
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnusual cysteine modifications in natural product biosynthesis
Yaojie
Gao
a,
Yuhao
Zhu
a,
Takayoshi
Awakawa
 ab and
Ikuro
Abe
*ac
ab and
Ikuro
Abe
*ac
aGraduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: abei@mol.f.u-tokyo.ac.jp
bRIKEN Center for Sustainable Resource Science, Wako, Saitama 351-0198, Japan
cCollaborative Research Institute for Innovative Microbiology, The University of Tokyo, Yayoi 1-1-1, Bunkyo-ku, Tokyo 113-8657, Japan
First published on 9th February 2024
Abstract
L-Cysteine is a highly reactive amino acid that is modified into a variety of chemical structures, including cysteine sulfinic acid in human metabolic pathways, and sulfur-containing scaffolds of amino acids, alkaloids, and peptides in natural product biosynthesis. Among the modification enzymes responsible for these cysteine-derived compounds, metalloenzymes constitute an important family of enzymes that catalyze a wide variety of reactions. Therefore, understanding their reaction mechanisms is important for the biosynthetic production of cysteine-derived natural products. This review mainly summarizes recent mechanistic investigations of metalloenzymes, with a particular focus on recently discovered mononuclear non-heme iron (NHI) enzymes, dinuclear NHI enzymes, and radical-SAM enzymes involved in unusual cysteine modifications in natural product biosynthesis.
 Yaojie Gao | Yaojie Gao received his MSc degree in biochemistry and microbiology from Wuhan University (2017) and his PhD degree in biology from Shanghai Jiao Tong University (2022). After that, he joined Prof. Ikuro Abe's group at The University of Tokyo as a postdoctoral research fellow. His research interest focuses on resolving the biosynthetic mechanisms of bioactive microbial natural products and understanding the structure-based catalytic mechanism of novel biosynthetic enzymes. |
 Yuhao Zhu | Yuhao Zhu received his BS degree (2019) and MSc degree (2021) from School of Pharmaceutical Sciences, Peking University. In 2022, he joined Prof. Ikuro Abe's group in the University of Tokyo as a PhD student. His research interest focuses on the function and mechanism study of biosynthetic enzymes based on the protein structure. |
 Takayoshi Awakawa | Takayoshi Awakawa received his BS degree from the Agricultural department, The University of Tokyo, in 2006, and PhD degree from the Agricultural and Life Sciences department, The University of Tokyo, in 2011. After obtaining PhD, he worked as an Assistant Professor (2011–2017), Lecturer (2018–2019), and Associate Professor (2019–2022), and was then appointed as a team leader in the RIKEN Center for Sustainable Resource Science (2022–current). He also carried out postdoctoral research in the laboratory of Prof. Bradley S. Moore at UC San Diego, Scripps Institute of Oceanography (2014–2016), as a visiting scholar. His research interest mainly focuses on the engineered biosynthesis and chemical biology of natural products. |
 Ikuro Abe | Ikuro Abe received his BS (1984) and PhD (1989) from The University of Tokyo under the guidance of Professors Ushio Sankawa and Yutaka Ebizuka, where he studied chemistry and biochemistry of natural product biosynthesis. After postdoctoral research with Professor Guy Ourisson at the CNRS Institut de Chimie des Substances Naturelles, and mostly with Professor Michel Rohmer at the Ecole Nationale Supérieure de Chimie de Mulhouse (1989–1991), he stayed in the United States to work with Professor Glenn D. Prestwich at the State University of New York at Stony Brook (1991–1996) and then at the University of Utah (1996–1998). In 1998, he moved back to Japan to join the faculty at University of Shizuoka (1998–2009), and then he was appointed as the Professor of Natural Products Chemistry at the Graduate School of Pharmaceutical Sciences, The University of Tokyo (2009–). His research interests mostly focus on exploring and engineering the natural product biosynthesis. He received the Japanese Society of Pharmacognosy Award (2017), Sumiki Umezawa Memorial Award (2017), The Pharmaceutical Society of Japan Award (2019), Prizes for Science and Technology by the Minister of Education, Culture, Sports, Science and Technology, Japan (2019), and The Society for Actinomycetes Japan, Omura Award (2023). He is a fellow of The Royal Society of Chemistry (FRSC) and a former President of The Japanese Society of Pharmacognosy. |
Introduction
L-Cysteine (L-Cys, 1) is a highly reactive amino acid that can be transformed into a variety of chemical structures because it contains sulfur, which can adopt a wide range of oxidation states, ranging from −2 to +6, with −2 (H2S, thiol, sulfide, and sulfonium ion), −1 (disulfide), 0 (elemental, sulfoxide, and sulfenic acid), +2 (sulfone and sulfinic acid), +4 (SO2, sulfonic acid, and sulfite ester), and +6 (SO3 and sulfate ester).1 Protein redox signaling studies revealed that the oxidation of L-Cys in proteins results in the generation of a range of sulfur-containing scaffolds, including thiyl radical, sulfenic acid, sulfonic acid, sulfenyl amide, sulfinamide, sulfonamide, S-nitrosothiol, disulfide, S-glutathione adduct, S-sufhydryl persulfide, thiosulfinate, and thiosulfate.2 In the biosynthesis of natural products, including nonproteinogenic amino acids, alkaloids, and peptides, the L-Cys modifications also lead to various unique chemical structures, such as 2-sulfamoylacetic aldehyde in the sulfonamide alkaloid altemicidin/SB-203208 biosynthesis,3 thiohydroximate in the copper chelating agent fluopsin C biosynthesis,4 nor-Cys in the nonproteinogenic amino acid 3-thiaglutamate (3-thiaGlu) biosynthesis,5 and enethiol in lantibiotic peptide biosynthesis.6,7 Therefore, understanding the catalytic mechanisms of such Cys-modifying enzymes is a prerequisite for expanding the structural complexity of natural products. In particular, metal-dependent enzymes are versatile catalysts that facilitate dynamic scaffold transformations, which would be challenging with purely chemical catalysts.8 These enzymes utilize metallocofactors that mediate electron transfers, enabling reactions with oxygen and the formation of organic radicals or anions which can react with 1 or 1-derived scaffolds, leading to various oxidative transformations.This review mainly focuses on the recent mechanistic investigations of three groups of Cys-modifying metalloenzymes in natural product biosynthesis. The first group comprises mononuclear nonheme iron (NHI) enzymes, which belong to the cupin enzyme superfamily and utilize a mononuclear metal ion to activate and transfer electrons to molecular oxygen, as exemplified by isopenicillin N synthase (IPNS)9 and cysteine dioxygenase (CDO).10,11 Recent studies of CDO and its novel homologs, including SbzM in alkaloid altemicidin/SB-203208 biosynthesis3 and OvoA/EgtB in mercaptohistidine ovothiol biosynthesis,12,13 will be described. The second group comprises dinuclear NHI enzymes, which utilize two iron atoms to activate molecular oxygen and generate diiron–peroxo intermediates.14 This group includes heme oxygenase diiron-like (HOD) enzymes such as FlcD and FlcE,12 and DUF692-type enzymes such as MbnBC in CuMbn biosynthesis,15 TglHI in 3-thiaGlu biosynthesis,5 TmoHI in 3-thia-α-amino acid biosynthesis,16 and ChrHI in the ribosomally-synthesized and post-translationally modified peptide (RiPP) ChrA* biosynthesis.17 The third group consists of radical-SAM enzymes that utilize a [4Fe–4S] cluster and SAM to generate a reactive 5′-deoxyadenosyl radical (5′-dA˙) that abstracts a hydrogen atom from substrates,18 including TmoD in 3-thia-α-amino acid biosynthesis16 and SycC in AviCamCys biosynthesis.7 This review summarizes recent mechanistic investigations of the striking variety of metalloenzyme-catalyzed reactions that enhance the complexity of Cys-derived natural products, highlighting potential applications for the creation of new molecules and supranatural artificial biocatalysts.
1. Cysteine dioxygenase (CDO)
CDO is a well-studied mononuclear NHI enzyme that plays an important role in human L-Cys (1) metabolism. The product of CDO, L-cysteine sulfinic acid (2), is catabolized by an aminotransferase or cysteine sulfinate decarboxylase in different pathways and transformed into taurine or pyruvate (Fig. 1).19 | ||
| Fig. 1 Metabolic pathway of L-cysteine sulfinic acid (2) generated from L-Cys (1) by CDO. | ||
The first CDO (from mouse) crystal structure was determined at a resolution of 1.75 Å in 2006.20 The protein core consists of a conserved overall β-barrel-like structure, which is characteristic of cupin superfamily proteins.10 In the crystal structure of rat CDO, the ferrous iron is coordinated by the three His facial triad motif (His86, His88, and His140), and additionally coordinated by one water molecule (Fig. 2(A)).21,22 In the crystal structure of human CDO solved in 2007, 1 was found in the substrate binding site.23 In addition, 1 was coordinated to the ferrous iron via an amino nitrogen and a thiol sulfur, and supported by several hydrogen bonds with surrounding residues (Fig. 2(B)).
 | ||
| Fig. 2 Crystal structures of CDOs. (A) Metal binding site in rat CDO. (B) Substrate and metal binding site and the Tyr-Cys cross-link in human CDO. (C) Substrate and metal binding site in BsCDO. Fe(II) is colored brown, and water is colored silver. Hydrogen bonds are shown with yellow dotted lines. | ||
A covalent linkage between Cys93 and Tyr157 near the metal binding site was found as a post-translational modification in human CDO (Fig. 2(B)).21–23 The covalent linkage between the corresponding Cys and Tyr residues is conserved in eukaryotic CDOs, whereas in bacteria, Cys93 is substituted by glycine, which precludes the formation of the cross-link in BsCDO (CDO from B. subtilis, Fig. 2(C)).24,25 The subsequent mutation study suggested that the covalent linkage enhances the activity by alleviating the inhibitory effect of Cys93 in human CDO. In addition to the Tyr–Cys linkage, other examples of naturally cross-linked Cys residues in enzymes have been reported. Wensien and his co-workers recently discovered a special lysine–cysteine redox switch with an N–O–S bridge in the transaldolase from Neisseria gonorrhoeae. The N–O–S bridge regulates the enzyme activity through a loaded-spring mechanism, changing the conformations of several key residues.26 A more recent study showed that Orf1, which is involved in the biosynthesis of BD-12, contains an N–O–S bridge that plays an important role in catalysis.27 Orf1 is a noncanonical FAD-dependent enzyme responsible for appending a glycine-derived N-formimidoyl group to glycinothricin, producing the antibiotic BD-12. The X-ray crystallographic analysis revealed that Orf1 forms an oximinoglycine with an N–O–S–Cys281 substrate–enzyme bridge, which mediates the nucleophilic addition of glycinothricin and decarboxylation, leading to the cleavage of the N–O bond and the production of a formimidoylated final product.27
Several mechanisms for the CDO reaction have been proposed, based on its X-ray crystal structures solved in early studies.20,21,23 Recently, to obtain experimental evidence for the catalytic mechanism, researchers have conducted experiments to mimic or capture potential intermediates during the CDO reaction. Nitric oxide (NO) was utilized as an O2 analog to investigate the binding order of substrates 1 and O2 through electron paramagnetic resonance (EPR) spectroscopy. In the absence of 1, CDO does not interact with NO, but in the presence of 1, CDO interacts with NO to generate a low-spin [FeNO]7 (S = 1/2) signal,28 suggesting that 1 binds to CDO prior to molecular oxygen. Substitutions of 1 with analogs, including cysteamine, 3-mercaptopropionic acid, and propane thiol, abolished this signal, indicating the strict substrate specificity of the CDO reaction. This [FeNO]7 (S = 1/2) signal was assumed to originate from the bidentate thiol/amine coordination of 1 to the iron–nitrosyl complex,28 suggesting the presence of an Fe(III)–superoxo intermediate (Fig. 3). In addition, azide was employed to mimic superoxide. As suggested by EPR and DFT calculations, azide directly binds to Fe(III), but not to Fe(II). In the DFT-optimized structure of this complex, the central nitrogen atom of the azide moiety is 3.12 Å away from the sulfur of 1, supporting the attack from the superoxide group of the putative Fe(III)–superoxo intermediate.29
 | ||
| Fig. 3 Proposed reaction mechanisms of CDO. | ||
Fe(III)-bound CDO was mixed with superoxide anions generated by xanthine oxidase, in an attempt to detect the characteristic signals of the Fe(III)–superoxo complex. The addition of superoxide anions generated a transient species observed at 565 nm by UV-vis spectroscopy. The decay of this 565 nm absorption kinetically matched the formation of 2. Further analyses by EPR spectroscopy suggested that this transient species is an Fe(III)–superoxo intermediate.30 These data provided additional evidence for the presence of the Fe(III)–superoxo intermediate.
By mixing CDO:Fe(II):1 with oxygenated buffer at a low temperature, characteristic signals for intermediates were captured through UV-vis and 57Fe-Mössbauer spectrum analyses. Transient absorption peaks at 500–640 nm formed and disappeared within 20 ms after mixing. This transient intermediate was further captured by a characteristic signal in the 57Fe-Mössbauer spectrum,31 and the DFT calculation suggested that this signal corresponds to the cyclic peroxo intermediate in Fig. 3.
Iron(II)–thiolate models were adopted for understanding the reaction mechanism of CDO. In the CDO mimic models, oxygen can only be activated by specific thiolate ligands, suggesting that they play a critical role. Further calculation results suggested that the thiolate ligand must transfer a charge to Fe(II), resulting in a lower redox potential, which is crucial for the reaction of oxygen with iron.32 Sulfur oxidation was also investigated in an [Fe(III)(LN3S)]+ model, in which LN3S represents a tetradentate ligand with a bis(imino)pyridyl scaffold and a pendant arylthiolate group. Furthermore, additional calculations suggested that oxygen preferentially binds to the iron center, instead of attacking the sulfur group directly. Together, these data suggested that the formation of the Fe(III)–superoxo complex initiates the catalysis, followed by the stepwise oxidation of the sulfur group.33
Consequently, the mechanism of the CDO reaction has been proposed as described below (Fig. 3). The molecular oxygen binds to Fe(II), leading to the formation of the Fe(III)–superoxo intermediate. The distal oxygen of the superoxo complex reacts with activated sulfur, generating a cyclic peroxo intermediate. Heterolytic O–O bond cleavage generates a sulfoxide and Fe(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species. The Fe(IV)O species reacts with the sulfoxide, and 2 is released from the enzyme in the final step of the catalysis. Another mechanism was also considered, involving a persulfenate intermediate detected in the X-ray crystal structure analysis.34 However, this pathway exhibited a high energy barrier in the DFT calculations, suggesting that the formation of such an intermediate is unlikely in the catalytic reaction of CDO.
O species. The Fe(IV)O species reacts with the sulfoxide, and 2 is released from the enzyme in the final step of the catalysis. Another mechanism was also considered, involving a persulfenate intermediate detected in the X-ray crystal structure analysis.34 However, this pathway exhibited a high energy barrier in the DFT calculations, suggesting that the formation of such an intermediate is unlikely in the catalytic reaction of CDO.
1.1. Unusual CDO homologs
 | ||
| Fig. 4 Reaction and proposed mechanism of SbzM. (A) Biosynthetic pathway of altemicidin and analogs. (B) Proposed reaction of SbzM. | ||
Our recent investigations on the metal dependency of the CDO homolog enzyme SbzM suggest that nickel and iron are potential active metals.3 A plausible mechanism was proposed for the SbzM reaction (Fig. 4(B)),3 in which 1 is transformed into (Z)-(2-aminovinyl)sulfanolate (9) through sulfur monooxygenation followed by decarboxylation, and then the sulfanolate is further oxidized to a second intermediate, (Z)-2-aminoethanone-1-sulfonate (10). The subsequent intramolecular rearrangement of the amino group on the sulfur atom, accompanied by the attack of water, results in the formation of the N–S bond in the final product 6. An alternative mechanism involving an Fe(IV)O complex,38 as in the cases of CDO and IPNS,9,39,40 is also possible, in which a mercapto group is oxidized by the superoxo complex. However, since the activity can be restored with the addition of Ni(II), and considering that nickel cannot readily form the Ni(IV)O intermediate, this hypothesis would be excluded. Detailed structural and mechanistic investigations are still in progress.
 | ||
| Fig. 5 Reaction and mechanism of OvoA. (A) C–S bond formation between 1 and L-histidine (12) catalyzed by OvoA. (B) Formation of 2 catalyzed by OvoA in the presence of 1 and 12. (C) L-Cystine (14) formation by OvoA in the presence of 1. (D) Proposed catalytic mechanism of OvoA to produce 2 or 13. | ||
OvoA catalyzes the oxidative coupling between 1 and 12 to generate the 5-histidylcysteine sulfoxide conjugate (13), which is the precursor of the final product 11 (Fig. 5(A)).12 OvoA is an NHI enzyme functionally similar to CDO, with the iron coordinated by the 2xHis–1xGlu triad motif within a conserved HX3HXE sequence in the N-terminal domain. Besides the main reaction, OvoA also catalyzes a CDO-type reaction to produce 2, as a minor reaction (Fig. 5(B)).47 OvoA generates L-cystine (14) when the substrate is only 1, and produces 13 as the main product along with 2 as a co-product when the substrates are both 1 and 12 (Fig. 5(C)).47 A study using an OvoA homolog from Methyloversatilis thermotolerans (OvoAmet) showed that only 1, but not 12, is required for CDO activity.48
Structure–function analyses of the homology model of OvoA and the functionally similar enzyme EgtB showed that Tyr417 of OvoA corresponds to Tyr377 of EgtB. By substituting Tyr417 with an unnatural amino acid, 2-amino-3-(4-hydroxy-3-(methylthio)phenyl) propanoic acid (MtTyr), the CDO-type activity was considerably enhanced.49 These data suggested that Tyr417 plays an important role in bifurcating the two pathways, oxidative C–S bond formation and CDO-type oxidation. The crystal structure of OvoATh2 (OvoA homolog from H. thermophila) in complex with two substrates, 12 and 1, was recently reported (PDB ID: 8KHQ). This structure provided critical information to explain the regioselectivity of C–S bond formation.50 In the substrate binding pocket, Co(II) is coordinated by three His residues (His71, His162, and His166), with 12 coordinating to Co(II) from the opposite side of His71 and 1 coordinating from the opposite side of His162 (Fig. 6(A)). The possible molecular oxygen binding site, situated opposite to His166, is occupied by a water molecule, and the conserved Tyr406 forms a hydrogen bond with it. A hydrogen bond exists between the amino group of 1 and the carboxyl group of 12, bringing them closer together.
 | ||
| Fig. 6 Crystal structures of OvoATh2 (homolog of OvoA) and EgtB. (A) The active site complexed with substrates 1 and 12 in OvoATh2. Tyr406 corresponds to Tyr417 in OvoA. (B) The active site complexed with substrates 15 and 16 in EgtB. Hydrogen bonds are shown as yellow dotted lines, the manganese ion is colored silver, and the chloride ion is shown as a small green ball. | ||
A catalytic mechanism for OvoA has been proposed that explains the generation of both 13 and 2 with Tyr417 (Fig. 5(D)).49 A ternary Fe(III)–superoxo radical complex is formed, and a hydrogen is transferred from Tyr417 to the superoxo species, resulting in a peroxo species. The radical on the sulfur, formed by single electron transfer to Fe(III), reacts with the imidazole to form a C–S bond between 1 and 12. Dehydrogenation from the peroxo species by Tyr417 and oxidation by the generated superoxo species lead to the production of the product 13. In the Y417MtTyr variant, the CDO-type reaction occurs, producing 2. An Fe(IV)O intermediate in the proposed mechanism was observed in OvoAmet through stopped-flow, 57Fe-Mössbauer spectra, and X-ray absorption near edge structure (XANES) analyses.51
In ergothioneine biosynthesis, the NHI enzyme EgtB, which is functionally related to OvoA, catalyzes the O2-dependent C–S bond formation between γ-glutamyl cysteine (γ-GC, 15) and N-α-trimethyl histidine (TMH, 16), producing 17 (Fig. 7(A)).13 To investigate the structural basis of EgtB catalysis, the crystal structure complexed with 15 and 16 was solved.52 Tyr377, located close to the binding site of 16, is assumed to be the key residue for oxygen binding (Fig. 6(B)). Interestingly, EgtB Y377F oxidized the thiol of 15, similar to CDO (Fig. 7(A)), indicating that the hydroxyl group of Tyr377 determines the fate of the reaction.53
 | ||
| Fig. 7 Reaction and mechanism of EgtB. (A) C–S bond formation between 15 and 16 by EgtB WT, and thiol oxidation of 15 by the EgtB Y377F variant. (B) Proposed mechanism of EgtB WT and the Y377F variant. | ||
Based on the crystal structure and the mutation experiments, the reaction mechanism of EgtB has been elucidated (Fig. 7(B)). The Fe(III)–superoxo species is transformed to Fe(III)–hydroperoxo species through a hydrogen radical transfer from Tyr377. The C–S bond is formed by the attack of the sulfur of 15 on C-2 of the imidazole of 16, followed by the removal ofH-2 of the imidazole by Tyr377. Finally, the sulfur group undergoes sulfoxidation to generate the final product 17. As observed in the case of Y377F, without the hydrogen radical transfer from the hydroxyl group of Tyr377, the Fe(III)–superoxo species will directly attack the thiol group of 15, generating a thiol-dioxygenated product via a CDO-like mechanism.53
2. FlcD and FlcE in fluopsin C biosynthesis
Fluopsin C (18) (Fig. 8) was first isolated in 1970 from environmental Pseudomonas aeruginosa and P. fluorescens, and also in 1972 from a Streptomyces strain,54,55 as a copper-containing, dark green complex with specific d–d transition UV-vis absorption for copper(II) in the range of 550–600 nm. 18 exhibits antibiotic activity against a broad spectrum of bacteria and fungi, and even inhibits multidrug-resistant bacterial pathogens, such as Acinetobacter baumannii and Staphylococcus aureus. However, the producer, P. aeruginosa, is resistant to 18, consistent with the ability of P. aeruginosa to tolerate high concentrations of copper.56 | ||
| Fig. 8 Biosynthetic pathway of fluopsin C (18). | ||
The author focused on the gene cluster flc (PA3515–PA3523), which is upregulated under copper stress, as a putative biosynthetic cluster for 18.57,73 The flc operon encodes five proteins, including the methyltransferase FlcA, two adenylosuccinate lyases (ASLs)58 FlcB and FlcC, and two HOD enzymes FlcD and FlcE, together with the copper chaperone CopZ1 and the efflux pumps PA3521-PA3523. Feeding experiments, heterologous expression in E. coli, and in vitro assays were employed to characterize each of the Flc biosynthetic reactions (Fig. 8). As the initial step, the ASL FlcB catalyzes the conjugation of 1 and fumarate to produce 19.
The HOD enzyme FlcE catalyzes both the oxidative decarboxylation and N-hydroxylation of 19 to produce 20 (Fig. 8). FlcE requires only Fe(II) for its activity, and does not require reductants, similar to other HOD enzymes, including Chlamydia protein associating with death domains (CADD) in folate biosynthesis and SznF in streptozotocin biosynthesis.59,60 Next, 20 is oxidized to 21 by the other HOD enzyme FlcD through a four-electron oxidation process, with one carbon being released as formic acid. An assay using L-[3-13C]-1 identified the lost carbon position in the reaction as C-2. In addition, like FlcE, FlcD does not require a reducing agent for the reaction.
The proposed carbon excision reaction by FlcD is given below (Fig. 9). FlcD's diiron center first binds to molecular oxygen, forming μ-peroxo-Fe2(III/III) (Fig. 9(i)). This μ-peroxo–Fe2(III/III) is then converted into the Fe2(IV/IV) complex (Fig. 9(ii)). Subsequently, Fe2(IV/IV) abstracts a hydrogen radical from the N-hydroxyl group (Fig. 9(iii)). The generated N-hydroxyl radical facilitates isomerization and hydroxylation at C-α (Fig. 9(iv)). After hydrogen atom transfer from CH2-β, transient C–N cleavage forms the NO radical (Fig. 9(v)), and further a 1,2-shift rearrangement constructs the C–N bond at C-β (Fig. 9(vi)), as proposed in the UndA reaction in undecene biosynthesis.61,62 A second hydroxylation at C-α regenerates the Fe2(II/II) center (Fig. 9(vii)). Finally, the generated gem-diol is cleaved to form formate and 21 (Fig. 9(viii)). The proposed sequential radical reaction mechanism, leading to carbon excision without any reducing agent, is intriguing. In the structural study of other di-iron oxygenases, such as myo-inositol oxygenase (MIOX),63 its di-iron center was shown to be coordinated by four His and two Asp. The substrate myoinositol is bound to one of the iron atoms in bidentate coordination via OH-1 and OH-6. Structural analysis of FlcD will provide a structural basis for this interesting reaction, as exemplified by the study for MIOX.
 | ||
| Fig. 9 Proposed mechanism of carbon excision catalyzed by FlcD. | ||
For the remaining two enzymes, the lyase FlcC converts 21 to the thiohydroxamate 22, and the SAM-dependent methyltransferase FlcA methylates 22 to form Cu-free fluopsin. Fluopsin chelates a Cu(II) ion to yield the final product, fluopsin C (18) (Fig. 8).
3. MbnBC in CuMbn biosynthesis
The copper-binding peptidic methanobactins (Mbns) chelate Cu(I) to form Cu(I)Mbn (23) as a siderophore,80,81 which was originally found in obligate methanotrophs64 under copper-limiting conditions (Fig. 10). Mbns are synthesized by ribosomes, and classified as RiPPs.70–72 During the biosynthesis of RiPPs, a cysteine residue in the peptide is typically cyclized to a thiazoline and further oxidized to a thiazole.65 In addition to the well-known thiazoline oxidation, L-Cys-related biosynthesis in RiPPs involves several interesting oxidation reactions, as described below. | ||
| Fig. 10 Biosynthetic pathway of Cu(I)Mbn (23). The scheme includes oxidation catalyzed by MbnBC, transamination at leucine catalyzed by the aminotransferase MbnN, and secretion by the multidrug exporter MbnM. | ||
The mbn operon was first identified as regulated in response to copper in the methanotroph Methylosinus trichosporium OB3b.66 The uncharacterized proteins MbnBC are encoded in mbn operons and are responsible for the post-translational modification of the precursor peptide MbnA. The oxazolone ring and thioamide group in mature Mbns are thought to originate from a Cys residue (Fig. 10),67,68 and are transformed into an oxazolone–thioamide moiety through a net four-electron oxidation catalyzed by MbnBC.
MbnB is a member of the DUF692 family and widely distributed in bacteria, but none of the DUF692 family enzymes has been functionally characterized, although the crystal structure of one (HsMbnB from Haemophilus somnus, PDB ID: 3BWW) was solved as a TIM barrel fold protein, which has metal-binding residues. MbnC is present exclusively in the mbn operons and Pseudomonas species, but its function is unknown. Co-expression of MbnBC resulted in soluble proteins, which were purified as a heterodimer. 57Fe-Mössbauer spectral analysis and native top-down mass spectrometry indicated that MbnBC has three Fe(II) in each subunit.69 The LC-MS/MS analysis of the products showed that MbnBC oxidizes Cys21 of MbnA to produce the oxazolone–thioamide moiety of MbnAoxa in the presence of Fe(II) and O2, and does not require any reducing agents.
The oxidation of MbnA by MbnBC was proposed as described below (Fig. 11). A hydrogen atom is abstracted from C-β of Cys21 by an Fe(III)–superoxo intermediate, forming a radical (i). A thioaldehyde is then formed via radical recombination (ii). The thioaldehyde is attacked by an amide nitrogen, aided by the deprotonation by the peroxide coordinated to Fe(III), forming a β-lactam (iii), as seen in the ring formation by IPNS.70 Base-mediated deprotonation from the other amide nitrogen forms an amidate (iv). The generated amidate reacts with the β-lactam to produce an oxazolone ring bound to a thioaminal (v).71 Abstraction of another hydrogen atom by Fe(IV)O lead to the formation of a C-β radical (vi). A hydroxide bound to Fe(III) then abstracts H-α to construct the thioamide group of MbnAoxa and reduce Fe(III) to Fe(II) (vii). This is the first example of an oxazolone and thioamide synthase, and an oxidation reaction catalyzed by a DUF692-type NHI enzyme.
 | ||
| Fig. 11 Proposed mechanism of oxazolone/thioamide formation catalyzed by MbnBC. | ||
A quite recent study presents the X-ray crystal structures of MbnABC complexes, revealing that the leader peptide of MbnA binds to MbnC and facilitating the recruitment of the MbnBC holoenzyme. Meanwhile, the core peptide of MbnA resides in the catalytic cavity formed by the interaction between MbnB and MbnC, which contains a unique tri-iron cluster. The thiol group of the substrate is ligated to this tri-iron center, leading to a dioxygen-dependent reaction for oxazolone–thioamide installation. This study also identified the conserved residue D240 as the base required for reaction step iv in Fig. 11.72
4. TglHI in 3-thiaGlu biosynthesis
In the biosynthesis of 3-thiaGlu 24, the second example of a DUF692-type NHI enzyme family member was reported. A complex of the DUF692-type NHI enzyme (TglH) and a leader peptide-binding protein (TglI) was shown to catalyze the unique C-β excision reaction of a Cys residue in the substrate peptide, producing the nor-cysteine scaffold of TglA-norCys (25), which is a precursor of 24 (Fig. 12A). TglHI is encoded in the RiPPs biosynthetic gene cluster (tgl cluster) in the plant pathogen P. syringae pv. maculicola ES432. The tgl cluster encodes a leader peptide (TglA), a peptide-aminoacyl tRNA ligase (PEARL,5 TglB), TglH, TglI, a carboxy-S-adenosyl-L-methionine (Cx-SAM73) synthase (TglE), and a Cx-SAM-dependent transferase (TglF). Each of the biosynthetic reactions catalyzed by Tgl enzymes was characterized through heterologous expression in E. coli and in vitro assays, as shown in Fig. 12A.73 | ||
| Fig. 12 Biosynthetic pathway of 3-thiaglutamate (24). (A) 3-thiaglutamate (24) formation catalyzed by Tgl enzymes. (B) Cysteine addition by the PEARL TglB to the leader peptide TglA. The reaction starts with phosphorylation of the C-terminal carboxylate of TglA, which then forms an amide bond with the amino group of Cys-tRNACys, followed by the hydrolysis of the tRNA to generate TglA-Cys.5 | ||
TglA-Cys is generated in the reaction catalyzed by the PEARL TglB, from TglA, Cys-tRNA, and ATP (Fig. 12B), and oxidized to 25 by TglHI without any reducing agent. In the TglHI assay with TglA-3-13C-Cys, 13C-labeled formate was detected as a by-product, suggesting that the β-carbon of Cys was removed from the substrate.
The proposed reaction mechanism of the β-carbon scission by TglHI is described below (Fig. 13). First, the activation of the Fe(II) complex by O2 lead to the formation of the Fe(III)–superoxo species (Fig. 13(i)). This Fe(III)–superoxo species abstracts the C-β hydrogen to form a radical intermediate (Fig. 13(ii)). The Fe(III)–hydroperoxo species reacts with the C-β radical to generate a thioacetal Fe(IV)O complex (Fig. 13(iii)), which abstracts a hydrogen from OH-β to form an oxygen radical (Fig. 13(iv)). This leads to the homolysis of Cα–Cβ, generating a C-α radical (Fig. 13(v)), through a mechanism similar to that reported for 2-hydroxyethylphosphonate dioxygenase (HEPD).74 The following radical recombination generates a new C–S bond in the S configuration as 25, with the reduction of the Fe(III) complex (Fig. 13(vi)), similar to thioether formation by IPNS.39 Finally, the thioformyl moiety is hydrolyzed by a hydroxide bound to Fe(II), producing both formate and 25 (Fig. 13(vii)). The TglHI-catalyzed methylene excision reaction is a novel type of post-translational modification in a natural product.
 | ||
| Fig. 13 Proposed mechanism of the TglHI reaction. | ||
Very recently, the X-ray crystal structures of TglHI complexed with and without iron ions were reported.75 TglH adopts a TIM barrel structure with β-sheets wrapped around helices, forming a stable complex with TglI, in a similar manner to MbnBC.72,76 These structures revealed the formation of a catalytic pocket between TglH and TglI, and two iron–iron binding sites in the pocket and a third auxiliary iron binding site were identified. The complex structure model of TglHI and TglA-Cys generated by AlphaFold277 identified the key amino acid Asn73, which could contribute to the H-β abstraction by orienting the thiol of TglA-Cys via hydrogen bonding.
After the TglHI reaction, TglF reacts with 25 and Cx-SAM, generating TglA-thiaGlu (L-3-thiaglutamate). Its absolute configuration was confirmed by microcrystal electron diffraction (MicroED) analyses with synthesized diastereoisomers (Fig. 12(A)). TglG, a membrane-bound protease, cleaves the amide bond of TglA-thiaGlu, releasing 3-thiaGlu.
5. TmoHI and TmoD in 3-thia-α-amino acid biosynthesis
In the biosynthesis of 3-thiahomoleucine (26), the DUF692 protein (TmoH, 67% sequence identity with TglH) and a vitamin B12-dependent radical-SAM (rSAM) enzyme (TmoD), were characterized as new L-Cys oxidases in Tistrella mobile. The biosynthetic cluster (tmo cluster) encodes a precursor peptide (TmoA), a PEARL (TmoB), TmoH, a SAM-dependent methyltransferase (TmoS), a RiPPs recognition element-containing protein (TmoI), TmoD, a metalloprotease (TmoG), and a transporter (TmoT). Each of the reactions by Tmo enzymes was identified in vitro and in an E. coli expression system (Fig. 14).16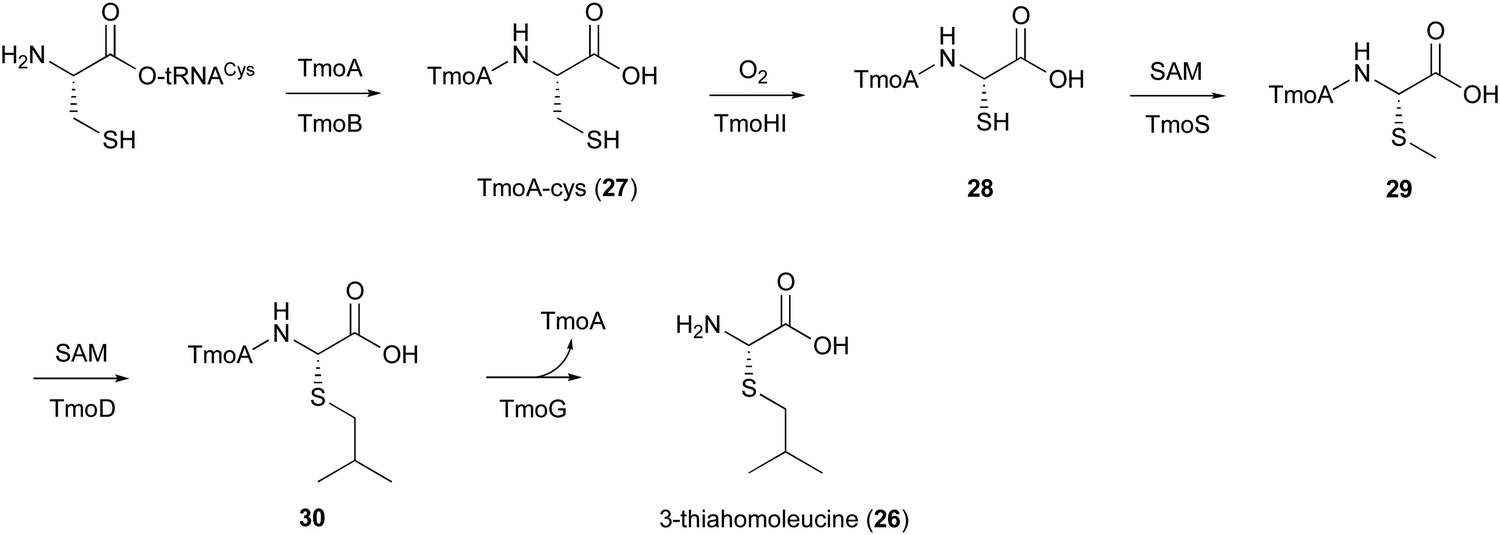 | ||
| Fig. 14 Biosynthetic pathway of 3-thiahomoleucine (26). | ||
TmoHI cleaves the β-carbon of TomA-Cys (27) to generate nor-Cys (28), in a similar manner to TglHI. TmoS catalyzes the S-methylation of 28 to produce 29. TmoD is classified as a class B vitamin B12-dependent rSAM enzyme. This rSAM enzyme family utilizes SAM, methylcobalamin, and a [4Fe–4S]2+ cluster to initiate methyl transfer onto a non-nucleophilic phosphorus or carbon atom. TmoD was anaerobically reconstituted with Na2S, (NH4)2Fe(SO4)2, and hydroxocobalamin (HOCbl), enabling the catalysis of the tandem trimethylation of the C-terminal residue of 29 to yield 30 (Fig. 15).
 | ||
| Fig. 15 Proposed mechanism of the methylation catalyzed by TmoD. | ||
The hypothetical reaction mechanism of TmoD is described below (Fig. 15). HOCbl is first reduced to cobalamin Co(I) (i) and then reacts with SAM to form methylcobalamin Me–Co(III) (ii). The [4Fe–4S]2+ cluster is reduced to [4Fe–4S]+ (iii), and generates 5′-dA˙ from another SAM (iv).78 The 5′-dA˙ abstracts a hydrogen from 29 to produce 29˙ (v), which then accepts a methyl group from Me–Co(III), forming a monomethylated compound 31 (vi). After repeating another two rounds of methylation, the final compound 30 is produced. CysS,79 ThnK,80 and TokK,81 which belong to the same group as TmoD in the similarity network of vitamin B12-dependent rSAM enzymes, have similar functions, adding methyl groups iteratively. For example, TokK catalyzes three tandem methylation reactions of the intermediate to generate the isopropyl group of the carbapenem asparenomycin.81 The trimethylation by TmoD, which construct an isopropyl group on the S-methyl group, expands the reaction diversity of the vitamin B12-dependent rSAM enzymes responsible for peptide modifications.
Finally, the membrane-bound TmoG cleaves the C-terminal 26 from 30 to regenerate TmoA. The substrate specificity of TmoG was tested with TmoA-Cys, TmoA-Leu, TmoA-Gln, and TmoA-Met, and only TmoA-Met was accepted. This preference is consistent with the recognition of the extended hydrophobic side chain of 30.
6. ChrHI in RiPP ChrA* biosynthesis
To discover novel DUF692 proteins, 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 108 DUF692 sequences were retrieved from the Sequence Similarity Network (SSN) and the Genomic Neighborhood Network (GNN).17 As a result, the DUF692 protein ChrH encoded in the gene cluster in Chryseobacterium sp. (chr cluster) was analyzed, and its product was identified as the peptide ChrA*. Heterologous expression in E. coli and in vitro assays revealed the details of the ChrHI reaction that converts ChrA to ChrA*, which has one additional sulfide and one additional methyl group, with the loss of a single thiol group compared to its substrate (Fig. 16).
108 DUF692 sequences were retrieved from the Sequence Similarity Network (SSN) and the Genomic Neighborhood Network (GNN).17 As a result, the DUF692 protein ChrH encoded in the gene cluster in Chryseobacterium sp. (chr cluster) was analyzed, and its product was identified as the peptide ChrA*. Heterologous expression in E. coli and in vitro assays revealed the details of the ChrHI reaction that converts ChrA to ChrA*, which has one additional sulfide and one additional methyl group, with the loss of a single thiol group compared to its substrate (Fig. 16).
 | ||
| Fig. 16 Modification of ChrA by ChrHI. Purposed macrocyclization catalyzed by ChrHI. R: the remaining sequence of ChrA after digestion of the C-terminal fragment with the endoproteinase LysC. | ||
Extensive 1D and 2D NMR analyses, including HSQC, dqCOSY, NOESY, and HMBC, coupled with an HR-MS/MS analysis showed the structural changes on C63 and C66 in ChrA*, produced from E. coli expressing ChrA and ChrHI. The reaction of ChrHI with ChrA as the substrate was reconstituted in vitro. The reaction with SAM yielded the same product ChrA*, while the reaction without SAM generated a new product whose m/z is 36 Da lower than ChrA (32, Fig. 17).
 | ||
| Fig. 17 Proposed mechanism of the macrocyclization catalyzed by ChrHI. | ||
Based on these data, the proposed reaction mechanism of ChrHI is described below (Fig. 17). First, the Fe(II) in the active site of ChrH reacts with molecular oxygen to form the Fe(III)–superoxo intermediate. Next, in a similar manner to TglH and MbnB, the Fe(III)–superoxo species abstracts H-β of Cys66 to produce a thioketyl radical (i), which forms a thioaldehyde and transfers one electron to Fe(III) (ii). The amide nitrogen of Gly67 then attacks the thioaldehyde to form a β-lactam (iii), similar to MbnBC and the mononuclear NHI enzyme, IPNS.39 A second nucleophilic attack by the amide nitrogen of Met68 to the β-lactam forms a bicyclic intermediate (iv). The generation of Fe(IV)O is expected to occur after one electron is transferred from Fe(II) to the Fe(III)-hydroperoxo species, although the exact timing has not been determined. The Fe(IV)O abstracts a hydrogen atom from the hemiaminal in the bicyclic intermediate, generating an oxygen radical (v). Subsequent ring opening leads to the C-α radical (vi), which can follow either pathway A [generation of the product via episulfide formation, and episulfide opening via nucleophilic attack by C63 thiol and methylation by SAM, (vii)] or pathway B [first oxidized to a cation (viii), followed by S-methylation of the hemiaminal by SAM (ix), then addition of the thiol of Cys63 to Cys66 (x), and MeS migration to Cys66 (xi)], to generate ChrA* (Fig. 17). In absence of SAM, the radical intermediate is reduced (pathway C). Subsequent imine formation (xii) and the addition of the thiol of Cys63 to Cys66 (xiii) lead to the generation of 32. ChrHI is the first DUF692-type protein that catalyzes reactions by using SAM as a cofactor, in addition to iron. The reaction bifurcation with or without SAM is intriguing.
7. SycC in AviCamCys biosynthesis
The LanD-like flavoproteins, belonging to the family of homo-oligomeric flavin-containing cysteine decarboxylases, catalyze the oxidative decarboxylation of 1 at the C-terminus of the peptide to form an enethiol (Fig. 18).82,83 This Cys-derived enethiol is highly nucleophilic and easily reacts with Dha or Dhb to form S-[2-aminovinyl]-cysteine (AviCys) and S-[2-aminovinyl]-3-methylcysteine (AviMeCys), as part of a macrocyclic ring containing 4–6 amino acids.84,85 | ||
| Fig. 18 Reaction catalyzed by the LanD-like protein, and the formation of AviCys and AviMeCys. | ||
Genome mining, using LanD as a query, was performed for the biosynthesis, including the generation of the Cys-derived enethiol. As a result, a novel radical SAM enzyme (SycC), which generates a radical to react with an enethiol, forming the sulfide ring of AviCamCys (33) (Fig. 19), was identified in S. ureilyticus YC419. SycC is encoded in the syc cluster with a 22-aa precursor peptide (SycA), a major facilitator superfamily transporter (SycB), and a LanD-like protein (SycD). Heterologous expression in E. coli and in vitro assays showed that SycD reacts with SycA to produce the C-terminal enethiol, and SycC oxidizes the enethiol intermediate to produce 33. Advanced Marfey's method86 and NOESY analyses identified the absolute configuration of C-3 as R.
SycC belongs to the radical SAM protein family, including NxxcB87 and CteB (PDB ID: 5WHY, contains three ion–sulfur cluster),88 which is known for forming saturated thioether bonds. Similar to NxxcB and CteB, SycC contains both the CxxxCxxC motif for binding to the canonical [4Fe–4S]+ cluster and the SPASM domain for binding to the auxiliary [4Fe–4S]+ cluster, which enhances the activity.89 Similar to the NxxcB-catalyzed process, the reaction of SycD was proposed as described below (Fig. 19). SycC first utilizes the canonical [4Fe–4S]+ cluster to provide an electron to SAM, generating the 5′-dA radical. The 5′-dA radical then abstracts the C-β hydrogen of Asn19, forming the C-β radical. The auxiliary [4Fe–4S]+ cluster either activates/conjugates the enethiol of the C-terminal Cys22 with the Asn19Cβ radical (route a) or deprotonates H-α to form an α,β-unsaturated diamide for the following enethiol addition (route b). In the absence of SycC, the intermediate 34 undergoes hydration-coupled dethiolation to form the shunt aldehyde 35.
 | ||
| Fig. 19 Proposed mechanism of the macrocyclization catalyzed by SycD and SycC. | ||
Previously, the knowledge about enethiol modification was limited to the generation of Avi(Me)Cys by dehydration or dethiolation.83 SycC is a novel enzyme that forms a radical at the upstream Asn residue and promotes sulfide formation between the generated radical and enethiol to produce the unsaturated thioether, AviCamCys. This finding enriches our knowledge about the enzymatic transformation of enethiol-containing peptides. Several radical SAM enzymes involved in the modification of Cys-containing peptides have also been reported, such as AlbA for thioether formation in subtilosin A biosynthesis,90 but this review presents the more recently discovered example involving the modification of a Cys-derived enethiol.
8. Conclusion
Several new Cys-modifying enzymes, including CDO homologs, dinuclear NHI-dependent enzymes, and radical SAM enzymes, have been identified in natural product biosynthesis, revealing unusual chemical reactions in L-Cys metabolism. These advances provide detailed insight into the novel secondary metabolism of L-Cys-related natural products, expand the repertoire of L-Cys modifying enzymes, and allow more efficient access to unique molecules. With growing interest in the biosynthesis of natural products related to amino acids, the biosynthetic study of peptides or alkaloids from nature has emerged as an important research direction. With significant progress in the study of L-Cys metabolism, such mechanistic elucidation based on structural, biochemical, and computational analyses will not only provide useful insights into the Cys-derived biosynthetic pathways, but also pave the way for enzyme engineering toward future drug discovery.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by a grant-in-aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (JSPS KAKENHI grant number JP20KK0173, JP21K18246, JP21H02636, JP22H05123, and JP23H00393), the New Energy and Industrial Technology Development Organization (NEDO, grant Number JPNP20011), AMED (grant Number JP21ak0101164) from Japan Science and Technology Agency, The Naito foundation, Nagase Science Technology Foundation, Yamada Science foundation, The Mitsubishi Foundation, the Chugai Foundation for Innovative Drug Discovery Science, and The Nakajima Foundation. Y. G. is a recipient of the JSPS Postdoctoral Fellowship for Foreign Researchers (ID No. P22405) and the Outstanding Doctoral Graduates Development Scholarship of Shanghai Jiao Tong University. We thank Drs Takahiro Mori and Richiro Ushimaru for critically reading the manuscript.References
- E. Block, Reactions of Organosulfur Compounds: Organic Chemistry: A Series of Monographs, Academic Press, 2013, vol. 37 Search PubMed.
- C. E. Paulsen and K. S. Carroll, Chem. Rev., 2013, 113, 4633–4679 CrossRef CAS PubMed.
- Z. Hu, T. Awakawa, Z. Ma and I. Abe, Nat. Commun., 2019, 10, 184 CrossRef PubMed.
- J. B. Patteson, A. T. Putz, L. Tao, W. C. Simke, L. H. Bryant III, R. D. Britt and B. Li, Science, 2021, 374, 1005–1009 CrossRef CAS PubMed.
- C. P. Ting, M. A. Funk, S. L. Halaby, Z. Zhang, T. Gonen and W. A. van der Donk, Science, 2019, 365, 280–284 CrossRef CAS PubMed.
- M. A. Ortega, Y. Hao, Q. Zhang, M. C. Walker, W. A. van der Donk and S. K. Nair, Nature, 2015, 517, 509–512 CrossRef CAS PubMed.
- B. Cheng, J. Huang, Y. Duan and W. Liu, Angew. Chem., Int. Ed., 2023, 62, e202308733 CrossRef CAS PubMed.
- K. S. Ryan and C. L. Drennan, Nat. Prod. Rep., 2018, 35, 612–614 RSC.
- P. Rabe, J. J. A. G. Kamps, C. J. Schofield and C. T. Lohans, Nat. Prod. Rep., 2018, 35, 735–756 RSC.
- C. A. Joseph and M. J. Maroney, Chem. Commun., 2007, 3338–3349 RSC.
- J. M. Dunwell, A. Culham, C. E. Carter, C. R. Sosa-Aguirre and P. W. Goodenough, Trends Biochem. Sci., 2001, 26, 740–746 CrossRef CAS PubMed.
- A. Braunshausen and F. P. Seebeck, J. Am. Chem. Soc., 2011, 133, 1757–1759 CrossRef CAS PubMed.
- F. P. Seebeck, J. Am. Chem. Soc., 2010, 132, 6632–6633 CrossRef CAS PubMed.
- A. J. Komor, A. J. Jasniewski, L. Que and J. D. Lipscomb, Nat. Prod. Rep., 2018, 35, 646–659 RSC.
- G. E. Kenney, L. M. Dassama, M.-E. Pandelia, A. S. Gizzi, R. J. Martinie, P. Gao, C. J. DeHart, L. F. Schachner, O. S. Skinner, S. Y. Ro, X. Zhu, M. Sadek, P. M. Thomas, S. C. Almo, J. M. J. Bollinger, C. Krebs, N. L. Kelleher and A. C. Rosenzweig, Science, 2018, 359, 1411–1416 CrossRef CAS PubMed.
- Y. Yu and W. A. van der Donk, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2205285119 CrossRef CAS PubMed.
- R. S. Ayikpoe, L. Zhu, J. Y. Chen, C. P. Ting and W. A. van der Donk, ACS Cent. Sci., 2023, 9, 1008–1018 CrossRef CAS PubMed.
- M. W. Ruszczycky, A. Zhong and H.-w Liu, Nat. Prod. Rep., 2018, 35, 615–621 RSC.
- M. H. Stipanuk, Annu. Rev. Nutr., 2004, 24, 539–577 CrossRef CAS PubMed.
- J. G. McCoy, L. J. Bailey, E. Bitto, C. A. Bingman, D. J. Aceti, B. G. Fox and G. N. J. Phillips, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3084–3089 CrossRef CAS PubMed.
- C. R. Simmons, Q. Liu, Q. Huang, Q. Hao, T. P. Begley, P. A. Karplus and M. H. Stipanuk, J. Biol. Chem., 2006, 281, 18723–18733 CrossRef CAS PubMed.
- C. R. Simmons, Q. Hao and M. H. Stipanuk, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2005, 61, 1013–1016 CrossRef CAS PubMed.
- S. Ye, X. a Wu, L. Wei, D. Tang, P. Sun, M. Bartlam and Z. Rao, J. Biol. Chem., 2007, 282, 3391–3402 CrossRef CAS PubMed.
- J. E. J. Dominy, C. R. Simmons, P. A. Karplus, A. M. Gehring and M. H. Stipanuk, J. Bacteriol., 2006, 188, 5561–5569 CrossRef CAS PubMed.
- C. M. Driggers, S. J. Hartman and P. A. Karplus, Protein Sci., 2015, 24, 154–161 CrossRef CAS PubMed.
- M. Wensien, F. R. von Pappenheim, L.-M. Funk, P. Kloskowski, U. Curth, U. Diederichsen, J. Uranga, J. Ye, P. Fang, K.-T. Pan, H. Urlaub, R. A. Mata, V. Sautner and K. Tittmann, Nature, 2021, 593, 460–464 CrossRef CAS PubMed.
- Y.-L. Wang, C.-Y. Chang, N.-S. Hsu, I.-W. Lo, K.-H. Lin, C.-L. Chen, C.-F. Chang, Z.-C. Wang, Y. Ogasawara, T. Dairi, C. Maruyama, Y. Hamano and T.-L. Li, Nat. Commun., 2023, 14, 2528 CrossRef CAS PubMed.
- B. S. Pierce, J. D. Gardner, L. J. Bailey, T. C. Brunold and B. G. Fox, Biochemistry, 2007, 46, 8569–8578 CrossRef CAS PubMed.
- E. J. Blaesi, B. G. Fox and T. C. Brunold, Biochemistry, 2014, 53, 5759–5770 CrossRef CAS PubMed.
- J. A. Crawford, W. Li and B. S. Pierce, Biochemistry, 2011, 50, 10241–10253 CrossRef CAS PubMed.
- E. P. Tchesnokov, A. S. Faponle, C. G. Davies, M. G. Quesne, R. Turner, M. Fellner, R. J. Souness, S. M. Wilbanks, S. P. De Visser and G. N. L. Jameson, Chem. Commun., 2016, 52, 8814–8817 RSC.
- X. Che, J. Gao, D. Zhang and C. Liu, J. Phys. Chem. A, 2012, 116, 5510–5517 CrossRef CAS PubMed.
- D. Kumar, G. N. Sastry, D. P. Goldberg and S. P. De Visser, J. Phys. Chem. A, 2012, 116, 582–591 CrossRef CAS PubMed.
- C. R. Simmons, K. Krishnamoorthy, S. L. Granett, D. J. Schuller, J. E. J. Dominy, T. P. Begley, M. H. Stipanuk and P. A. Karplus, Biochemistry, 2008, 47, 11390–11392 CrossRef CAS PubMed.
- A. Takahashi, S. Kurasawa, D. Ikeda, Y. Okami and T. Takeuchi, J. Antibiot., 1989, 42, 1556–1561 CrossRef CAS PubMed.
- A. L. Stefanska, R. Cassels, S. J. Ready and S. R. Warr, J. Antibiot., 2000, 53, 357–363 CrossRef CAS PubMed.
- L. Barra, T. Awakawa, K. Shirai, Z. Hu, G. Bashiri and I. Abe, Nature, 2021, 600, 754–758 CrossRef CAS PubMed.
- L. A. McNeill, T. J. Brown, M. Sami, I. J. Clifton, N. I. Burzlaff, T. D. Claridge, R. M. Adlington, J. E. Baldwin, P. J. Rutledge and C. J. Schofield, Chem. – Eur. J., 2017, 23, 12815–12824 CrossRef CAS PubMed.
- E. Tamanaha, B. Zhang, Y. Guo, W.-c Chang, E. W. Barr, G. Xing, J. St. Clair, S. Ye, F. Neese, J. M. J. Bollinger and C. Krebs, J. Am. Chem. Soc., 2016, 138, 8862–8874 CrossRef CAS PubMed.
- R. B. Hamed, J. R. Gomez-Castellanosa, L. Henry, C. Ducho, M. A. McDonough and C. J. Schofield, Nat. Prod. Rep., 2013, 30, 21–107 RSC.
- T. Pholler and P. B. Hopkins, Meth. Enzymol., 1995, 252, 115–123 CrossRef PubMed.
- H. S. C. Spies and D. J. Steenkamp, Eur. J. Biochem., 1994, 224, 203–213 CrossRef CAS PubMed.
- I. Castellano and F. P. Seebeck, Nat. Prod. Rep., 2018, 35, 1241–1250 RSC.
- K. H. Weaver and D. L. Rabenstein, J. Org. Chem., 1995, 60, 1904–1907 CrossRef CAS.
- M. R. Ariyanayagam and A. H. Fairlamb, Mol. Biochem. Parasitol., 2001, 115, 189–198 CrossRef CAS PubMed.
- C. Jacob, Nat. Prod. Rep., 2006, 23, 851–863 RSC.
- H. Song, A. S. Her, F. Raso, Z. Zhen, Y. Huo and P. Liu, Org. Lett., 2014, 16, 2122–2125 CrossRef CAS PubMed.
- R. Cheng, A. C. Weitz, J. Paris, Y. Tang, J. Zhang, H. Song, N. Naowarojna, K. Li, L. Qiao, J. Lopez, M. W. Grinstaff, L. Zhang, Y. Guo, S. Elliott and P. Liu, Chem. Sci., 2022, 13, 3589–3598 RSC.
- L. Chen, N. Naowarojna, H. Song, S. Wang, J. Wang, Z. Deng, C. Zhao and P. Liu, J. Am. Chem. Soc., 2018, 140, 4604–4612 CrossRef CAS PubMed.
- X. Wang, S. Hu, J. Wang, T. Zhang, K. Ye, A. Wen, G. Zhu, A. Vegas, L. Zhang, W. Yan, X. Liu and P. Liu, ACS Catal., 2023, 13, 15417–15426 CrossRef CAS PubMed.
- J. C. Paris, S. Hu, A. Wen, A. C. Weitz, R. Cheng, L. B. Gee, Y. Tang, H. Kim, A. Vegas, W.-C. Chang, S. J. Elliott, P. Liu and Y. Guo, Angew. Chem., Int. Ed., 2023, 62, e202309362 CrossRef CAS PubMed.
- K. V. Goncharenko, A. Vit, W. Blankenfeldt and F. P. Seebeck, Angew. Chem., Int. Ed., 2015, 54, 2821–2824 CrossRef CAS PubMed.
- K. V. Goncharenko and F. P. Seebeck, Chem. Commun., 2016, 52, 1945–1948 RSC.
- Y. Egawa, K. Umino, S. Awataguchi, Y. Kawano and T. Okuda, J. Antibiot., 1970, 23, 267–270 CrossRef CAS PubMed.
- S. Miyamura, N. Ogasawara, H. Otsuka, S. Niwayama, H. Tanaka, T. Take, T. Uchiyama, H. Nakazawa, K. Abe and K. Koizumi, J. Antibiot., 1972, 25, 369–370 CrossRef CAS PubMed.
- G. M. Teitzel and M. R. Parsek, Appl. Environ. Microbiol., 2003, 69, 2313–2320 CrossRef CAS PubMed.
- J. T. Thaden, S. Lory and T. S. Gardner, J. Bacteriol., 2010, 192, 2557–2568 CrossRef CAS PubMed.
- E. A. Toth and T. O. Yeates, Structure, 2000, 8, 163–174 CrossRef CAS PubMed.
- R. Schwarzenbacher, F. Stenner-Liewen, H. Liewen, H. Robinson, H. Yuan, E. Bossy-Wetzel, J. C. Reed and R. C. Liddington, J. Biol. Chem., 2004, 279, 29320–29324 CrossRef CAS PubMed.
- M. J. McBride, S. R. Pope, K. Hu, C. D. Okafor, E. P. Balskus, J. M. Bollinger Jr and A. K. Boal, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2015931118 CrossRef CAS PubMed.
- O. M. Manley, R. Fan, Y. Guo and T. M. Makris, J. Am. Chem. Soc., 2019, 141, 8684–8688 CrossRef CAS PubMed.
- B. Zhang, L. J. Rajakovich, D. Van Cura, E. J. Blaesi, A. J. Mitchell, C. R. Tysoe, X. Zhu, B. R. Streit, Z. Rui and W. Zhang, J. Am. Chem. Soc., 2019, 141, 14510–14514 CrossRef CAS PubMed.
- P. M. Brown, T. T. Caradoc-Davies, J. M. Dickson, G. J. Cooper, K. M. Loomes and E. N. Baker, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15032–15037 CrossRef CAS PubMed.
- H. J. Kim, D. W. Graham, A. A. DiSpirito, M. A. Alterman, N. Galeva, C. K. Larive, D. Asunskis and P. M. A. Sherwood, Science, 2004, 305, 1612–1615 CrossRef CAS PubMed.
- E. S. Sattely, M. A. Fischbach and C. T. Walsh, Nat. Prod. Rep., 2008, 25, 757–793 RSC.
- B. D. Krentz, H. J. Mulheron, J. D. Semrau, A. A. DiSpirito, N. L. Bandow, D. H. Haft, S. Vuilleumier, J. C. Murrell, M. T. McEllistrem, S. C. Hartsel and W. H. Gallagher, Biochemistry, 2010, 49, 10117–10130 CrossRef CAS PubMed.
- G. E. Kenney and A. C. Rosenzweig, BMC Biol., 2013, 11, 1–17 CrossRef PubMed.
- G. E. Kenney, A. W. Goering, M. O. Ross, C. J. DeHart, P. M. Thomas, B. M. Hoffman, N. L. Kelleher and A. C. Rosenzweig, J. Am. Chem. Soc., 2016, 138, 11124–11127 CrossRef CAS PubMed.
- Z. Liu, X. Chen, S. Yang, R. Tian and F. Wang, Curr. Opin. Chem. Biol., 2023, 74, 102305 CrossRef CAS PubMed.
- H. Lechner, D. Pressnitz and W. Kroutil, Biotechnol. Adv., 2015, 33, 457–480 CrossRef CAS PubMed.
- J. Grzetic and J. Oomens, J. Am. Soc. Mass Spectrom., 2011, 23, 290–300 CrossRef PubMed.
- C. Dou, Z. Long, S. Li, D. Zhou, Y. Jin, L. Zhang, X. Zhang, Y. Zheng, L. Li, X. Zhu, Z. Liu, S. He, W. Yan, L. Yang, J. Xiong, X. Fu, S. Qi, H. Ren, S. Chen, L. Dai, B. Wang and W. Cheng, Cell Res., 2022, 32, 302–314 CrossRef CAS PubMed.
- J. Kim, H. Xiao, J. B. Bonanno, C. Kalyanaraman, S. Brown, X. Tang, N. F. Al-Obaidi, Y. Patskovsky, P. C. Babbitt, M. P. Jacobson, Y.-S. Lee and S. C. Almo, Nature, 2013, 498, 123–126 CrossRef CAS PubMed.
- S. C. Peck, C. Wang, L. M. Dassama, B. Zhang, Y. Guo, L. J. Rajakovich, J. M. Bollinger Jr, C. Krebs and W. A. van der Donk, J. Am. Chem. Soc., 2017, 139, 2045–2052 CrossRef CAS PubMed.
- Y. Zheng, X. Xu, X. Fu, X. Zhou, C. Dou, Y. Yu, W. Yan, J. Yang, M. Xiao and W. A. van der Donk, Structure, 2023, 31, 1220–1232 CrossRef CAS PubMed.
- Y. J. Park, R. J. Jodts, J. W. Slater, R. M. Reyes, V. J. Winton, R. A. Montaser, P. M. Thomas, W. B. Dowdle, A. Ruiz, N. L. Kelleher, J. M. J. Bollinger, C. Krebs, B. M. Hoffman and A. C. Rosenzweig, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2123566119 CrossRef CAS PubMed.
- M. Mirdita, K. Schütze, Y. Moriwaki, L. Heo, S. Ovchinnikov and M. Steinegger, Nat. Methods, 2022, 19, 679–682 CrossRef CAS PubMed.
- M. N. Lundahl, R. Sarksian, H. Yang, R. J. Jodts, A. Pagnier, D. F. Smith, W. A. van der Donk, B. M. Hoffman, W. E. Broderick and J. B. Broderick, J. Am. Chem. Soc., 2022, 144, 5087–5098 CrossRef CAS PubMed.
- Y. Wang and T. P. Begley, J. Am. Chem. Soc., 2020, 142, 9944–9954 CrossRef CAS PubMed.
- D. R. Marous, E. P. Lloyd, A. R. Buller, K. A. Moshos, T. L. Grove, A. J. Blaszczyk, S. J. Booker and C. A. Townsend, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10354–10358 CrossRef CAS PubMed.
- H. L. Knox, E. K. Sinner, C. A. Townsend, A. K. Boal and S. J. Booker, Nature, 2022, 602, 343–348 CrossRef CAS PubMed.
- C. S. Sit, S. Yoganathan and J. C. Vederas, Acc. Chem. Res., 2011, 44, 261–268 CrossRef CAS PubMed.
- E. S. Grant-Mackie, E. T. Williams, P. W. Harris and M. A. Brimble, JACS Au, 2021, 1, 1527–1540 CrossRef CAS PubMed.
- Y. Qiu, J. Liu, Y. Li, Y. Xue and W. Liu, Cell Chem. Biol., 2021, 28, 675–685 CrossRef CAS PubMed.
- J. Lu, Y. Wu, Y. Li and H. Wang, Angew. Chem., Int. Ed., 2021, 60, 1951–1958 CrossRef CAS PubMed.
- K. Fujii, Y. Ikai, H. Oka, M. Suzuki and K.-I. Harada, Anal. Chem., 1997, 69, 5146–5151 CrossRef CAS.
- A. Caruso, L. B. Bushin, K. A. Clark, R. J. Martinie and M. R. Seyedsayamdost, J. Am. Chem. Soc., 2018, 141, 990–997 CrossRef PubMed.
- T. L. Grove, P. M. Himes, S. Hwang, H. Yumerefendi, J. B. Bonanno, B. Kuhlman, S. C. Almo and A. A. Bowers, J. Am. Chem. Soc., 2017, 139, 11734–11744 CrossRef CAS PubMed.
- N. Mahanta, G. A. Hudson and D. A. Mitchell, Biochemistry, 2017, 56, 5229–5244 CrossRef CAS PubMed.
- L. Flühe, T. A. Knappe, M. J. Gattner, A. Schäfer, O. Burghaus, U. Linne and M. A. Marahiel, Nat. Chem. Biol., 2012, 8, 350–357 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2024 |