fluorescently labelled binding proteins. Such methods can readily use FACS for facile ultrahigh-throughput screening (Fig. 7). For example, polysialyltransferase activity can be screened by FACS through use of a GFP-fused inactive polysialic acid hydrolase for product detection.104 Similarly, ultrahigh-throughput screening for enhanced production of eukaryotic N- and O-glycans in E. coli has also been explored by FACS.105,106 In these methods, a fluorescently labelled lectin or antibody is used to probe cell surface glycosylation.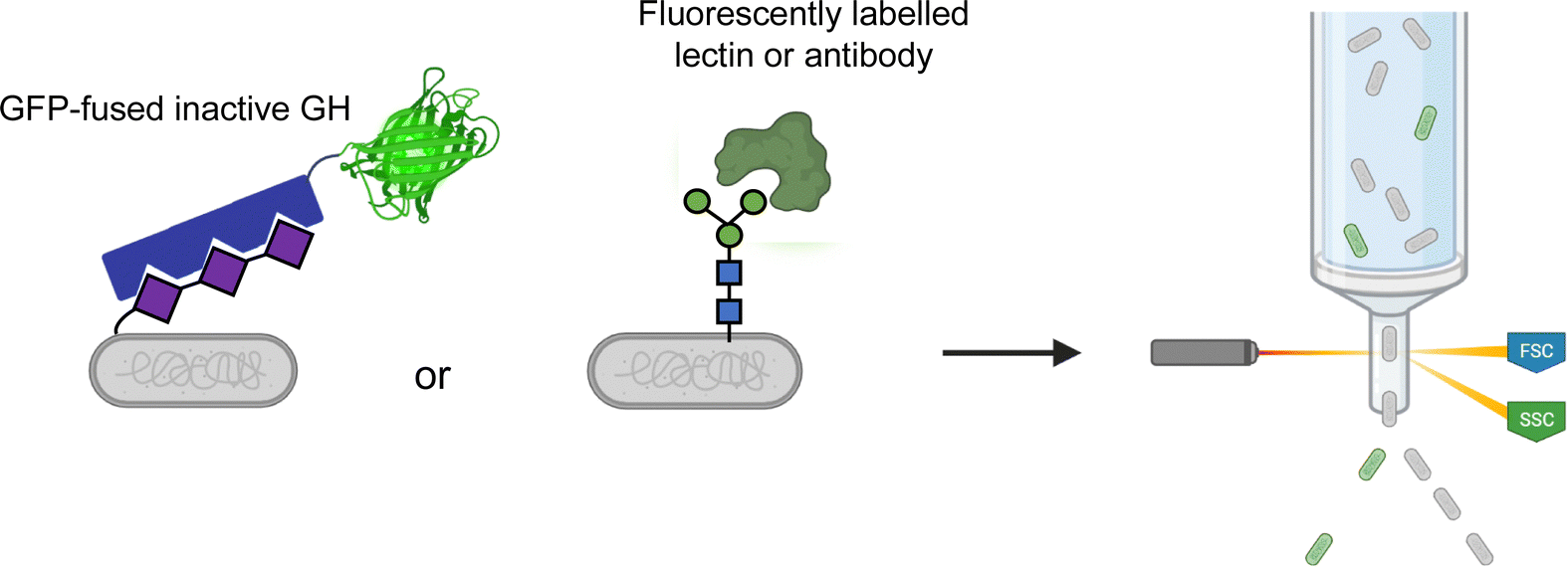 | ||
| Fig. 7 Use of glycan-binding proteins for FACS-based screening. Inactivated GHs with appended GFP tags or fluorescently labeled lectins or antibodies can be used to label cells carrying the glycans of interest. These cells can then be sorted via FACS at ultrahigh-throughputs. | ||
The DeLisa group has also developed a method for improvement of bacterial oligosaccharyltransferases (OSTs) wherein secreted glycoproteins are detected by immunoblotting.107 This offers a potential improvement over cell-based screens as the assay is not confounded by the build-up of lipid-linked oligosaccharides which may be present in the cell membrane. Bacterial OSTs are useful for en bloc transfer of a number of different glycan structures, including eukaryotic N-glycans,108 to Asn residues within a D/E-X-N-X-S/T consensus sequence.109 Relaxation of this specificity is desirable as it allows for use of OSTs with more diverse protein targets including eukaryotic proteins. Ollis et al. were able to apply this immunoblot-based method for the directed evolution of an OST from Campylobacter jejuni (PglB) such that it had a more relaxed sequence specificity.107 This was done through site-saturation mutagenesis of two Arg residues thought to form salt bridges with the D/E in the consensus sequence.107 In applying this, they were able to engineer PglB such that it recognized the eukaryotic N-X-S/T sequon and could carry out effective glycosylation of RNase A with limited activity against the canonical bacterial sequon.107 This same immunoblotting approach has since been used to identify in-depth how insertion of different glycosylation sites (via inclusion of short sequons) affected the function and stability of a number of diverse proteins in a process they coined as “shotgun scanning glycomutagenesis”.110 Intriguingly, Li et al. showed that when shotgun scanning glycomutagenesis was applied to the scFv of an anti-HER2 antibody, a number of sites could be glycosylated in a manner that readily increased the affinity of the scFV for HER2 and/or increased the stability of the protein.110 Such results and methods open the door to new avenues in the glyco-optimization of therapeutics.
In an interesting case of combining different approaches, Keys and colleagues engineered a polysialyltransferase for the synthesis of well-defined polysialylated proteins with low dispersity.111 Decreased product dispersity is a difficult enzymatic property to screen for as time- intensive HPLC analysis of the products is the only method with sufficient resolution for the task. To overcome this, Keys et al. employed an ingenious neutral drift approach which combined high-throughput screening with lower-throughput characterization by HPLC.111 Neutral drift is a method for protein engineering in which after the introduction of mutations (often, but not necessarily, at a relatively high rate111,112) – rather than screening for variants with higher activity – the experimenter only screens for variants with similar activity to the WT enzyme.112 Through rounds of neutral drift, one can thus generate a pool of variants that are both highly divergent but still functional – enabling efficient evolution with decreased screening burden.112 As well, the produced protein scaffolds are often highly evolvable as the high mutational load frequently results in the enrichment of stabilizing mutations.111–113 For each round of their campaign, Keys et al. employed a previously developed colony immunoblotting assay to identify active polysialyltransferases from screens of >104 clones in each round.114 This method does not allow for differentiation of polydispersity, and so for each round, they then analyzed the product profiles of just 100–200 clones by HPLC.111 Ultimately, in their third round of screening, they characterized just 51 clones by HPLC and identified a single residue which was able to dictate whether the polysialyltransferases produced products with small or large dispersity. This single residue switch was key in modulating the ability of the enzyme to bind elongated polysialic acid polymers – thus shifting the enzyme between processive and distributive modes of action (and high dispersity and low dispersity product profiles respectively).111
Screens using glycan-binding proteins have also been utilized in microtitre plate-based formats. For example, Hancock et al. carried out the directed evolution of glycosphingolipid synthase with product detection by use of an ELISA.115 In this work, they sought to expand the lipid specificity of an endoglycoceramidase glycosynthase mutant originally derived from Rhodococcus sp. Strain M-777 (EGC-II). While the naïve glycosynthase is largely sphingosine-specific, they sought to expand this activity towards phytosphingosine, for which the native enzyme has 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold less activity. This enhancement in activity could then enable use of the enzyme for synthesis of diverse gangliosides. The authors used the GM1-specific Cholera toxin B subunit as the binding protein in the assay. The Cholera toxin B subunit, while specific for the glycan portion of the ganglioside, is promiscuous with respect to the lipid and so ideal for this purpose. Binding of the Cholera toxin B subunit could then be detected by antibody binding coupled to horse radish peroxidase activity. From a screen of 10000 clones, a single mutation adjacent to the active site drove an 8100-fold increase in its activity towards phytosphingosine while maintaining activity against its native substrate. As a result, the mutant enzyme had near equal activity against sphingosine and phytosphingosine.115
000-fold less activity. This enhancement in activity could then enable use of the enzyme for synthesis of diverse gangliosides. The authors used the GM1-specific Cholera toxin B subunit as the binding protein in the assay. The Cholera toxin B subunit, while specific for the glycan portion of the ganglioside, is promiscuous with respect to the lipid and so ideal for this purpose. Binding of the Cholera toxin B subunit could then be detected by antibody binding coupled to horse radish peroxidase activity. From a screen of 10000 clones, a single mutation adjacent to the active site drove an 8100-fold increase in its activity towards phytosphingosine while maintaining activity against its native substrate. As a result, the mutant enzyme had near equal activity against sphingosine and phytosphingosine.115
High-throughput substrate screening
While not a traditional aspect of high-throughput screening for CAZymes, the high-throughput identification of preferred substrates for CAZymes is an important component of better understanding the mechanisms and specificities of CAZymes. As well, it opens the possibility of engineering substrates such that they are better recognized by the desired enzymes. For GHs, it is generally easy to identify the preferred non-reducing end glycoside (−1 subsite and beyond), but considerably more difficult to determine the identity and linkage of the leaving group at the reducing end (+1 subsite and beyond). At a low-throughput, different oligosaccharides can simply be assayed for activity. However, this depends upon the availability of diverse substrates with differing linkages and substituents; thus, introducing a challenge in terms of scope and scalability.A useful method for identifying preferred substrates of retaining GHs is through the use of 2F-sugar GH inactivators (Fig. 8a).116,117 In this approach, introduction of electronegative fluorine at the C2 position of the substrate results in a great reduction in the rates of both steps of the reaction through destabilization of the oxocarbenium ion-like transition states via inductive effects.117 When an activated leaving group is also incorporated to accelerate the first step, reaction of these inactivators with a GH results in accumulation of the covalent glycosyl-enzyme intermediate, thus inactivation of the enzyme. This intermediate is resistant to hydrolysis, but, remarkably, in the presence of a productively bound glycan acceptor, can be reactivated via transglycosylation.116 The identity of the preferred reducing end substituent can thus be identified by screening for (rates of) reactivation with libraries of different potential glycan acceptors in a method that is readily done in a plate-based format.116 Moreover, the identity of the linkage can also be determined through subsequent analysis of the rescue product.116 The initial study profiled the aglycone specificities of a number of GHs against a panel of glycosides and free sugars with the goal of using this information to guide GH-mediated glycoside synthesis.116 The approach has also been applied to study the substrate scope of E. coli glucuronidase and to better understand which non-carbohydrate small molecule substrates it acts on.118 Taking this even further, a synthetic gene library of GH1s was screened for their ability to cleave a library of glycosides modified individually with azide and amine substituents at the 3-, 4- and 6- positions.119 The aglycone specificities of the top eight hits were then profiled at high-throughput using a total of 83 different potential acceptors, allowing the identification of optimal enzyme/substrate combinations for synthesis of specific glycosides using the glycosynthase technology.119
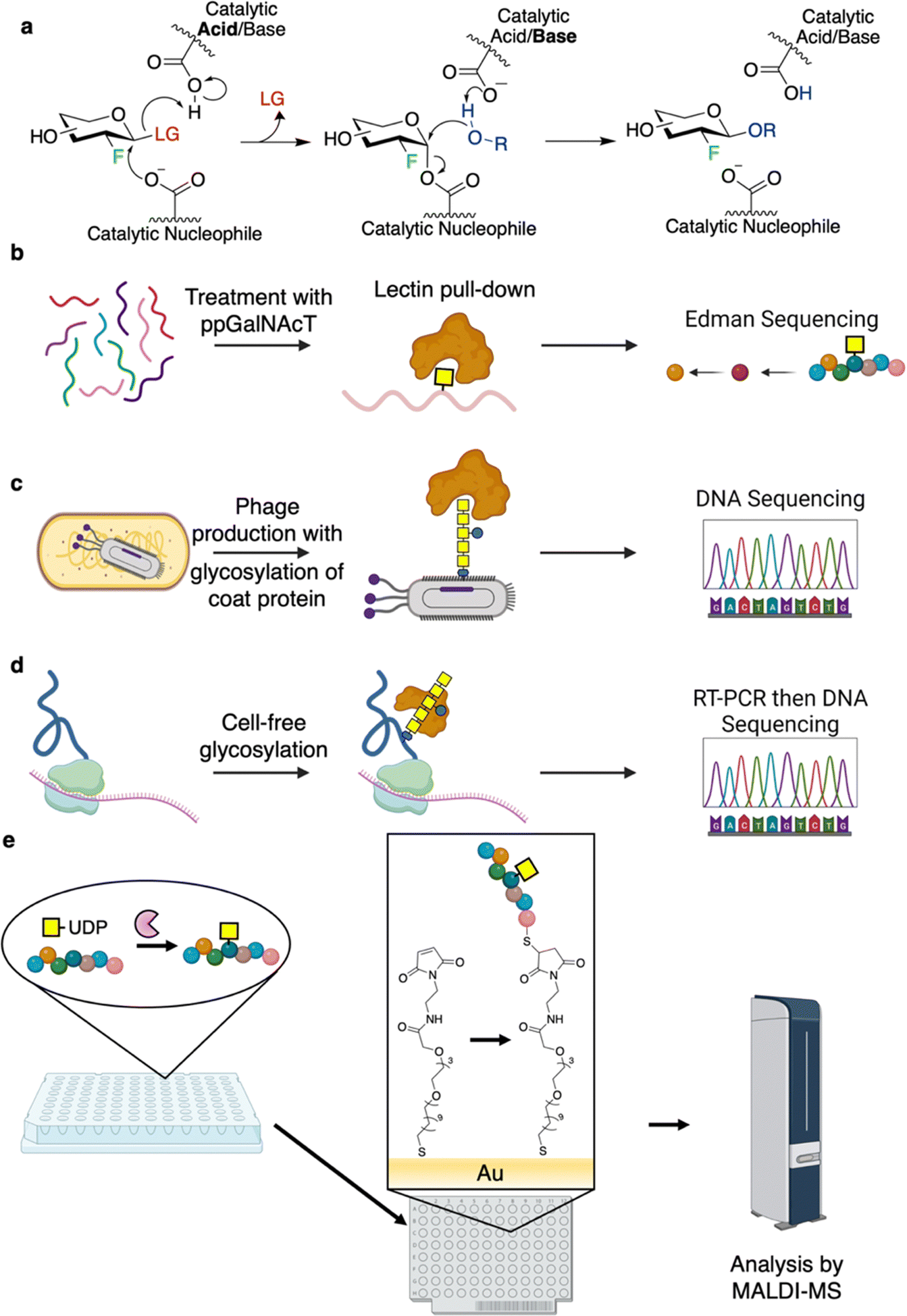 | ||
| Fig. 8 Methods for high-throughput substrate screening. (a) Use of a 2F-sugar inactivator to identify preferred reducing end substituent. In this assay, the enzyme is inactivated using a 2F-sugar inactivator, through accumulation of a glycosyl-enzyme intermediate. Enzyme activity can be recovered by transglycosylation in the presence of acceptors that bind productively in the aglycone site. (b)–(d) Affinity-based methods to identify preferred substrates for polypeptide glycosylating proteins. In (b) a pool of random peptides is glycosylated by a ppGalNAcT. The glycopeptides are then enriched using lectin affinity chromatography and analyzed by Edman sequencing. (c) and (d) Show phage display and mRNA display approaches for determination of preferred glycosylation sites/sequons. (e) Application of SAMDI-MS for identification of preferred sequons for GTs. | ||
The substrate preferences of GTs that act on polypeptides are of interest, especially with respect to the production of glycoproteins. Of particular interest have been the polypeptide N-acetylgalactosaminyltransferases (ppGalNAcTs), O-linked GlcNAc transferase (OGT), OSTs, and N-glycosyltransferases (NGTs). In seminal studies on ppGalNAcTs, sequence specificities were assessed by incubation of the enzymes with UDP-GalNAc and randomized pools of peptides. Subsequent purification of the modified glycopeptides by lectin affinity chromatography followed by Edman sequencing enabled facile identification of preferred substrate sequences (Fig. 8b).120–126 Results obtained have informed the software, IsoGlyP, which predicts ppGalNAcT glycosylation for a given sequence.127 Correspondingly, IsoGlyP has found considerable use in the design of synthetic sequons to target ppGalNAcT activity.128,129
Phage and mRNA display offer new pathways for determining sequence preferences and engineering bacterial N-glycosylation.130,131 In 2010, the Aebi group developed a method known as “GlycoPhage” (Fig. 8c).130 In this, a bacterial N-glycoprotein (AcrA from Campylobacter jejuni) is genetically linked to a phage coat protein (pIII). The AcrA-pIII fusion is exported to the periplasm upon expression where it can be glycosylated by OST. It is then incorporated into the phage progeny to make glycophage. The glycophage can then be detected and/or pulled down by a number of different methods. In the proof of concept study, the authors were able to enrich for sequences that could be glycosylated by C. jejuni OST by varying the N-glycosylation sequons and panning for binding using a glycan-specific antibody.130 This method should, in principle, be applicable to evolving any enzyme involved in glycan biosynthesis.130 In 2022, the DeLisa group described an mRNA display screen for glycoprotein sequon engineering (Fig. 8d).131 In this method, the protein of interest is fused to a short sequence which stalls the ribosome such that the nascent polypeptide and mRNA are still bound within the ribosome.131 The polypeptide can then be glycosylated through cell-free glycoprotein synthesis, the glycoprotein enriched by affinity purification, and the sequence of the RNA within the stalled ribosome determined by sequencing of the RT-PCR product.131 While not described within the paper, this method should be applicable to engineering/determination of glycosylation sequons and other aspects of protein glycosylation and affords a much higher throughput than cell-based methods (>109 clones per screen).131
A number of studies have applied self-assembled monolayers for matrix-assisted desorption/ionization mass spectrometry (SAMDI-MS) for determination of sequence specificities (Fig. 8e). The workflows for the campaigns outlined here are as follows: glycosylation reactions are carried out in microtitre plates, and the reaction mixtures are applied to a plate with a coating that immobilizes the substrate/products (e.g. reactive maleimide to couple to Cys-containing peptides or Ni-NTA for His-tagged protein binding).132–134 Following this, the plates are washed and then analyzed by MALDI-TOF MS.132–134 A series of studies by the Jewett and Mrksich groups have shown the utility of SAMDI for characterizing the specificities of OGT, ppGalNAcTs, and NGTs.132–134 These experiments have enabled the identification of a number of “GlycTags” for modification using NGTs.133 In fact, by profiling a number of different NGTs, and determining sequence preferences of these enzymes, semi-orthogonal GlycTags were identified. With this knowledge in hand, site-selective protein glycosylation was then carried out using these NGT-GlycTag pairs.133 Such an approach is particularly useful since introduction of the Glc or GlcN handles by NGTs can allow for subsequent extension to produce a glycan of interest.28,133,135 In this way, through cycles of handle attachment and glycan installation, proteins of defined glycosylation at different sites can be readily prepared.133
Transcription factor biosensors for enzymatic activities
In principle, transcription factor-based biosensors offer a flexible method for monitoring the synthesis or degradation of different compounds.136 Such approaches rely on co-opting the abilities of organisms to sense different small molecules. This is then often used to induce expression of a fluorescent protein, with subsequent sorting of active clones by FACS at ultrahigh-throughput.136 In practice so far, most established screens for CAZymes actually detect the release of different artificial leaving groups.137,138 The benefit of applying these methods over non-transcription factor-based screens, is that one can conceivably use less material compared to that needed for plate-based screens (including both substrates and consumables such as plates) while screening many more clones with the same substrate. For example, in a general method to screen for hydrolases, Choi et al. applied an engineered phenol-sensitive transcription factor to screen for release of nitrophenols.137 Many available chromogenic glycoside substrates produce nitrophenols upon cleavage. And so, by using a GFP reporter under control of phenol-induced transcription, the authors were able to show that addition of pNP-cellotriose to the culture media enabled ready differentiation of cellulase-expressing cells from non-expressers.137 While the authors were able to apply this method for functional metagenomic screening for phosphatases, further work is required to show its applicability for GHs.137Catalyze or die – selections for better CAZymes
Owing to central roles of sugars in primary metabolism, the selection of clones on the basis of survival and/or improved growth when certain carbohydrates are used as the sole carbon source can be used to identify novel and improved enzyme variants.139–141 Such selections are often used in conjunction with more quantitative methods in order to further distinguish improved clones.80,142,143However, there have been a number of creative implementations of selections in the engineering of CAZymes beyond simple growth screens. As an example, the Cornish laboratory has shown the utility of chemical complementation for engineering of glycosynthases144 and cellulases.145 In brief, this method relies on the use of proteins that can recognize different chemical moieties. These are then fused to DNA-binding proteins such that gene expression is either activated or blocked when the small molecule of interest is present. While this approach has been used for glycosynthases and cellulases, and should be highly scalable, it has seen limited use since its first implementations.144,145 Of recent note is also the use of a combined antibiotic selection and auxotroph complementation strategy by Bennet and colleagues for the evolution of a sialidase to specifically cleave deaminated neuraminic acid (Kdn).146 In this, activity against Kdn was selected for in a tyrosine auxotroph line of E. coli by supplementation of the culture media with Kdn-caged tyrosine. Meanwhile, the native Neu5Acase activity of the sialidase was selected against by inclusion of Neu5Ac-α2,6-Gal-chloramphenicol. In applying this they were able to identify variants with up to 20-fold enhancements in Kdnase activity and up to 60-fold change in specificity.146
Into parts unknown
Enzyme discovery and engineering campaigns have offered not only improved enzymes, but also new understandings and insights into how known enzymes work and the evolutionary paths that have been undertaken in Nature. Progress has also come from computational approaches, especially in light of the success of programs such as AlphaFold in the sequence-based prediction of protein structures.147 Design of enzymes has been a long sought-after goal that should allow the development of enzymes with precisely tuned reactivity.148 Computational enzyme design, using programs for structure prediction such as RosETTA, especially when used in combination with directed evolution have scored a number of successes.148 Recent advances have brought this goal closer to general use for CAZymes, as described below.Artificial intelligence has yielded CAZymes that are not as yet found in Nature.149 In a remarkable case, Madani et al. used a large language model – trained on sequence data from >19000 PFAM families with further tuning on lysozyme sequences from CAZy families GH24, GH73, GH108, and others not yet classified within CAZyDB – to design a suite of lysozymes. These designed lysozymes were found to have similar activity profiles – both in the number of enzymes with activity (73% of designed enzymes) and the level of activity – to those of a set of random natural enzymes from the lysozyme families used for training. These screens used fluorescence-quenched fluorescein-labeled cell wall from Micrococcus lysodeikticus as a substrate. Further characterization showed that sequences with identity as low as 31% to any known enzyme were found to have activities and structural folds similar to those of known lysozymes.149 While this does not yet offer the ability to explicitly encode/discover new activities, it does offer a means for exploration of sequences not found in Nature.
The inherent reactivities of metals have been utilized in a number of natural and engineered enzymatic activities.148,150,151 For many hydrolytic enzymes, metals such as Zn2+ act as Lewis acids and activate a water molecule for nucleophilic attack.152 However, metals are not generally used for catalysis in this manner by GHs. In the closest instance, families GH127 and GH146 utilize a Zn2+ ion to activate the thiol of a Cys residue that then acts as the nucleophile.153,154 This leaves one to wonder whether there is a reason why Nature has not utilized “metalloglycosidases” more broadly. In recent work, β-glucosidase activity was introduced into OmpF (a porin) via introduction of a Zn2+ binding site with an open coordination site.155 Through rounds of directed evolution using a simple X-Glc and cellobiose-growth dependent agar plate assay, the authors were able to improve the activity of this enzyme against β-glucosides by two orders of magnitude.155 Curiously, these enzymes show a strong but incomplete dependence upon Zn2+ for hydrolysis with a competing non-metal dependent reaction.155 While the best enzymes were still relatively slow (kcat/KM of 10 min−1 M−1), it will be interesting to see how well metalloenzymes can be adapted to glycoside hydrolysis. Given the “late” transition states associated with glycosyl transfer via oxocarbenium ion-like transition states5 it may be that there is little advantage to increasing the nucleophilicity of water in this way. Analogous work on phosphotriesterases has shown that promiscuous enzymes that operate by different mechanisms than those found in Nature, can be evolved to have the same catalytic efficiency as the natural enzymes.156 Albeit in the case of phosphotriesterases, the natural enzymes are metal-dependent while the enzyme evolved in the laboratory does not use a metal for catalysis.156
Recent advances in ultrahigh-throughput screening and beyond
Functional metagenomics combined with droplet microfluidics has proven to be a potent combination for enzyme discovery. If the given activity exists, identification of this activity via functional metagenomics is often a matter of screening a sufficient number of clones. With the appropriate source material, hit rates in fosmid screens for common activities such as cellulases are ∼1 hit/384 fosmids (or ∼15000 genes),43 but for more complex activities such as endo-O-glycan degradation, hit rates can drop to ∼1 hit/5000 fosmids (or ∼184000 genes).157 Thus, the ability to rapidly screen through the swathes of inactive clones is key. Droplet-based microfluidics offers a method to achieve the needed throughputs with minimal expenditure of material. Excellent work on this matter has been carried out by the Hollfelder laboratory and its collaborators.59,158,159 For example, a single GH3 glucuronidase with distant relation to any characterized GH3 (37% sequence identity) was identified by screening a small insert library of ∼106 clones.158 This was the first time that glucuronidase activity had been identified in the GH3 family despite its extensive study.158 Indeed, such work highlights the ability of functional metagenomics to identify highly active catalysts which would not otherwise be explored.10,49,158,160
Directed evolution and deep characterization of GHs can also be carried out in droplets.161 Romero and colleagues performed deep mutational scanning of a GH1 from Streptomyces sp. (Bgl3) in droplets and characterized millions of mutants in terms of activity and thermostability.161 This was the first deep mutational scan of this class of enzyme. Curiously, it revealed residues essential for function which were distant from the active site and also non-conserved in other GH1 family members.161 Such results provide clear indications that, even though highly conserved sites are indispensable for function, proteins acquire local adaptations and networks of residues required for enzyme function over the course of evolution.63 The authors further show the utility of droplet microfluidics for characterizing variants by heat-treating the droplets and then screening for thermostable variants.161
In 2023, Lipsh-Sokolik et al. were able to show the utility of activity-based protein profiling for identification of highly active designed GH10 xylanases.162 Activity based probes based upon epoxide and aziridine derivatives of glycosides have been developed extensively by the Overkleeft group.163 These probes enable capture of retaining Koshland GHs via formation of an irreversibly linked cyclitol upon attack of the epoxide/aziridine by the enzyme's catalytic nucleophile (Fig. 9a).163 In this screen, the probes carried a fluorophore and thus linkage of the probes to a yeast surface displayed enzyme allowed for separation of active variants via FACS (Fig. 9b).162 Diverse xylanases were generated in silico via recombination of natural GH10 sequences with further re-design and optimization to eliminate incompatibilities due to epistasis. By applying this FACS-based approach, libraries of >105 variants could be readily screened, with 103–104 active designed xylanases being identified in these screens. While the screen itself only requires the enzymes to carry out the first step in the retaining Koshland mechanism, most of the identified enzymes were capable of carrying out the full catalytic cycle. Moreover, by using different probes, the authors were able to further separate variants with activity on xylan, cellulose, or both. Further work using these activity-based probes for CAZyme engineering seems likely in addition to their previously established utility in identification of enzymes from complex mixtures,164 and inhibitor discovery,165 amongst a number of other applications.163 While there may be concerns about the ability of the method to identify highly active variants – as only a single, partial turnover can be achieved – a number of other successfully applied methodologies for enzyme engineering also only allow for a single turnover of the screening substrate.166,167
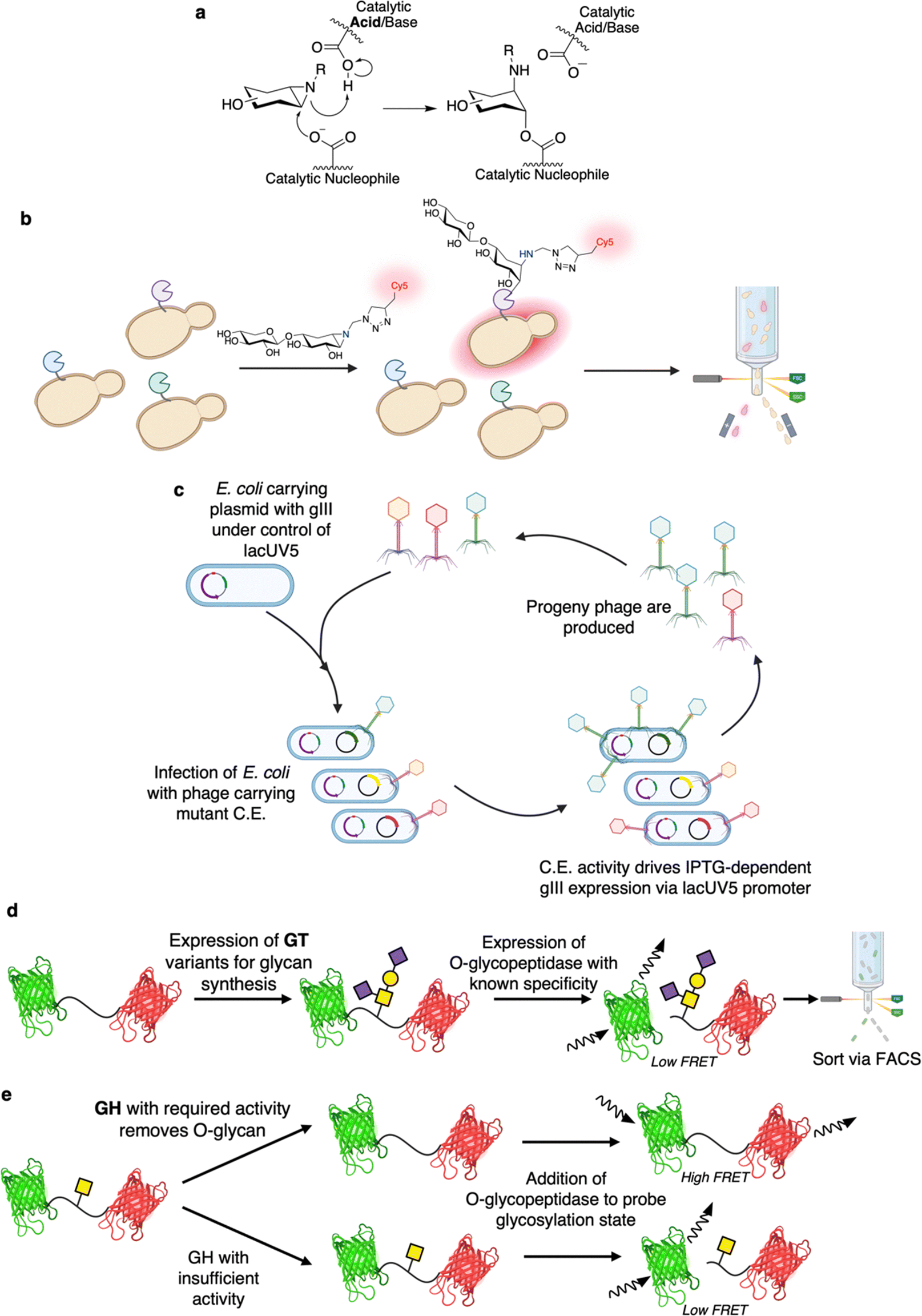 | ||
| Fig. 9 Recent advances in ultrahigh-throughput screening for CAZymes. (a) Mechanism for inactivation and labeling of retaining Koshland GHs by aziridine bearing activity-based probes. (b) Use of fluorescent aziridine-bearing activity-based probes for screening of xylanases via yeast surface display and FACS. In this screen, active xylanase variants are fluorescently labeled as they are covalently inactivated by the aziridine substrate upon nucleophilic attack at the pseudo-anomeric centre. (c) Workflow for application of PACS in the directed evolution of CEs. Note that PACS differs from PACE only in that an additional mutagenesis plasmid is included in PACE compared to PACS. MELiORA can be applied for the engineering of GTs (panel (d)) and GHs (panel (e)). In brief, this method relies on linking of changes to the glycosylation state (through the activity of GHs or GTs) of a fluorescent protein FRET pair to proteolytic cleavage via the activity of O-glycopeptidases (peptidases that require certain O-glycan structures on the substrate for activity). | ||
Phage assisted continuous evolution (PACE) has been a largely unexplored method for CAZyme engineering. One of the great benefits of continuous evolution strategies such as PACE is that they enable rapid, unsupervised, explorations of sequence space.168 Indeed, depending upon the experimental setup, almost 200 rounds of evolution can be carried out in a single week whereas many directed evolution campaigns would only finish a single round in that time.169 The basis on which PACE was developed is that the activity of interest must be linked to successful synthesis of the pIII coat protein (Fig. 9c).170 In brief, E. coli are used as hosts for phage which lack the pIII gene (gIII) but carry the gene of interest.170 These E. coli host cells carry gIII under the control of a genetic circuit that is activated by the activity of interest.170 Replication of the phage is thus dependent upon the activity of interest.170 The E. coli also carry a plasmid for increased rate of mutagenesis that enables continual diversification of the library.170 And so, over time, phage encoded enzyme variants capable of driving pIII expression (and thus phage replication) are continually selected for and continually diversified.170 PACE has been shown to be useful for evolution of for a number of enzymes including proteases, base editors, RNA polymerases, and aminoacyl-tRNA synthetases.170 With respect to CAZymes, in the closest available example, PACE has been shown to theoretically enable screening for CEs capable of de-esterifying IPTG (Fig. 9c).171 Note though that, while esterases with improved activity could be found by PACE, the authors actually had greater success with a non-continuous evolution approach (phage assisted continuous selection (PACS)). In PACS, mutants are mutagenized in vitro prior to introduction into phage rather than relying upon a mutagenesis-enhancing plasmid for continuous mutagenesis in vivo.171 The study also identified a number of different esterified substrates with which one could readily apply PACE or PACS.171 In principle, many more substrates could be employed as long as transcription factor-activator pairs with requisite specificities are known. And so, better understandings of the transcriptional activation in microbial carbohydrate degradation pathways (such as polysaccharide utilization loci) could offer further avenues for exploration.172
The Withers laboratory has developed the mucinase/O-glycopeptidase enabled linking of O-glycosylation and related activities (MELiORA) method for use in engineering of enzymes active on mucin-type O-glycoproteins (Fig. 9d and e).173 With respect to CAZyme engineering, MELiORA uses O-glycopeptidases (a type of peptidase with absolute specificity for mucin-type O-glycosylated protein substrates174) to identify the glycosylation state of a fluorescent protein FRET probe. Using this, enzymes that add or remove glycans, such as GTs (Fig. 9d) and GHs (Fig. 9e), can be assayed with a read-out based upon the change in FRET arising from O-glycopeptidase-catalysed cleavage of the probe. Because all of the assay components can be expressed within the cell (i.e. the probe, the glycosyltransferases and other enzymes required for glycan synthesis,128,175 the O-glycopeptidase, etc.), enzyme variants can also be readily screened at ultrahigh-throughput via FACS. Even for plate-based assays, the approach still enables screening using complex mucin-type O-glycoprotein substrates with minimal costs; as the glycoprotein substrates – with diverse, defined glycan structures – can be readily produced in E. coli with high yields.128,173,175 In many ways MELiORA is ideal as the protein substrate is likely similar, at least locally, to that on which one would want the enzymes to ultimately act. When used in screens for glycan biosynthetic enzymes, MELiORA also offers the same benefits as in the coupled enzyme screen devised by Mayer and co-workers.88 That is, with the use of the appropriate O-glycopeptidase, it is only when the glycan of interest is produced that there is any change in fluorescence. Moreover, the method is not limited to a FRET probe as initially described. Rather, almost any screening system for peptidase activities should be readily amenable to MELiORA, thus these screens can be converted such that they enable screening for CAZymes.176 While MELiORA was only applied to the directed evolution of an O-glycopeptidase in the initial study, the broader applicability of the approach should be borne out by further work in the field.173
Concluding remarks and the future of the field
Nature has evolved CAZymes to carry out diverse reactions with a high degree of specificity. In light of this, it is perhaps then even more remarkable that these enzymes can be so readily engineered to better suit human purposes. The implementation of high-throughput screens is a vital step in the engineering of these enzymes. However, as highlighted in this review, there are clear benefits and drawbacks to individual screens with respect to their scalability and applicability. The scalability of many methods is likely to improve over time as miniaturization using techniques such as droplet microfluidics becomes more widespread.57 What is a remaining hurdle then is the fact that substantial discrepancies in screened activity as compared to desired activity have been observed in a number of CAZyme engineering and discovery campaigns.87,99,157,177 As such, a few approaches stand out for their ability to use substrates with structures as close as possible to the natural/ultimate desired substrate, and be carried out in a manner similar to that ultimately required.A great benefit of using natural substrates in screening is that the observed activity improvements should be readily translatable to the desired task and conditions. However, many of these screens suffer from the need for processing steps (e.g. addition of developing reagents, additional incubation steps, or elevated incubation temperatures to drive chemical reactions) that limit throughput and add difficulty. The creativity and ingenuity displayed within the field, particularly in the use of coupled assays and highly relevant substrates (or ideally the desired substrate) to detect activity stand as highlights and portend future developments. In moving this forward there is likely to be synergy between the engineering of CAZymes and the engineering of the coupling enzymes used (whether they be oxidases,79,81 dehydrogenases,78,86O-glycopeptidases173 or whatever).
As is evident from their absence from this review, but widespread use within the field, there are a number of enzymes for which little high-throughput engineering has been carried out. Prominent examples of this are the ENGases – incredibly powerful enzymes for remodeling of N-glycans to produce designer glycoproteins.178 The workflow for such glycoengineering often employs both a hydrolytic enzyme (to remove the native N-glycans) and then glycosynthase mutants to install the desired glycan structure. Serious engineering work could generate new enzymes of improved specificity, perhaps allowing simple site-selective glycosylations to be performed. In other enzyme classes, limited work has been performed on engineering enzymes such as PLs and lytic polysaccharide monooxygenases (LPMOs) (which are classified in the CAZy database in a number of auxiliary activity families179 and have attracted attention due to their ability to initiate degradation of otherwise recalcitrant crystalline cellulose and chitin6,179). Indeed, there are only two reported LPMO directed evolution campaigns.180,181 In a rather impressive study, Jensen et al. employed high-throughput mass spectrometry-based screening to almost completely shift an LMPO's substrate specificity from cellulose to chitin.180 In light of these examples, it is clear that further work is needed within the field to expand the available assays for these enzymes and thereby enable improvement of what are already highly useful and in-demand enzymes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. F. W. was supported by a four-year doctoral fellowship from UBC during the writing of this review. We would like to further thank the Canadian Glycomics Network (GlycoNet), the Natural Sciences and Engineering Research Council of Canada and the Canadian Institutes of Health Research for additional financial support. The table of contents graphic and Fig. 5–9 were created in part or in whole with Biorender.com.References
- B. A. Stankiewicz, D. E. G. Briggs, R. P. Evershed, M. B. Flannery and M. Wuttke, Science, 1997, 276, 1541–1543 CrossRef CAS.
- S. L. Richter, A. H. Johnson, M. M. Dranoff, B. A. LePage and C. J. Williams, Geochim. Cosmochim. Acta, 2008, 72, 2744–2753 CrossRef CAS.
- R. A. Laine, Glycobiology, 1994, 4, 759–767 CrossRef CAS PubMed.
- M. Critcher, A. A. Hassan and M. L. Huang, Trends Biochem. Sci., 2022, 47, 492–505 CrossRef CAS PubMed.
- D. L. Zechel and S. G. Withers, Acc. Chem. Res., 2000, 33, 11–18 CrossRef CAS PubMed.
- G. R. Hemsworth, G. Déjean, G. J. Davies and H. Brumer, Biochem. Soc. Trans., 2016, 44, 94–108 CrossRef CAS PubMed.
- S. Chakraborty, A. Rani, A. Dhillon and A. Goyal, in Current Developments in Biotechnology and Bioengineering, eds. A. Pandey, S. Negi and C. R. Soccol, Elsevier, 2017, pp. 527–539 Search PubMed.
- Y. Tan, Y. Zhang, Y. Han, H. Liu, H. Chen, F. Ma, S. G. Withers, Y. Feng and G. Yang, Sci. Adv., 2019, 5, eaaw8451 CrossRef CAS PubMed.
- T. Verhaeghe, K. De Winter, M. Berland, R. De Vreese, M. D’Hooghe, B. Offmann and T. Desmet, Chem. Commun., 2016, 52, 3687–3689 RSC.
- P. Rahfeld, L. Sim, H. Moon, I. Constantinescu, C. Morgan-Lang, S. J. Hallam, J. N. Kizhakkedathu and S. G. Withers, Nat. Microbiol., 2019, 4, 1475–1485 CrossRef CAS PubMed.
- D. H. Kwan, I. Constantinescu, R. Chapanian, M. A. Higgins, M. P. Kötzler, E. Samain, A. B. Boraston, J. N. Kizhakkedathu and S. G. Withers, J. Am. Chem. Soc., 2015, 137, 5695–5705 CrossRef CAS PubMed.
- Q. P. Y. P. Liu, G. Sulzenbacher, H. P. Yuan, E. P. Bennett, G. Pietz, K. Saunders, J. Spence, E. Nudelman, S. B. Levery, T. White, J. M. Neveu, W. S. Lane, Y. Bourne, M. L. Olsson, B. Henrissat and H. Clausen, Nat. Biotechnol., 2007, 25, 454–464 CrossRef CAS PubMed.
- A. Wang, R. V. P. Ribeiro, A. Ali, E. Brambate, E. Abdelnour-Berchtold, V. Michaelsen, Y. Zhang, P. Rahfeld, H. Moon, H. Gokhale, A. Gazzalle, P. Pal, M. Liu, T. K. Waddell, C. Cserti-Gazdewich, K. Tinckam, J. N. Kizhakkedathu, L. West, S. Keshavjee, S. G. Withers and M. Cypel, Sci. Transl. Med., 2022, 14, eabm7190 CrossRef CAS PubMed.
- T. Lindhout, U. Iqbal, L. M. Willis, A. N. Reid, J. Li, X. Liu, M. Moreno and W. W. Wakarchuk, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 7397–7402 CrossRef CAS PubMed.
- A. Geissner, L. Baumann, T. J. Morley, A. K. O. Wong, L. Sim, J. R. Rich, P. P. L. So, E. M. Dullaghan, E. Lessard, U. Iqbal, M. Moreno, W. W. Wakarchuk and S. G. Withers, ACS Cent. Sci., 2021, 7, 345–354 CrossRef CAS PubMed.
- T. B. Parsons, W. B. Struwe, J. Gault, K. Yamamoto, T. A. Taylor, R. Raj, K. Wals, S. Mohammed, C. V. Robinson, J. L. P. Benesch and B. G. Davis, Angew. Chem., Int. Ed., 2016, 55, 2361–2367 CrossRef CAS PubMed.
- C. Li, G. Chong, G. Zong, D. A. Knorr, S. Bournazos, A. H. Aytenfisu, G. K. Henry, J. V. Ravetch, A. D. MacKerell and L.-X. Wang, J. Am. Chem. Soc., 2021, 143, 7828–7838 CrossRef CAS PubMed.
- J. F. Wardman, R. K. Bains, P. Rahfeld and S. G. Withers, Nat. Rev. Microbiol., 2022, 20, 542–556 CrossRef CAS PubMed.
- M. Critcher, T. O’Leary and M. L. Huang, Biochem. J., 2021, 478, 703–719 CrossRef CAS PubMed.
- M. Taupp, K. Mewis and S. J. Hallam, Curr. Opin. Biotechnol, 2011, 22, 465–472 CrossRef CAS PubMed.
- F. H. Arnold, Angew. Chem., Int. Ed., 2019, 58, 14420–14426 CrossRef CAS PubMed.
- E. Drula, M.-L. Garron, S. Dogan, V. Lombard, B. Henrissat and N. Terrapon, Nucleic Acids Res., 2022, 50, D571–D577 CrossRef CAS PubMed.
- A. Levasseur, E. Drula, V. Lombard, P. M. Coutinho and B. Henrissat, Biotechnol. Biofuels, 2013, 6, 41 CrossRef CAS PubMed.
- A. B. Boraston, D. N. Bolam, H. J. Gilbert and G. J. Davies, Biochem. J., 2004, 382, 769–781 CrossRef CAS PubMed.
- S. A. K. Jongkees and S. G. Withers, Acc. Chem. Res., 2014, 47, 226–235 CrossRef CAS PubMed.
- L. L. Lairson, B. Henrissat, G. J. Davies and S. G. Withers, Annu. Rev. Biochem., 2008, 77, 521–555 CrossRef CAS PubMed.
- B. Bissaro, P. Monsan, R. Fauré and M. J. O’Donohue, Biochem. J., 2015, 467, 17–35 CrossRef CAS PubMed.
- P. M. Danby and S. G. Withers, ACS Chem. Biol., 2016, 11, 1784–1794 CrossRef CAS PubMed.
- H. Nakai, M. Kitaoka, B. Svensson and K. Ohtsubo, Curr. Opin. Chem. Biol., 2013, 17, 301–309 CrossRef CAS PubMed.
- E. C. O’Neill and R. A. Field, Carbohydr. Res., 2015, 403, 23–37 CrossRef PubMed.
- L. F. Mackenzie, Q. Wang, R. A. J. Warren and S. G. Withers, J. Am. Chem. Soc., 1998, 120, 5583–5584 CrossRef CAS.
- M. Jahn, J. Marles, R. A. J. Warren and S. G. Withers, Angew. Chem., Int. Ed., 2003, 42, 352–354 CrossRef CAS PubMed.
- M. Noguchi, T. Tanaka, H. Gyakushi, A. Kobayashi and S. I. Shoda, J. Org. Chem., 2009, 74, 2210–2212 CrossRef CAS PubMed.
- J. Müllegger, M. Jahn, H. M. Chen, R. A. J. Warren and S. G. Withers, Protein Eng., Des. Sel., 2005, 18, 33–40 CrossRef PubMed.
- Z. Armstrong, S. Reitinger, T. Kantner and S. G. Withers, ChemBioChem, 2010, 11, 533–538 CrossRef CAS PubMed.
- Y. W. Kim, A. L. Lovering, H. Chen, T. Kantner, L. P. McIntosh, N. C. J. Strynadka and S. G. Withers, J. Am. Chem. Soc., 2006, 128, 2202–2203 CrossRef CAS PubMed.
- M. Nieto-Domínguez, B. Fernández de Toro, L. I. de Eugenio, A. G. Santana, L. Bejarano-Muñoz, Z. Armstrong, J. A. Méndez-Líter, J. L. Asensio, A. Prieto, S. G. Withers, F. J. Cañada and M. J. Martínez, Nat. Commun., 2020, 11, 4864 CrossRef PubMed.
- Y. W. Kim, R. Zhang, H. Chen and S. G. Withers, Chem. Commun., 2010, 46, 8725–8727 RSC.
- C. Li, H. J. Ahn, J. H. Kim and Y. W. Kim, Carbohydr. Polym., 2014, 99, 39–46 CrossRef CAS PubMed.
- M. Jahn, H. Chen, J. Müllegger, J. Marles, R. A. J. Warren and S. G. Withers, Chem. Commun., 2004, 274–275 RSC.
- H. M. Chen, Z. Armstrong, S. J. Hallam and S. G. Withers, Carbohydr. Res., 2016, 421, 33–39 CrossRef CAS PubMed.
- Z. Armstrong, K. Mewis, F. Liu, C. Morgan-Lang, M. Scofield, E. Durno, H. M. Chen, K. Mehr, S. G. Withers and S. J. Hallam, ISME J., 2018, 12, 2757–2769 CrossRef CAS PubMed.
- K. Mewis, Z. Armstrong, Y. C. Song, S. A. Baldwin, S. G. Withers and S. J. Hallam, J. Biotechnol., 2013, 167, 462–471 CrossRef CAS PubMed.
- S. S. Macdonald, A. Patel, V. L. C. Larmour, C. Morgan-Lang, S. J. Hallam, B. L. Mark and S. G. Withers, J. Biol. Chem., 2018, 293, 3451–3467 CrossRef CAS PubMed.
- D. K. Hansen, J. R. Winther and M. Willemoës, Carbohydr. Res., 2019, 480, 54–60 CrossRef CAS PubMed.
- D. K. Hansen, A. L. Hansen, J. M. Koivisto, B. Shuoker, M. Abou Hachem, J. R. Winther and M. Willemoës, Arch. Biochem. Biophys., 2022, 725, 109280 CrossRef CAS PubMed.
- I. P. Wood, A. Elliston, P. Ryden, I. Bancroft, I. N. Roberts and K. W. Waldron, Biomass Bioenergy, 2012, 44, 117–121 CrossRef CAS.
- J. Franceus, S. Dhaene, H. Decadt, J. Vandepitte, J. Caroen, J. Van Der Eycken, K. Beerens and T. Desmet, Chem. Commun., 2019, 55, 4531–4533 RSC.
- L. Chuzel, M. B. Ganatra, E. Rapp, B. Henrissat and C. H. Taron, J. Biol. Chem., 2018, 293, 18138–18150 CrossRef CAS PubMed.
- P. Gacesa and F. S. Wusteman, Appl. Environ. Microbiol., 1990, 56, 2265–2267 CrossRef CAS PubMed.
- F. Meddeb-Mouelhi, J. K. Moisan and M. Beauregard, Enzyme Microb. Technol., 2014, 66, 16–19 CrossRef CAS PubMed.
- G. Bastien, G. Arnal, S. Bozonnet, S. Laguerre, F. Ferreira, R. Fauré, B. Henrissat, F. Lefèvre, P. Robe, O. Bouchez, C. Noirot, C. Dumon and M. O’Donohue, Biotechnol. Biofuels, 2013, 6, 78 CrossRef CAS PubMed.
- L. Tasse, J. Bercovici, S. Pizzut-Serin, P. Robe, J. Tap, C. Klopp, B. L. Cantarel, P. M. Coutinho, B. Henrissat, M. Leclerc, J. Doré, P. Monsan, M. Remaud-Simeon and G. Potocki-Veronese, Genome Res., 2010, 20, 1605–1612 CrossRef CAS PubMed.
- A. I. Solbak, T. H. Richardson, R. T. McCann, K. A. Kline, F. Bartnek, G. Tomlinson, X. Tan, L. Parra-Gessert, G. J. Frey, M. Podar, P. Luginbühl, K. A. Gray, E. J. Mathur, D. E. Robertson, M. J. Burk, G. P. Hazlewood, J. M. Short and J. Kerovuo, J. Biol. Chem., 2005, 280, 9431–9438 CrossRef CAS PubMed.
- F. M. T. Koné, M. Le Béchec, J. P. Sine, M. Dion and C. Tellier, Protein Eng., Des. Sel., 2009, 22, 37–44 CrossRef PubMed.
- G. Yang and S. G. Withers, ChemBioChem, 2009, 10, 2704–2715 CrossRef CAS PubMed.
- M. Gantz, S. Neun, E. J. Medcalf, L. D. van Vliet and F. Hollfelder, Chem. Rev., 2023, 123, 5571–5611 CrossRef CAS PubMed.
- A. Autour and M. Ryckelynck, Micromachines, 2017, 8, 128 CrossRef.
- A. S. Tauzin, M. R. Pereira, L. D. Van Vliet, P.-Y. Colin, E. Laville, J. Esque, S. Laguerre, B. Henrissat, N. Terrapon, V. Lombard, M. Leclerc, J. Doré, F. Hollfelder and G. Potocki-Veronese, Microbiome, 2020, 8, 141 CrossRef CAS PubMed.
- J. M. Holstein, C. Gylstorff and F. Hollfelder, ACS Synth. Biol., 2021, 10, 252–257 CrossRef CAS PubMed.
- H. M. Chen, S. A. Nasseri, P. Rahfeld, J. F. Wardman, M. Kohsiek and S. G. Withers, Org. Biomol. Chem., 2021, 19, 789–793 RSC.
- G. Woronoff, A. El Harrak, E. Mayot, O. Schicke, O. J. Miller, P. Soumillion, A. D. Griffiths and M. Ryckelynck, Anal. Chem., 2011, 83, 2852–2857 CrossRef CAS PubMed.
- C. J. Markin, D. A. Mokhtari, F. Sunden, M. J. Appel, E. Akiva, S. A. Longwell, C. Sabatti, D. Herschlag and P. M. Fordyce, Science, 2021, 373, eabf8761 CrossRef CAS PubMed.
- E. J. Medcalf, M. Gantz, T. S. Kaminski and F. Hollfelder, Anal. Chem., 2023, 95, 4597–4604 CrossRef CAS PubMed.
- F. Gielen, R. Hours, S. Emond, M. Fischlechner, U. Schell and F. Hollfelder, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E7383–E7389 CrossRef CAS PubMed.
- A. Zinchenko, S. R. A. Devenish, B. Kintses, P. Y. Colin, M. Fischlechner and F. Hollfelder, Anal. Chem., 2014, 86, 2526–2533 CrossRef CAS PubMed.
- S. A. Nasseri, L. Betschart, D. Opaleva, P. Rahfeld and S. G. Withers, Angew. Chem., Int. Ed., 2018, 130, 11529–11534 CrossRef.
- X. Chen and B. Li, Curr. Opin. Chem. Biol., 2023, 76, 102377 CrossRef CAS PubMed.
- S. A. Nasseri, A. C. Lazarski, I. L. Lemmer, C. Y. Zhang, E. Brencher, H.-M. Chen, L. Sim, L. Betschart, L. J. Worrall, N. C. J. Strynadka and S. G. Withers, bioRxiv, 2024, preprint DOI:10.1101/2024.03.25.586180.
- J. Franceus, J. Lormans, L. Cools, M. D’Hooghe and T. Desmet, ACS Catal., 2021, 11, 6225–6233 CrossRef CAS.
- S. S. Macdonald, Z. Armstrong, C. Morgan-Lang, M. Osowiecka, K. Robinson, S. J. Hallam and S. G. Withers, Cell Chem. Biol., 2019, 26, 1001–1012 CrossRef CAS PubMed.
- M. R. M. De Groeve, G. H. Tran, A. Van Hoorebeke, J. Stout, T. Desmet, S. N. Savvides and W. Soetaert, Anal. Biochem., 2010, 401, 162–167 CrossRef CAS PubMed.
- S. S. MacDonald, J. H. Pereira, F. Liu, G. Tegl, A. Degiovanni, J. F. Wardman, S. Deutsch, Y. Yoshikuni, P. D. Adams and S. G. Withers, ACS Cent. Sci., 2022, 8, 430–440 CrossRef CAS PubMed.
- L. C. MacDonald and B. W. Berger, J. Biol. Chem., 2014, 289, 312–325 CrossRef CAS PubMed.
- C. S. Rye and S. G. Withers, J. Am. Chem. Soc., 2002, 124, 9756–9767 CrossRef CAS PubMed.
- C. S. Rye, A. Matte, M. Cygler and S. G. Withers, ChemBioChem, 2006, 7, 631–637 CrossRef CAS PubMed.
- D. F. Day and W. E. Workman, Anal. Biochem., 1982, 126, 205–207 CrossRef CAS PubMed.
- S. Ladeveze, P. J. Zurek, T. S. Kaminski, S. Emond and F. Hollfelder, ACS Catal., 2023, 13, 10232–10243 CrossRef CAS PubMed.
- C. Pitzler, G. Wirtz, L. Vojcic, S. Hiltl, A. Böker, R. Martinez and U. Schwaneberg, Chem. Biol., 2014, 21, 1733–1742 CrossRef CAS PubMed.
- M. R. M. De Groeve, M. De Baere, L. Hoflack, T. Desmet, E. J. Vandamme and W. Soetaert, Protein Eng., Des. Sel., 2009, 22, 393–399 CrossRef CAS PubMed.
- N. Lülsdorf, C. Pitzler, M. Biggel, R. Martinez, L. Vojcic and U. Schwaneberg, Chem. Commun., 2015, 51, 8679–8682 RSC.
- A. P. Mattey, W. R. Birmingham, P. Both, N. Kress, K. Huang, J. M. Van Munster, G. S. Bulmer, F. Parmeggiani, J. Voglmeir, J. E. R. Martinez, N. J. Turner and S. L. Flitsch, ACS Catal., 2019, 9, 8208–8212 CrossRef CAS.
- J. B. Rannes, A. Ioannou, S. C. Willies, G. Grogan, C. Behrens, S. L. Flitsch and N. J. Turner, J. Am. Chem. Soc., 2011, 133, 8436–8439 CrossRef CAS PubMed.
- L. Sun, T. Bulter, M. Alcalde, I. P. Petrounia and F. H. Arnold, ChemBioChem, 2002, 3, 781–783 CrossRef CAS PubMed.
- A. Debon, M. Pott, R. Obexer, A. P. Green, L. Friedrich, A. D. Griffiths and D. Hilvert, Nat. Catal., 2019, 2, 740–747 CrossRef CAS.
- M. Penner, O. J. Klein, M. Gantz, S. Boss, P. Barker, P. Dupree and F. Hollfelder, bioRxiv, 2023, preprint DOI:10.1101/2023.11.22.568356.
- V. N. Perna, K. Barrett, A. S. Meyer and B. Zeuner, Glycobiology, 2023, 33, 396–410 CrossRef CAS PubMed.
- C. Mayer, D. L. Jakeman, M. Mah, G. Karjala, L. Gal, R. A. J. Warren and S. G. Withers, Chem. Biol., 2001, 8, 437–443 CrossRef CAS PubMed.
- Y. W. Kim, S. S. Lee, R. A. J. Warren and S. G. Withers, J. Biol. Chem., 2004, 279, 42787–42793 CrossRef CAS PubMed.
- M. N. Namchuk and S. G. Withers, Biochemistry, 1995, 34, 16194 CrossRef CAS PubMed.
- J. B. Kempton and S. G. Withers, Biochemistry, 1992, 31, 9961–9969 CrossRef CAS PubMed.
- L. L. Lairson, W. W. Wakarchuk and S. G. Withers, Chem. Commun., 2007, 365–367 RSC.
- G. J. Williams and J. S. Thorson, Nat. Protoc., 2008, 3, 357–362 CrossRef CAS PubMed.
- G. J. Williams, C. Zhang and J. S. Thorson, Nat. Chem. Biol., 2007, 3, 657–662 CrossRef CAS PubMed.
- J. K. Roy, H.-W. Ahn, J. Lee, J.-H. Kim, S.-H. Yoo and Y.-W. Kim, Food Chem., 2024, 437, 137898 CrossRef CAS PubMed.
- S. M. Forget, S. B. Shepard, E. Soleimani and D. L. Jakeman, J. Org. Chem., 2019, 84, 11482–11492 CrossRef CAS PubMed.
- R. W. Gantt, P. Peltier-Pain, W. J. Cournoyer and J. S. Thorson, Nat. Chem. Biol., 2011, 7, 685–691 CrossRef CAS PubMed.
- R. W. Gantt, P. Peltier-Pain, S. Singh, M. Zhou and J. S. Thorson, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7648–7653 CrossRef CAS PubMed.
- A. Aharoni, K. Thieme, C. P. C. Chiu, S. Buchini, L. L. Lairson, H. Chen, N. C. J. Strynadka, W. W. Wakarchuk and S. G. Withers, Nat. Methods, 2006, 3, 609–614 CrossRef CAS PubMed.
- G. Yang, J. R. Rich, M. Gilbert, W. W. Wakarchuk, Y. Feng and S. G. Withers, J. Am. Chem. Soc., 2010, 132, 10570–10577 CrossRef CAS PubMed.
- C. Bachert and A. D. Linstedt, Traffic, 2013, 14, 47–56 CrossRef CAS PubMed.
- L. Song, C. Bachert, K. T. Schjoldager, H. Clausen and A. D. Linstedt, J. Biol. Chem., 2014, 289, 30556–30566 CrossRef CAS PubMed.
- S. J. Hendel and M. D. Shoulders, Nat. Methods, 2021, 18, 346–357 CrossRef CAS PubMed.
- B. Janesch, L. Baumann, A. Mark, N. Thompson, S. Rahmani, L. Sim, S. G. Withers and W. W. Wakarchuk, Glycobiology, 2019, 29, 588–598 CrossRef CAS PubMed.
- C. J. Glasscock, L. E. Yates, T. Jaroentomeechai, J. D. Wilson, J. H. Merritt, J. B. Lucks and M. P. DeLisa, Metab. Eng., 2018, 47, 488–495 CrossRef CAS PubMed.
- A. Natarajan, T. Jaroentomeechai, M. Cabrera-Sánchez, J. C. Mohammed, E. C. Cox, O. Young, A. Shajahan, M. Vilkhovoy, S. Vadhin, J. D. Varner, P. Azadi and M. P. DeLisa, Nat. Chem. Biol., 2020, 16, 1062–1070 CrossRef CAS PubMed.
- A. A. Ollis, S. Zhang, A. C. Fisher and M. P. Delisa, Nat. Chem. Biol., 2014, 10, 816–822 CrossRef CAS PubMed.
- J. D. Valderrama-Rincon, A. C. Fisher, J. H. Merritt, Y. Y. Fan, C. A. Reading, K. Chhiba, C. Heiss, P. Azadi, M. Aebi and M. P. DeLisa, Nat. Chem. Biol., 2012, 8, 434–436 CrossRef CAS PubMed.
- W. Kightlinger, K. F. Warfel, M. P. DeLisa and M. C. Jewett, ACS Synth. Biol., 2020, 9, 1534–1562 CrossRef CAS PubMed.
- M. Li, X. Zheng, S. Shanker, T. Jaroentomeechai, T. D. Moeller, S. W. Hulbert, I. Koçer, J. Byrne, E. C. Cox, Q. Fu, S. Zhang, J. W. Labonte, J. J. Gray and M. P. DeLisa, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2107440118 CrossRef CAS PubMed.
- T. G. Keys, H. L. S. Fuchs, J. Ehrit, J. Alves, F. Freiberger and R. Gerardy-Schahn, Nat. Chem. Biol., 2014, 10, 437–442 CrossRef CAS PubMed.
- R. D. Gupta and D. S. Tawfik, Nat. Methods, 2008, 5, 939–942 CrossRef CAS PubMed.
- S. Bershtein, K. Goldin and D. S. Tawfik, J. Mol. Biol., 2008, 379, 1029–1044 CrossRef CAS PubMed.
- T. G. Keys, M. Berger and R. Gerardy-Schahn, Anal. Biochem., 2012, 427, 60–68 CrossRef CAS PubMed.
- S. M. Hancock, J. R. Rich, M. E. C. Caines, N. C. J. Strynadka and S. G. Withers, Nat. Chem. Biol., 2009, 5, 508–514 CrossRef CAS PubMed.
- J. E. Blanchard and S. G. Withers, Chem. Biol., 2001, 8, 627–633 CrossRef CAS PubMed.
- S. G. Withers, K. Rupitz and I. P. Street, J. Biol. Chem., 1988, 263, 7929–7932 CrossRef CAS PubMed.
- S. M. Wilkinson, C. W. Liew, J. P. Mackay, H. M. Salleh, S. G. Withers and M. D. McLeod, Org. Lett., 2008, 10, 1585–1588 CrossRef CAS PubMed.
- Z. Armstrong, F. Liu, H.-M. Chen, S. J. Hallam and S. G. Withers, ACS Catal., 2019, 9, 3219–3227 CrossRef CAS.
- C. L. Perrine, A. Ganguli, P. Wu, C. R. Bertozzi, T. A. Fritz, J. Raman, L. A. Tabak and T. A. Gerken, J. Biol. Chem., 2009, 284, 20387–20397 CrossRef CAS PubMed.
- J. Raman, T. A. Fritz, T. A. Gerken, O. Jamison, D. Live, M. Liu and L. A. Tabak, J. Biol. Chem., 2008, 283, 22942–22951 CrossRef CAS PubMed.
- M. De Las Rivas, E. J. Paul Daniel, H. Coelho, E. Lira-Navarrete, L. Raich, I. Compañón, A. Diniz, L. Lagartera, J. Jiménez-Barbero, H. Clausen, C. Rovira, F. Marcelo, F. Corzana, T. A. Gerken and R. Hurtado-Guerrero, ACS Cent. Sci., 2018, 4, 1274–1290 CrossRef CAS PubMed.
- M. F. Festari, F. Trajtenberg, N. Berois, S. Pantano, L. Revoredo, Y. Kong, P. Solari-Saquieres, Y. Narimatsu, T. Freire, S. Bay, C. Robello, J. Bénard, T. A. Gerken, H. Clausen and E. Osinaga, Glycobiology, 2017, 27, 140–153 CrossRef CAS PubMed.
- T. A. Gerken, K. G. Ten Hagen and O. Jamison, Glycobiology, 2008, 18, 861–870 CrossRef CAS PubMed.
- T. A. Gerken, O. Jamison, C. L. Perrine, J. C. Collette, H. Moinova, L. Ravi, S. D. Markowitz, W. Shen, H. Patel and L. A. Tabak, J. Biol. Chem., 2011, 286, 14493–14507 CrossRef CAS PubMed.
- E. J. P. Daniel, M. Las Rivas, E. Lira-Navarrete, A. García-García, R. Hurtado-Guerrero, H. Clausen and T. A. Gerken, Glycobiology, 2020, 30, 910–922 CrossRef CAS PubMed.
- J. E. Mohl, T. A. Gerken and M. Y. Leung, Glycobiology, 2021, 31, 168–172 CrossRef CAS PubMed.
- T. Du, N. Buenbrazo, L. Kell, S. Rahmani, L. Sim, S. G. Withers, S. DeFrees and W. W. Wakarchuk, Cell Chem. Biol., 2019, 26, 203–212 CrossRef CAS PubMed.
- N. K. Thompson, L. T. N. Leclaire, S. R. Perez and W. W. Wakarchuk, Biochem. J., 2021, 478, 3527–3537 CrossRef CAS PubMed.
- C. Durr, H. Nothaft, C. Lizak, R. Glockshuber and M. Aebi, Glycobiology, 2010, 20, 1366–1372 CrossRef PubMed.
- S. S. Chung, E. J. Bidstrup, J. M. Hershewe, K. F. Warfel, M. C. Jewett and M. P. DeLisa, ACS Synth. Biol., 2022, 11, 3892–3899 CrossRef CAS PubMed.
- W. Kightlinger, L. Lin, M. Rosztoczy, W. Li, M. P. Delisa, M. Mrksich and M. C. Jewett, Nat. Chem. Biol., 2018, 14, 627–635 CrossRef CAS PubMed.
- L. Lin, W. Kightlinger, S. K. Prabhu, A. J. Hockenberry, C. Li, L. X. Wang, M. C. Jewett and M. Mrksich, ACS Cent. Sci., 2020, 6, 144–154 CrossRef CAS PubMed.
- J.-M. Techner, W. Kightlinger, L. Lin, J. Hershewe, A. Ramesh, M. P. DeLisa, M. C. Jewett and M. Mrksich, Anal. Chem., 2020, 92, 1963–1971 CrossRef CAS PubMed.
- W. Kightlinger, K. E. Duncker, A. Ramesh, A. H. Thames, A. Natarajan, J. C. Stark, A. Yang, L. Lin, M. Mrksich, M. P. DeLisa and M. C. Jewett, Nat. Commun., 2019, 10, 5404 CrossRef PubMed.
- J. Zhang, M. K. Jensen and J. D. Keasling, Curr. Opin. Chem. Biol., 2015, 28, 1–8 CrossRef PubMed.
- S. L. Choi, E. Rha, S. J. Lee, H. Kim, K. Kwon, Y. S. Jeong, Y. H. Rhee, J. J. Song, H. S. Kim and S. G. Lee, ACS Synth. Biol., 2014, 3, 163–171 CrossRef CAS PubMed.
- C. K. Bandi, K. S. Skalenko, A. Agrawal, N. Sivaneri, M. Thiry and S. P. S. Chundawat, ACS Synth. Biol., 2021, 10, 682–689 CrossRef CAS PubMed.
- D. A. Cecchini, E. Laville, S. Laguerre, P. Robe, M. Leclerc, J. Doré, B. Henrissat, M. Remaud-Siméon, P. Monsan and G. Potocki-Véronèse, PLoS One, 2013, 8, e72766 CrossRef CAS PubMed.
- S. M. Lee, T. Jellison and H. S. Alper, Appl. Environ. Microbiol., 2012, 78, 5708–5716 CrossRef CAS PubMed.
- Y.-S. Kim, H.-C. Jung and J.-G. Pan, Appl. Environ. Microbiol., 2000, 66, 788–793 CrossRef CAS PubMed.
- W. Liu, J. Hong, D. R. Bevan and Y. H. P. Zhang, Biotechnol. Bioeng., 2009, 103, 1087–1094 CrossRef CAS PubMed.
- B. A. Van Der Veen, G. Potocki-Véronèse, C. Albenne, G. Joucla, P. Monsan and M. Remaud-Simeon, FEBS Lett., 2004, 560, 91–97 CrossRef CAS PubMed.
- H. Lin, H. Tao and V. W. Cornish, J. Am. Chem. Soc., 2004, 126, 15051–15059 CrossRef CAS PubMed.
- P. Peralta-Yahya, B. T. Carter, H. Lin, H. Tao and V. W. Cornish, J. Am. Chem. Soc., 2008, 130, 17445–17452 CrossRef PubMed.
- S. S. K. Abadi, M. C. Deen, J. N. Watson, F. S. Shidmoossavee and A. J. Bennet, Glycobiology, 2020, 30, 325–333 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- S. L. Lovelock, R. Crawshaw, S. Basler, C. Levy, D. Baker, D. Hilvert and A. P. Green, Nature, 2022, 606, 49–58 CrossRef CAS PubMed.
- A. Madani, B. Krause, E. R. Greene, S. Subramanian, B. P. Mohr, J. M. Holton, J. L. Olmos, C. Xiong, Z. Z. Sun, R. Socher, J. S. Fraser and N. Naik, Nat. Biotechnol., 2023, 41, 1099–1106 CrossRef CAS PubMed.
- S. Studer, D. A. Hansen, Z. L. Pianowski, P. R. E. Mittl, A. Debon, S. L. Guffy, B. S. Der, B. Kuhlman and D. Hilvert, Science, 2018, 362, 1285–1288 CrossRef CAS PubMed.
- S. Basler, S. Studer, Y. Zou, T. Mori, Y. Ota, A. Camus, H. A. Bunzel, R. C. Helgeson, K. N. Houk, G. Jiménez-Osés and D. Hilvert, Nat. Chem., 2021, 13, 231–235 CrossRef CAS PubMed.
- M. L. Zastrow and V. L. Pecoraro, Biochemistry, 2014, 53, 957–978 CrossRef CAS PubMed.
- N. G. S. McGregor, J. Coines, V. Borlandelli, S. Amaki, M. Artola, A. Nin-Hill, D. Linzel, C. Yamada, T. Arakawa, A. Ishiwata, Y. Ito, G. A. Van Der Marel, J. D. C. Codée, S. Fushinobu, H. S. Overkleeft, C. Rovira and G. J. Davies, Angew. Chem., Int. Ed., 2021, 60, 5754–5758 CrossRef CAS PubMed.
- T. Ito, K. Saikawa, S. Kim, K. Fujita, A. Ishiwata, S. Kaeothip, T. Arakawa, T. Wakagi, G. T. Beckham, Y. Ito and S. Fushinobu, Biochem. Biophys. Res. Commun., 2014, 447, 32–37 CrossRef CAS PubMed.
- W. J. Jeong and W. J. Song, Nat. Commun., 2022, 13, 6844 CrossRef CAS PubMed.
- J. D. Schnettler, O. J. Klein, T. S. Kaminski, P.-Y. Colin and F. Hollfelder, J. Am. Chem. Soc., 2023, 145, 1083–1096 CrossRef CAS PubMed.
- J. F. Wardman, P. Rahfeld, F. Liu, C. Morgan-Lang, L. Sim, S. J. Hallam and S. G. Withers, ACS Chem. Biol., 2021, 16, 2004–2015 CrossRef CAS PubMed.
- S. Neun, P. Brear, E. Campbell, T. Tryfona, K. El Omari, A. Wagner, P. Dupree, M. Hyvönen and F. Hollfelder, Nat. Chem. Biol., 2022, 18, 1096–1103 CrossRef CAS PubMed.
- P.-Y. Colin, B. Kintses, F. Gielen, C. M. Miton, G. Fischer, M. F. Mohamed, M. Hyvönen, D. P. Morgavi, D. B. Janssen and F. Hollfelder, Nat. Commun., 2015, 6, 10008 CrossRef CAS PubMed.
- P. Rahfeld, J. F. Wardman, K. Mehr, D. Huff, C. Morgan-Lang, H. M. Chen, S. J. Hallam and S. G. Withers, J. Biol. Chem., 2019, 294, 16400–16415 CrossRef CAS PubMed.
- P. A. Romero, T. M. Tran and A. R. Abate, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7159–7164 CrossRef CAS PubMed.
- R. Lipsh-Sokolik, O. Khersonsky, S. P. Schröder, C. de Boer, H. S. Overkleeft and S. J. Fleishman, Science, 2023, 379, 195–201 CrossRef CAS PubMed.
- L. Wu, Z. Armstrong, S. P. Schro, C. D. Boer, M. Artola, J. M. F. G. Aerts, H. S. Overkleeft and G. J. Davies, Curr. Opin. Chem. Biol., 2019, 53, 25–36 CrossRef CAS PubMed.
- Y. Chen, Z. Armstrong, M. Artola, B. I. Florea, C.-L. Kuo, C. De Boer, M. S. Rasmussen, M. Abou Hachem, G. A. Van Der Marel, J. D. C. Codée, J. M. F. G. Aerts, G. J. Davies and H. S. Overkleeft, J. Am. Chem. Soc., 2021, 143, 2423–2432 CrossRef CAS PubMed.
- Z. Armstrong, C. Kuo, D. Lahav, B. Liu, R. Johnson, T. J. M. Beenakker, C. D. Boer, C. Wong, E. R. V. Rijssel, M. F. Debets, B. I. Florea, R. G. Boot, P. P. Geurink, H. Ovaa, M. V. D. Stelt, G. A. V. D. Marel, J. D. C. Codée, J. M. F. G. Aerts, L. Wu, H. S. Overkleeft and G. J. Davies, J. Am. Chem. Soc., 2020, 142, 13021–13029 CrossRef CAS PubMed.
- I. Chen, B. M. Dorr and D. R. Liu, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11399–11404 CrossRef CAS PubMed.
- P. Knyphausen, M. Rangel Pereira, P. Brear, M. Hyvönen, L. Jermutus and F. Hollfelder, Nat. Commun., 2023, 14, 768 CrossRef CAS PubMed.
- R. S. Molina, G. Rix, A. A. Mengiste, B. Álvarez, D. Seo, H. Chen, J. E. Hurtado, Q. Zhang, J. D. García-García, Z. J. Heins, P. J. Almhjell, F. H. Arnold, A. S. Khalil, A. D. Hanson, J. E. Dueber, D. V. Schaffer, F. Chen, S. Kim, L. Á. Fernández, M. D. Shoulders and C. C. Liu, Nat. Rev. Methods Primer, 2022, 2, 36 CrossRef CAS.
- K. M. Esvelt, J. C. Carlson and D. R. Liu, Nature, 2011, 472, 499–503 CrossRef CAS PubMed.
- M. S. Morrison, C. J. Podracky and D. R. Liu, Nat. Chem. Biol., 2020, 16, 610–619 CrossRef CAS PubMed.
- K. A. Jones, H. M. Snodgrass, K. Belsare, B. C. Dickinson and J. C. Lewis, ACS Cent. Sci., 2021, 7, 1581–1590 CrossRef CAS PubMed.
- J. M. Grondin, K. Tamura, G. Déjean, D. W. Abbott and H. Brumer, J. Bacteriol., 2017, 199, e00860–e00816 CrossRef CAS PubMed.
- J. F. Wardman, L. Sim, J. Liu, T. A. Howard, A. Geissner, P. M. Danby, A. B. Boraston, W. W. Wakarchuk and S. G. Withers, Nat. Chem. Biol., 2023, 19, 1246–1255 CrossRef CAS PubMed.
- D. J. Shon, A. Kuo, M. J. Ferracane and S. A. Malaker, Biochem. J., 2021, 478, 1585–1603 CrossRef CAS PubMed.
- L. Sim, N. Thompson, A. Geissner, S. G. Withers and W. W. Wakarchuk, Glycobiology, 2022, 32, 429–440 CrossRef CAS PubMed.
- R. P. Dyer and G. A. Weiss, Cell Chem. Biol., 2022, 29, 177–190 CrossRef CAS PubMed.
- E. Hardiman, M. Gibbs, R. Reeves and P. Bergquist, Appl. Biochem. Biotechnol., 2010, 161, 301–312 CrossRef CAS PubMed.
- A. J. Fairbanks, Chem. Soc. Rev., 2017, 46, 5128–5146 RSC.
- K. S. Johansen, K. E. H. Frandsen, L. Lo Leggio, T. Tandrup and J.-G. Berrin, Biochem. Soc. Trans., 2018, 46, 1431–1447 CrossRef PubMed.
- M. S. Jensen, G. Klinkenberg, B. Bissaro, P. Chylenski, G. Vaaje-Kolstad, H. F. Kvitvang, G. K. Nærdal, H. Sletta, Z. Forsberg and V. G. H. Eijsink, J. Biol. Chem., 2019, 294, 19349–19364 CrossRef CAS PubMed.
- C. Cheng, J. Haider, P. Liu, J. Yang, Z. Tan, T. Huang, J. Lin, M. Jiang, H. Liu and L. Zhu, J. Agric. Food Chem., 2020, 68, 15257–15266 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |