
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Granular polyrotaxane microgels as injectable hydrogels for corneal tissue regeneration†
Antonio J.
Feliciano
a,
Yousra
Alaoui Selsouli
b,
Pamela
Habibovic
 b,
Zeinab Niloofar Tahmasebi
Birgani
ab,
Lorenzo
Moroni
a and
Matthew B.
Baker
*ab
b,
Zeinab Niloofar Tahmasebi
Birgani
ab,
Lorenzo
Moroni
a and
Matthew B.
Baker
*ab
aDepartment of Complex Tissue Regeneration, Maastricht University, MERLN Institute for Technology-Inspired Regenerative Medicine, Maastricht, the Netherlands. E-mail: m.baker@maastrichtuniversity.nl
bDepartment of Instructive Biomaterial Engineering, Maastricht University, MERLN Institute for Technology-Inspired Regenerative Medicine, Maastricht, the Netherlands
First published on 22nd August 2024
Abstract
Corneal diseases, a leading cause of global vision impairment, present challenges in treatment due to corneal tissue donor scarcity and transplant rejection. Hydrogel biomaterials in the form of corneal implants for tissue regeneration, while promising, have faced obstacles related to cellular and tissue integration. This study develops and investigates the potential of granular polyrotaxane (GPR) hydrogels as a scaffold for corneal keratocyte growth and transparent tissue generation. Employing host–guest driven supramolecular interactions, we developed injectable, cytocompatible hydrogels. By optimizing cyclodextrin (CD) concentrations in thiol–ene crosslinked PEG microgels, we observed improved mechanical properties and thermoresponsiveness while preserving injectability. These microgels, adaptable for precise defect filling, 3D printing or tissue culture facilitate enhanced cellular integration with corneal keratocytes and exhibit tissue-like structures in culture. Our findings demonstrate the promise of GPR hydrogels as a minimally invasive avenue for corneal tissue regeneration. These results have the potential to address transplantation challenges, enhance clinical outcomes, and restore vision.
Introduction
The cornea, a vital ocular tissue, plays a pivotal role in vision, and any damage to it can lead to severe visual impairment or blindness. Despite being the established gold standard for addressing corneal blindness, corneal transplantation often encounters obstacles such as limited donor availability and rejection rates.1 Recent advances in tissue engineering have led to several innovative strategies to overcome these limitations. 3D printing has emerged as a state-of-the-art technology with notable achievements, including the successful 3D printing of corneas using collagen-based materials among others.2–5Injectable hydrogels have gained prominence in the field of 3D printing and bioengineering, primarily due to their compatibility with nozzle-based techniques, enabling the creation of complex tissue structures.6 Among these hydrogels, supramolecular gels stand out as versatile injectable materials with a promising role in enhancing tissue integration.7 Supramolecular gels have shown promise in corneal repair, exemplified by hydrogels employing collagen mimetic peptides8 and host–guest interaction.9 However, the inherent weakness of these interactions has led to challenges related to structural stability, low stiffness, and resistance to erosion.10 To tackle these issues, secondary crosslinking strategies, such as bio-orthogonal crosslinking,11 have been employed to enhance construct stability and modulate mechanical properties.
Granular hydrogels, comprised of water-swelled microgels, represent an emerging and versatile class of hydrogel materials with wide-ranging biomedical applications, including wound healing, cardiac repair, tissue engineering, and cell delivery.12–14 Within these formulations, hydrogel microparticles constitute the solid phase, while the liquid phase, characterized by significant porosity, facilitates the flow of nutrients and cells within the interstitial spaces. Microgels are created by crosslinking polymers into a network that forms the solid phase, which, when densely packed, undergoes interparticle crosslinking, leading to “jamming” that gives rise to a dense yet flexible granular hydrogel scaffold. These dual-scale matrix structures are injectable owing to their remarkable self-healing capabilities. For instance, through interparticle crosslinking facilitated by supramolecular or non-covalent interactions, microgels can undergo shear-thinning to flow through a needle and subsequently reassemble due to their innate self-healing properties.
Polyethylene glycol (PEG) microgels have been fabricated using various methods such as templating,15 electrospraying,16,17 and microfluidics.18,19 Microgels have been synthesized using diverse polymeric backbones beyond PEG including alginate20–22 and hyaluronic acid23–25 while utilizing a variety of crosslinking chemistries to mechanically reinforce the particle or suprastructure.26 Recently, intraparticle crosslinking has been achieved through robust chemistries such as thiol–ene,17 acrylate27 or maleimide28 chemistry, which utilize covalent linkages to maintain particle geometry and prevent dissolution. On the other hand, interparticle crosslinking is often based on supramolecular or dynamic interactions that can be disrupted by shear stress. For instance, a granular hydrogel bioink developed in Burdick's lab relies on dynamic-covalent bonds formed through aldehyde- and hydrazide-modified microgels. These dynamic interactions not only influence mechanical properties, but also enhance the structural integrity of the granular material, facilitating cellular spreading into the interstitial spaces between microgels.29 This innovative framework holds promise for designing injectable materials for diverse 3D printing applications to reproduce tissue complexity, improve 3D cell cultures and localized drug delivery.30
An under-explored architecture for microgels is the polyrotaxane architecture. Polyrotaxanes represent molecular assemblies formed between long-chain polymers like PEG and macrocycles such as cyclodextrin (CD), creating a necklace-like structure through van der Waals, host–guest, and hydrogel bonding forces.31–33 This unique architecture allows the CD rings to shuttle back and forth while simultaneously rotating around the polymer axis. Access to metastable states of polypseudorotaxanes has been enabled through temperature control and crystallization at the molecular level, facilitating the fabrication of 3D-printed actuators with tunable network architectures.34 When the PEG chains are capped to prevent dethreading, both PEG and αCD can spontaneously form biocompatible and tunable hydrogels. The degree of molecular mobility within these hydrogels has been observed to be inversely correlated with the density of CD/PEG chains.35,36 The mobility of these molecules could be harnessed to activate cellular mechanosignaling, thus changing the cells’ function.37,38 Polyrotaxane biomaterials have found applications in various regenerative therapies, including corneal regeneration, cartilage tissue regeneration, bone tissue regeneration, angiogenesis enhancement, and even neuronal differentiation.39–42 More specifically, the development of polyrotaxane multiple aldehyde crosslinkers has shown promise in enhancing the mechanical and optical properties of collagen membranes for corneal repair, thereby demonstrating their potential for corneal regeneration.43,44 These collagen membranes were implanted in rabbits and successfully promoted corneal reepithelialization. Over a period of two months, the membranes facilitated the reorganization and healing of both the epithelium and stroma. However, due to the dynamic nature of the crosslinkers, the packing between CDs was poor, making it more difficult to form crystalline domains for structural support, which could destabilize the threading of CDs. In terms of microgels, polyrotaxanes are being actively considered for granular hydrogels due to their unique adaptive sliding crosslinks45 and their thermoresponsiveness.46
In this paper, we introduce a novel approach to address the challenges of corneal tissue regeneration via the use of polyrotaxane granular hydrogels (GPR) microgels. These microgels are produced using a microfluidic chip, from readily available biocompatible and non-toxic materials such as PEG and αCD. The polyrotaxanes are capped by a thiol–ene click reaction, preventing CD dethreading, and then annealed. This annealing process leads to enhanced mechanical properties and improved microgel interactions. Importantly, the microgels exhibit the ability to interact with each other through physical crosslinking mechanisms, which are triggered by the inherent thermoresponsiveness of the microgel constituents. Immortalized corneal keratocytes were grown on the hydrogels to recreate transparent and tissue-like corneal stroma.
Materials and methods
Materials
PEG dithiol (Mw: 20 kg mol−1), PEG 4 arm norbornene (Mw: 10 kg mol−1), and PEG 8 arm norbornene (Mw: 20 kg mol−1) were purchased from Creative PEGworks. Alpha-cyclodextrin (αCD, Cyclodextrin Shop), and peptide sequences of CGGGRGDS and CGGGRGDSK-FITC (Chinapeptides) were used as received. Lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP, Sigma, purity ≥95%), DMSO (Sigma), diethyl ether (Sigma), deuterium chloride (DCl, Sigma, Purity >99%), deuterated water (D2O) with 1% (w/w) 3-(trimethylsilyl)-1-propanesulfonic acid, sodium salt (DSS) standard (Sigma) were used as purchased reagents. Additionally, PEG 20 kg mol−1 (Sigma) was utilized in this study. Thincert cell culture inserts designed for 6-well plates (Greiner Bio-One) served as the casting platform for bulk PR gels.Preparation of polyrotaxane hydrogel precursor solution
An appropriate amount of αCD (15% w/v, 150 mg, 154 μmol) was dissolved in 900 μL MilliQ water at 60 °C and allowed to dissolve for 1 hour. The solution was then cooled back to room temperature (RT). PEGdithiol (MW: 20 kg mol−1, 5 wt%, 50 mg, 2.5 μmol) was added to the αCD solution. The mixture was thoroughly mixed overnight at RT to ensure supramolecular complex formation. A stock solution of the water-soluble photoinititator, LAP, was prepared at a concentration of 20 mM by dissolving 2.9 mg of LAP in 0.5 mL of water. To the αCD-PEGdithiol mixture, 100 μL of the 20 mM LAP stock solution was added, resulting in a final LAP concentration of 2 mM in the polypseudorotaxane solution. Lastly 12.5 mg (1.25 μmol) of 4 arm PEG norbornene (Mw: 10 kg mol−1) powder was mixed into the solution to act as a capping agent to cap the PEG ends and stabilize the hydrogel structure. The resulting polyrotaxane precursor solution was used for subsequent experiments.For bulk gel preparation, 30 μL of the prepared polyrotaxane mixture was placed in Thincert (Greiner Bio-One) culture inserts. UV crosslinking was carried out using a UV crosslinking oven (CL-1000 UVP Ultraviolet Crosslinker) equipped with a 365 nm wavelength bulb, maintaining a 10 cm distance between the sample and the light source. The gels were exposed to UV light at an intensity of 10 mW cm−2 for 120 s to facilitate crosslinking, ensuring uniformity in gel formation.
Addition of CGGGRGDS and CGGGRGDSK-FITC required new formulations for robust hydrogelation. The formulations listed in Table 1 were tested in bulk with rheology to determine optimal network formation. The concentration of thiol to norbornene functional groups was kept at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
:1.
| Formulation # | CGGGRGDS (μM) | Crosslinker |
|---|---|---|
| 1 | 1500 | 4 arm norbornene (10 kg mol−1) |
| 2 | 750 | 4 arm norbornene (10 kg mol−1) |
| 3 | 1500 | 8 arm norbornene (20 kg mol−1) |
For example, formulation 3 was synthesized according to the following protocol. Appropriate amount of αCD (i.e. 15% w/v, 225 mg, 231 μmol) was dissolved in 1.35 mL MilliQ water at 60 °C and allowed to dissolve for 1 hour. The solution was then cooled back to RT and then PEGdithiol (MW: 20 kg mol−1, 5 wt%, 75 mg, 3.75 μmol) was added to the αCD solution. The mixture was thoroughly mixed overnight at RT to ensure supramolecular formation. A stock solution of the water-soluble photoinitiator (LAP) was prepared at a concentration of 20 mM by dissolving 2.9 mg of LAP in 0.5 mL of water. To the αCD-PEGdithiol mixture, 150 μL of the 20 mM LAP stock solution was added, resulting in a final LAP concentration of 2 mM in the polypseudorotaxane solution. Lastly, 24.6 mg of 8 arm PEG-Norbornene (MW: 20 kg mol−1, 1.23 μmol) and 1.65 mg of CGGGRGDS (2.33 μmol) was added to the solution to cap the chain ends and add RGD functionality.
NMR
1H NMR spectra were acquired on a 700 MHz Bruker spectrometer at 299.7 K with water suppression sequence applied. NMR samples for evidence of inclusion were prepared by mixing αCD and PEG 20 kg mol−1 (non-thiolated) overnight in appropriate ratios, 1 mole of αCD to 1 mole of PEG 20 kg mol−1 (1:1) and 2 moles of αCD to 1 mole of PEG 20 kg mol−1 (2:1) in 0.6 mL of D2O.
For hydrolyzed bulk polyrotaxane hydrogel samples, we first swelled the gels in water to remove residual LAP, CD, and PEG chains. We washed the gels once in DMSO and then again in water. The gels were then freeze-dried. The dried disk shaped polymers were then digested in 10% v/v DCl in D2O. The 10% and 15% w/v αCD had to be heated to about 70 °C for only a few minutes to see complete dissolution. The dissolved samples were loaded into NMR tubes and tested with a DSS standard.
Bulk hydrogel rheology
The rheological measurements were conducted using a DHR-2 rheometer from TA Instruments, which was equipped with a Peltier heating element and solvent trap. Bulk hydrogel characterization was carried out employing an 8 mm plate-plate geometry, for bulk gels (0 to 15% w/v αCD) in Fig. 1. To ensure consistency, all measurements were conducted at 25 °C, and the solvent trap contained sufficient water to prevent dry-out during the experiments. | ||
| Fig. 1 Characterization of bulk polyrotaxane hydrogels. (a) Schematic representation of the photocrosslinking process during polyrotaxane synthesis employing thiol–ene click chemistry. (b) Rheological assessment revealing the stiffness variations of bulk gels as a function of increasing αCD concentration, n = 3. (c) 1H NMR characterization of hydrolyzed polyrotaxane bulk gels depicting increased CD threading onto PEG chains with higher αCD feed. (d) Self-healing of 15% w/v bulk hydrogel after slicing with blade and pressing back together. (e) Investigation of thermoresponsive behavior following a heat treatment (50 °C for 15 minutes, under constant humidity to prevent dryout). (f) Bulk polyrotaxane gels with αCD concentrations exceeding 10% exhibited pronounced thermoresponsiveness, resulting in significant stiffening. Mean and SD with n = 3 shown in b and d with statistical significance tested by two way ANOVA followed by Sidak's multiple comparison test. *, **, **** indicate p < 0.05, p < 0.01, p < 0.0001. | ||
Frequency sweeps were executed to determine the stiffness at 1 rad s−1, with strain set to 1% and the frequency sweep range from 100 rad s−1 to 0.01 rad s−1. The storage modulus (G′) and loss modulus (G′′) were recorded at 1 rad s−1 and subsequently compared across different samples.
Following the oscillatory frequency sweep, two sample-conditioning steps were implemented. After the first frequency sweep, a 15-minute treatment at 50° C was applied to assess thermoresponsivity. Subsequently, the sample was allowed to equilibrate for 15 minutes at 25° C before conducting the second frequency sweep. The same 1% strain oscillatory frequency sweep was performed on the heat-treated sample, and the G′ at 1 rad s−1 was compared to that of the pre-treated sample.
To investigate UV crosslinking of bulk hydrogels in with addition of RGD-thiol (formulations 1–3), we attached a UV attachment (TA UV accessory), 365 nm light source (Thor Labs) and UV window to the rheometer. A 20 mm cone-plate geometry (a truncation of 2.002 with gap = 53 μm) was placed on the polypseudorotaxane solutions at different compositions as described. An oscillatory time sweep was performed with 1% strain, 10 rad s−1 frequency for 240 s. The sample was exposed to UV light for 60 s (10 mW cm−2) and the rate of network formation for the different formulations were compared.
Microgel preparation
:curing agent = 10:1, RTV-615, Permacol B.V.) was poured onto the mold and cured at 80 °C for 45 min. Then, the PDMS layer was peeled off, and punched with a 25-gauge punch (Syneo Co.) to create inlet and outlets. The PDMS pieces were plasma-treated (Femto PCCE, Diener) in oxygen at 0.30 mBar for 30 s and thermally bonded to plasma treated glass slides at 80 °C for 12 hours. The replication of SU-8 structures onto PDMS pieces was inspected using a confocal laser scanning microscope-based profilometer (VK-X250, KEYENCE) integrated with MultiFileAnalyzer image analysis software (KEYENCE).
To produce water-in-oil droplets, syringe pumps (Chemyz, N3000) were used to inject the oil and pre-gel solutions through the Tygon ® tubes into the flow-focusing chip's inlets. The syringe pump that controlled the oil phase was set to 60 μL min−1 while the pre-gel phase syringe pump varied between 5–8 μL min−1. The outlet of the chips was connected to another Tygon® tube to collect the droplets in a petri-dish to be crosslinked using a UV lamp set to 365 nm for at least 60 seconds. Microgels were washed twice in diethyl ether to remove the oil, and then with ethanol and water, and finally dried under vacuum overnight at RT.
Chemical composition and microstructure of the microgels
The chemical composition of the microgels were analyzed using attenuated total reflection Fourier-transform infrared spectroscopy (ATR-FTIR, Nicolet iS50, ThermoFisher Scientific) with the wavenumber range of 400–4000 cm−1 and a step size of 0.482 cm−1. Size distribution of the microgels was calculated by Motic Camera software. We measured 3 diameters across a microgel and averaged them to obtain the average diameter of one microgel. The (sub)microstructure of the microgels were inspected using scanning electron microscopy (SEM, JSM-IT200, Jeol) at an accelerating voltage of 10 keV and magnifications of 500–10000 X.
Thermoresponsiveness of the microgels
Microgels (50 mg, 15% w/v αCD) were placed into 1 mL of MilliQ water and allowed to equilibrate/swell at room temperature overnight. Images were taken by Motic Camera prior to temperature changes. Average diameters were calculated by taking 3 diameters across the microgel and averaging them. We incubated the 1 mL of MilliQ water with microgels at 37 °C for 2 hours and took images of the microgels to calculate the average microgel diameter. We then incubated the same sample (after heat treatment) at 4 °C to see if the microgels were thermoreversible at lower temperature for 2 hours. We saw no increase in size and left the sample at 24 hours and then took the average diameter measurements. We plotted N = 33 microgels to get the size distribution.X-ray diffraction
X-ray diffraction (XRD) was used to determine the crystallinity changes with the addition of CD. The microgel samples were dried under vacuum (40 °C for two hours) and pressed onto a sample holder to form a thin film. The XRD patterns of gels with different amounts of CD were obtained using an X-ray diffractometer(D2 PHASER, Bruker) using a Cu Kα radiation with a wavelengths of 1.5406 Å at RT with a diffraction angle (2θ) range of 6–30°, a scan rate of 1° min−1 and a step size of 0.03°.Injectability of microgels
After microgel formation and purification, the microgels were loaded into a syringe and passed through various nozzles by hand. The best performance, in terms of retaining the geometry of microgels and limiting microgel destruction by shear stress was found with conically shaped nozzeles with inner diameters ≥1.2 mm.Microgel rheology
Flow curves to understand the injectability profile for GPR were obtained from the aforementioned rheometer (using a 20 mm cone-plate geometry with a truncation of 2.002 and gap = 53 μm). The curves were obtained by performing a logarithmic sweep at 25 °C and increasing shear rate from 0.1–500 s−1 on purified microgels.To understand the mechanical properties of GPR, we placed purified microgels on the rheometer with 8 mm parallel plate geometry. Typically, the plate was pressed down onto the sample to ensure 0.1 N of normal force (gap between 500–800 μm). The rheometer was equipped with a solvent trap and filled with water to prevent dry-out. The microgels were annealed at 50 °C for 15 minutes and allowed to equilibrate to RT for 15 additional minutes. Because the polyrotaxanes with >10% w/v αCD exhibited shrinkage, the gap was decreased to a point where the normal force was 0.1 N. Then, oscillatory frequency sweeps and amplitude sweeps were performed on the annealed sampled.
Cell culture
Flasks used for cell culture were initially coated with FNC coating mix® (AssayCell Technologies) to facilitate the initial cell adhesion post-thawing. Subsequently, immortalized human corneal keratocytes (at passage 3) from Applied Biological Materials were cultured in keratocyte expansion medium which consisted of DMEM F12 GlutaMax™ (ThermoFisher), 5% v/v FBS (Fetal Bovine Serum), 1 mM ascorbic acid supplemented with 100 IU mL−1 penicillin (Gibco) and 1 mg mL−1 streptomycin (Gibco) at 37 °C until the cells reached confluence. Upon reaching 80–90% confluence, cells were detached from the culture plates by incubating the cells with TrypLE™ (Gibco) for 1–2 minutes at 37 °C. Cells were seeded at a density of 6000 cells per cm2 for LIVE/DEAD in a 96-well plate. Typically, after 3 days in expansion medium, the cells were subjected to differentiation medium to promote keratocyte phenotype. The differentiation medium formulation comprised DMEM, 0.01 mg mL−1 human insulin, 55 μg mL−1 human transferrin, 50 ng mL−1 sodium selenite, 1 mM ascorbic acid, 2 mM GlutaMax (Gibco), 2 mg mL−1D-glucose, and 2.5 mg mL−1D-mannitol. Additionally, 1% 100 IU mL−1 penicillin (Gibco) and 1 mg mL−1 streptomycin (Gibco) was included in the differentiation medium.Cytotoxicity
To assess cell viability, a live/dead assay was conducted on samples exposed to microgel supernatant (n = 3). The microgel supernatant was created by combining 50% w/v (300 mg of 15% αCD microgels) in 600 μL of differentiation medium. Microgels were permitted to swell in the differentiation media for a 24-hour exposure period. Subsequently, the supernatant was collected through centrifugation to remove the microgels. Following culture in expansion medium for 3 days, cells were washed with phosphate buffered saline (PBS) to remove the medium, and 120 μL of microgel supernatant was administered to each well. Cells were cultured in this medium for 24 hours before the LIVE/DEAD staining procedure. The control wells contained of 120 μL of differentiation medium without microgel supernatant.The medium was removed from the cells and the cells were washed twice with PBS. The staining solution, comprising 4 μM Calcein-AM and 6 μM ethidium homodimer in PBS, was applied in each well (500 μL) and the culture plate was allowed to incubate for 30 minutes. Post-incubation, imaging was performed in five random areas using the Nikon Ti–S/L100 microscope, and the live and dead cells were quantified.
Microtissue formation
Polyrotaxane (15% αCD) microgels (300 mg) were placed in a mold which was machined to have a corneal-like shape (n = 4) and sealed to prevent water loss. The microgels were heat treated at 50 °C for 15 minutes and one microgel monolith was placed into each well. The monoliths were exposed to UV for 15 minutes before cell culture. Immortalized human corneal keratocytes were seeded on top of the microgel monoliths at 6000 cells per cm2. The cells were allowed to infiltrate the materials and grow in expansion medium for three days before switching to differentiation medium. The differentiation medium was refreshed every 2–3 days for a total of 14 days.At day 14, the cells were washed once in PBS and fixed with 4% PFA for 15 minutes at RT. Samples were then permeabilized with 0.1% Triton X-100 for 15 minutes at RT. Phalloidin conjugated with Alexa568 (0.5 μM) and DAPI (0.25 μg ml−1) were incubated with the cells for 1 hour at RT. After three 5-minute washes, samples were imaged. Imaging was performed on an inverted Nikon Ti–S/L100 microscope, equipped with a Nikon DS-Ri2 camera, a Lumencor Sola SE II for fluorescence, and a CoolLED pE100 system for diascopic white light. For fluorescent imaging, the DS-Ri2 was set to monochromatic mode and three images at 4× and three images at 10× were taken per well.
Transparency measurements
Optical transparency was assessed using a Clariostar microplate reader. The cell-laden constructs, after fixing the cells, were placed into the microplate reader and the absorbance was measured between 220 and 1000 nm. The absorbance was then transformed to % transmittance by eqn (1).| Transmittance (%) = 1/(10Abs) × 100 | (1) |
Degradation of GPR microgels
To assess the degradability of GPR hydrogels, we conducted basic-hydrolysis tests. We first let purified GPR microgels (200 mg) swell in PBS overnight. The swollen microgels were then placed in a cell strainer (VWR, 100 μm pore size), weighed, and sequentially exposed to alkaline water at increasing pH levels from 8 to 13, each for 15 minutes. After each exposure, we removed excess water and weighed the microgels again. By comparing these weights to the initial weight at pH 7, we determined the extent of degradation at each pH level.Statistical analysis
All statistical analyses were performed and graphed with GraphPad Prism software. Experiments with n ≥ 3 replicates were subjected to a one or two-way ANOVA analysis or an unpaired t-test as described. Sidak's or Tukey's multiple comparisons tests were used as described following the ANOVA analysis with a significance threshold of 0.05 or 95% confidence interval.Results and discussion
Bulk polyrotaxane synthesis and characterization
Polyrotaxanes, hold great promise for various biomedical applications due to their unique structural characteristics. These hydrogels are composed of PEG chains threaded through αCD rings in a manner similar to beads on a string, held together by van der Waals forces, host–guest interactions, and hydrogen forces.32 We first started with a comprehensive characterization of bulk polyrotaxane hydrogels which served as the basis of the microgels design and production. Fig. 1 serves as a visual overview of the characterization efforts.The bulk polyrotaxanes were synthesized by first threading αCD onto PEGdithiol chains (MW: 20 kg mol−1). We achieved the formation of polypesudorotaxanes (Fig. S1a†) at varying αCD concentrations (0, 5, 10, 15% w/v) through overnight mixing. With 0% and 5%, we did not observe any physical changes in the solution upon mixing. However, at concentrations of 10 and 15% an immediate sol–gel transition, was observed, rendering the material gelatinous, opaque and non-flowing (Fig. S1b,† 10% w/v αCD), which is emblematic of supramolecular inclusion complex formation between αCD and PEGdithiol.35
1H NMR spectroscopy is a valuable tool for the investigation of host–guest complexation, particularly when dealing with CDs. In the context of our study, we focused on the examination of H3 and H5 protons, located within the macrocycle of αCD (Fig. S2†). Our findings, as summarized in Table S1,† reveal a significant decrease in chemical shifts for these protons when αCD is combined with PEG 20 kg mol−1, suggesting the formation of an inclusion complex where the PEG polymer is encapsulated inside the CD cavity. Such an upfield shift typically signifies increased shielding from the magnetic field, suggesting a change in the local environment of these protons due to interactions like hydrogen bonding with the guest PEG molecule.47 This is further corroborated by a similar shift in the H6 proton, located on the CD cavity rim, which supports the occurrence of complexation.
Furthermore, a comparison between 1:1 and 2:1 αCD:PEG molar ratios showed an even greater upfield shift for the H3, H5, and H6 protons with the increased αCD concentration. This suggests that adding more αCD enhances the shielding effect, likely due to a higher probability of interaction or complexation between αCD and PEG. This behavior is indicative of a more pronounced change in the molecular environment surrounding these protons, possibly, because the additional αCD molecules contribute to a more substantial encapsulation of the PEG chain.
To ensure structural integrity and prevent ring dethreading, we introduced a 4 arm PEG norbornene crosslinker into the hydrogel formulation (Fig. 1a). This robust crosslinking strategy affords the use of thiol-norbornene photocoupling, a widely recognized reaction in drug delivery and tissue engineering applications16,48–50 to freeze the polypseudorotaxane structure. The norbornene-thiol chemistry is well known and produces stable covalently crosslinked gels when the thiol to norbornene are at a 1:1 ratio.48,51 Indeed, we found that adding an eqimolar amount of norbonene containing crosslinker to the thiol polyrotaxane solution enabled stable hydrogel formation. Capping was performed inside a UV oven (365 nm) with 2 mM LAP in solution. The photocrosslinking process was successfully employed to synthesize the polyrotaxane bulk hydrogels, establishing a network structure.
Stark differences were observed among hydrogels with varying αCD concentrations (0, 5, 10, 15% w/v). Visual analysis following a 2-day immersion in PBS, aimed at removing any unbound CD, revealed opaqueness and loss of transparency with increasing αCD (Fig. S3†). This loss of transparency can be attributed to the highly crystalline domains formed between threaded αCDs.52 Swelling tests further validated the presence of secondary crosslinks, with a distinct threshold observed between 5% and 10% αCD concentration. At this threshold, secondary crosslinks offered sufficient strength to reduce hydrogel swelling in PBS (Fig. S4†), allowing the swelling to decrease from 600% in the absence of CD to 200% with 15% w/v αCD.
Rheological analysis (Fig. 1b) of the bulk polyrotaxane hydrogels unveiled a direct correlation between stiffness and αCD concentration. For example, the PEG hydrogel (0% CD) had a mean storage modulus (G′) of 84 Pa when measured by oscillatory frequency sweeps at 1 rad s−1 and increased to 280 Pa with 15 w/v % αCD: over a 3× increase. The choice of αCD concentration in the feedstock enabled control over the stiffness of the hydrogel. Polyrotaxane formation has been shown to enhance rheological properties53 attributed to microcrystalline domains54 formed by αCD, and thus acts as supramolecular crosslinkers. Here, we observed an increase in storage moduli with increasing αCD concentration, which suggests a decrease in the mesh size between thiol-norbornene crosslinks and the presence of dynamic secondary crosslinks.
To determine the amount of bound αCD on a PEG chain in the different formulations, 1H NMR analysis of degradation products (Fig. 1c) was performed. First, we subjected fabricated bulk hydrogels to a two-day wash in PBS to remove unbound CDs, PEG and LAP followed by immersion in acidic conditions (10% v/v DCl in D2O) to facilitate ester bond hydrolysis and subsequent disintegration of the gel into its constituent parts. Then, 1H NMR analysis of the degraded hydrogel was used to determine (Fig. S5†) the ratio of CDs threaded onto the PEG post-capping. The NMR analysis indicated that, for a feed concentration of 15% w/v αCD, approximately 50 αCD rings were threaded onto each PEG dithiol (20 kg mol−1) chain (as shown in Table S2†). This result aligns with the trend that an increase in αCD concentration in the feedstock leads to a higher number of αCD rings threaded on the PEG chains, demonstrating that the degree of αCD threading can be controlled by adjusting the feed concentration.
The self-healing property of hydrogels is a critical feature that enhances their durability and functionality, particularly in biomedical applications where longevity and injectability of the materials are essential. Given that our hydrogels exhibited secondary crosslinking through the CDs, we went on to explore whether these dynamic crosslinks conferred self-healing abilities to the materials. To this end, a self-healing experiment on the 15% w/v bulk hydrogel was conducted. After incising the hydrogel with a blade into several pieces, we manually compressed the fragments together and observed that the bulk hydrogel was capable of self-repair (Fig. 1d). Although the cut lines remained faintly discernible, the strength of the adhesion mediated by the polyrotaxane network was sufficient to maintain the integrity of the structure.
Previous studies have demonstrated thermoresponsivity in 3D printed monoliths of polyrotaxanes.35,55 Here, the examination of thermoresponsive behavior via a heat treatment on a rheometer showed increased responsiveness in hydrogels with αCD concentrations surpassing 10% w/v (Fig. S6†). Upon reaching 30 °C, a marked stiffening effect was observed. To characterize the differences before and after heating, a brief 15-minute heat treatment of 50 °C (Fig. 1e and f) using a Peltier plate on the rheometer with solvent trap to keep the humidity constant was performed. During these measurements, we found that a stiffness response could be accurately measured and repeated (n = 3). Bulk gel stiffness proved to be thremoresponsive with more pronounced changes observed at higher αCD concentrations. For instance, the stiffness of PEG bulk hydrogels with no αCD on average remained around 96 Pa (no change after heat treatment), compared to an increase of 7230 Pa with the 15% w/v αCD hydrogels—an increase of about 26×.
This intriguing thermoresponsive behavior is corroborated with observations from prior studies35 on methylated α-CD polyrotaxanes, elucidating the temperature-induced aggregation of αCD rings within the polyrotaxane structure. Commonly, these αCDs exhibit dynamic shuttling along the PEG polymer chain. The authors suggest as the temperature rises, dehydration phenomena surrounding the αCDs prompt ring aggregation within the polyrotaxane, shifting them from dynamic shuttling to stationary residence. This collective aggregation at the hydrogel level leads to a rapid, temperature-dependent alteration in elastic moduli, offering promising prospects for thermally-triggered actuators. The methylated α-CD polyrotaxanes exhibited thermoreversibility due to the methyl groups acting as spacers to separate the dextran rings at lower temperatures. We did not observe the same reversibility; which is plausibly due to highly crystallinity and highly strengthened interaction between α-CD. Of note, we also observed a marked shrinking of the hydrogels with increasing temperature and expulsion of water when performing rheological testing perhaps due to the enhanced supramolecular crosslinking. This unique behavior adds an intriguing dimension to the versatility of polyrotaxane hydrogels and further highlights their potential for responsive tissue engineering applications.
Microgel polyrotaxane synthesis and characterization
Conventional viscoelastic hydrogels often create tightly knit nanoscale networks that can hinder diffusion of molecules and restrict cell movement, whereas granular hydrogels, with their loosely packed microgel particles, offer a structure with large, interconnected pores conducive to cell migration and fluid transport. Jammed microgels present a contrasting architecture featuring interconnected pores of micron scale that mirror the size of cells, thereby promoting unimpeded cell migration and efficient mass transport. Such microporous structures have already been documented to support cell spreading and migration better than their nanoporous counterparts.56 Because of this distinctive property, coupled with the rapid UV crosslinking and enhancement of mechanical properties by CD inclusion, we went on to explore granular polyrotaxane (GPR) hydrogels.To develop granular hydrogels dynamically crosslinked through polyrotaxanes, while simultaneously incorporating covalent crosslinks via thiol–ene PEG networks, we employed a flow-focusing microfluidic chip. Within this microfluidic setup, water-in-oil microdroplets were created with the hydrogel precursor phase as a droplet core (Fig. 2a). Flow-focusing microfluidics has earned considerable attention due to its high throughput and operational efficiency, especially for production of microgels.57 Upon formation, microdroplets were exposed to UV radiation (365 nm). This photo-initiated thiol–ene click reaction effectively capped the polypseudorotaxanes, preserving both the shape and size of the resulting spherical particles, ultimately yielding GPR microgels (as illustrated in Fig. 2a and b).
 | ||
| Fig. 2 Microgel fabrication and characterization (a) chip setup and photograph of the chip design (b) schematic image of the intraparticle crosslinking of GPR microgels. (c) Scanning electron microscopy (SEM) images comparing PEG microgels (0% αCD) and polyrotaxane microgels (15% αCD) post-purification in ethanol. Scale bar = 20 μm (d) images of microgels in water, revealing distinct size differences attributed to swelling. Scale bar = 800 μm. (e) Statistical analysis demonstrating the changes in the average diameter (n = 3) of microgels with increasing αCD content. One-way ANOVA analysis followed by Tukey multiple comparisons, N = 33. **** indicate p < 0.0001. (f) Evaluation of thermoresponsive behavior concerning the average diameter (n = 3) of polyrotaxane microgels with a 15% αCD concentration, N = 33. No indications of reversibility were observed after shrinkage. Scale bar = 800 μm. | ||
After optimization of the flow rates and composition of the hydrogel precursor phase, we were able to reliably produce a steady flow of stable droplets. Fig. 2c depicts the scanning electron microscopy (SEM) images of PEG microgels (0% w/v αCD) and polyrotaxane microgels (15% w/v) post-purification in ethanol. The smooth and unembellished surface of PEG microgels contrasts starkly with the surface of the polyrotaxane microgels that exhibit a rougher surface morphology. Furthermore, an increase in the prevalence of these rough-edged structures and overall surface roughness was observed with αCD addition (Fig. S7†). These structures likely represent crystalline domains within the polyrotaxane structure. X-ray diffraction (XRD) has been employed to explore the crystallinity of polyrotaxane materials, revealing distinct diffraction peaks that elucidated varying degrees of crystallinity.52 Others have also demonstrated the appearance of highly ordered solid-state superstructures with increased temperature, attributed to the enhanced CD packing at higher temperatures.35 Thus, a discernible structural crystalline component may exist within our polyrotaxane microgels, potentially enhancing surface interactions between microgels.
When submerged, the microgels demonstrated a swelling behavior similar to that of bulk hydrogels. PEG microgels (0% αCD) exhibited swelling a more pronounced swelling than microgels containing αCD, suggesting that the polyrotaxane structure may inhibit swelling (Fig. 2d and e). Specifically, the average diameter of microgels with higher αCD content (10% and 15%) was reduced, from 579 μm in 0% αCD microgels to 460 μm in 10 and 15% w/v αCD microgels, likely due to the αCD's constraining effect on water absorption. This pattern mirrors the reduced swelling observed in bulk polyrotaxane hydrogels with similar αCD concentrations, suggesting that the composition of the granular polyrotaxane (GPR) hydrogels dictates their swelling response in aqueous environments.
We further investigated the response of the microgels to environmental changes, particularly focusing on thermoresponsiveness (see Fig. 2f). When subjected to heat, the polyrotaxane hydrogels displayed a notable stiffening in bulk (Fig. 1f). To investigate whether this structural alteration also occurs in the microgel form, we conducted experiments while ensuring the microgels were submerged in water to prevent desiccation. These experiments revealed a thermoresponsive behavior in 15% αCD microgels immersed in water. Initially, these microgels exhibited an average diameter of 460 μm, which significantly decreased to 235 μm upon heating. However, it is worth noting that this thermoresponsiveness did not exhibit any indications of reversibility after shrinkage. Even when the microgels were stored at 4 °C overnight, the dimensions of the microgels remained relatively constant—the average diameter of the microgels remained around 250 μm. This could be due to the crystalline structure formed by the αCDs through hydrogen bonding and hydrophobic interactions, which, once established by heating, does not readily disassemble at lower temperatures.
Literature suggests that structural modifications, such as methylation of CDs, have been utilized to reverse such crystallization in polyrotaxane systems rendering them thermoreversible.35 Further studies have shown that the crystalline structures can be kinetically trapped into meta-stable polyrotaxane inks and annealing these polyrotaxane structures results in more stable conformers, with increased melting temperatures required to disrupt the hydrogen bonds between CDs.34 In our study, we did not explore the melting temperature of the CD microcrystalline structures. This does not necessarily mean that the structures produced here are inherently non-thermoreversible, but rather that they exhibit stability under the physiological conditions tested.
Injectable properties of GPR cellular-adhesive microgels
In pursuit of promoting cellular adhesion and interaction within the microgel matrix, we incorporated the RGD peptide sequence—a well-established peptide promoting cell adhesion. This addition becomes particularly significant in the context of PEG hydrogels, as they typically require cell adhesive peptides to render them bioactive, even for granular hydrogels.29Fig. 3 serves as an illustrative representation of our efforts to add the RGD peptide to the GPR structure by adjusting the valency of PEG norbornene. In our investigations with RGD-thiol, we observed that 4 arm PEG norbornene resulted in microgels that tended to disassemble and break apart in water (Fig. 3d). To overcome this, hydrogels of varying compositions were subjected to gelation kinetics studies using a photorheometer equipped with a UV light source. Formulations utilizing 4 arm norbornene with concentrations of RGD at 750 μM and 1500 μM yielded hydrogels with very low moduli (<40 Pa) with the latter displaying particularly slow gelation kinetics (see Fig. S8†).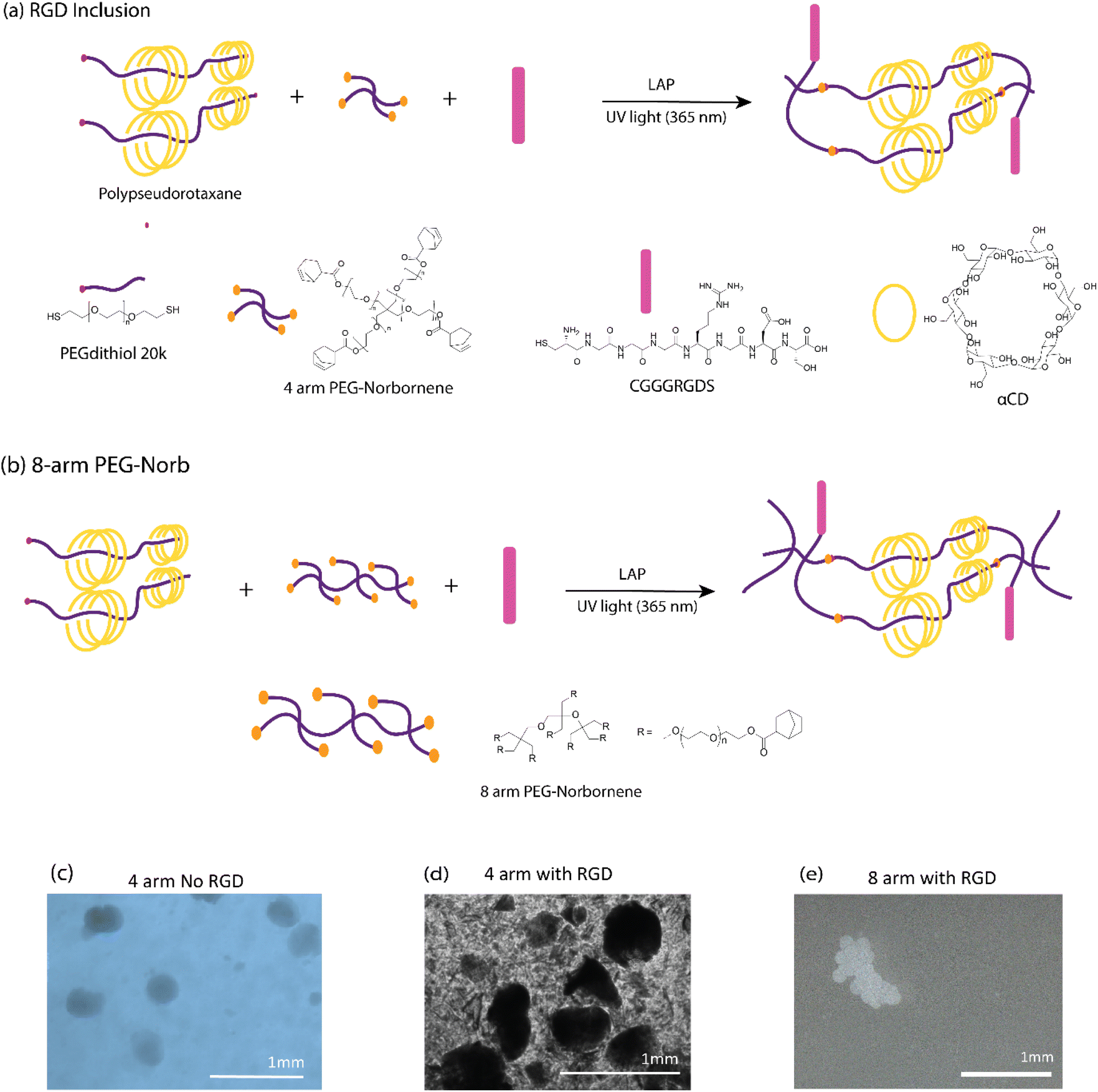 | ||
| Fig. 3 Increasing valency of PEG norborne enhances shape fidelity, resistance to dissolution and gelation where RGD-thiol is added. Proposed schematic representation of photo-initiated click reaction with (a) RGD and optimized reaction with (b) 8-arm PEG-Norb. Microscopic images of the GPR microgels submerged in water according to each of the proposed mechanisms. Initial studies with no RGD (c) produced robust GPR. RGD inclusion (d) into the GPR structure resulted in poorly stable GPR microgels in water. (e) An 8 arm PEG norbornene as crosslinker produced stable GPR with RGD. | ||
To obtain stable microgels in water with improved stiffness, we explored the use of an 8 arm PEG norbornene crosslinker (Fig. 3b). This modification yielded substantial enhancements in both microgel fabrication and resultant hydrogel properties. With the incorporation of thiol-RGD and the 8 arm norbornene crosslinker, we achieved well-defined shapes and stability in an aqueous environment (Fig. 3e and S9† for more pictures). It is plausible that when using the 4 arm PEG norbornene, we formed a network structure lacking sufficient interconnectivity or with dangling polymer ends. Previous work has shown that the deliberate incorporation of dangling polymer ends into thiol norbornene hydrogels incorporated structural defects that led to increased swelling and reduced mechanical properties.58 Given the quality of the microgels formed with 8 arm norbornene crosslinker, we used formulation 3 in the remainder of the studies. To prepare these microgels for subsequent experiments, we refined them through a series of purification steps, including double washing with diethyl ether, a single ethanol rinse, and a final water wash to eliminate any residual mineral oil (Fig. S10†).
To validate the presence of crystalline structures within the newly developed GPR microgels, we conducted X-ray diffraction (XRD) analysis (refer to Fig. S11†). In the case of PEG microgels without αCD, no discernible diffraction patterns were observed, confirming the amorphous nature of the hydrogel. However, microgels with αCD concentrations of 5%, 10%, and 15% exhibited well-pronounced diffraction patterns. In these cases, diffraction peaks and crystalline patterns were observed at 2θ° = 11, 13, 19.5, 20, and 23.5, which is in accordance with earlier studies showing – αCD's contribution to the polyrotaxane crystallinity.35,52 Ito and Ke provide a detailed description of the crystalline features,34 clarifying the unit cell parameters and the molecular packing arrangement. The offset packing they suggest could thus allow for more segmented crystallization of αCD from adjacent polyrotaxanes. These findings offer compelling evidence for the persistent presence of the polyrotaxane crystalline structure within our new 8-arm norbornene and RGD formulations containing αCD.
Granular hydrogels, as delineated by seminal studies13,59 exhibit strain-yielding and self-healing capabilities, which enable individual microgels to slide past one another during shear and re-assemble upon force removal. This dynamic behavior endows granular hydrogels with exceptional injectability, making them highly suitable for minimally invasive in vivo tissue regeneration procedures and as extrudable inks in the domain of bioprinting.60 To explore this functionality further, we conducted shear-thinning tests on microgels with varying concentrations of αCD, ranging from 0 to 15% w/v (Fig. 4a). The objective was to assess their flow characteristics under stress, simulating the conditions experienced during injection or extrusion through a bioprinting nozzle. The flow curves obtained from these tests revealed a consistent shear-thinning behavior across all tested formulations, including both the GPR microgels and PEG microgels (0% αCD). This behavior is characterized by a decrease in viscosity with an increase in shear rate. Fig. 4 presents an assessment of the injectability of our granular hydrogels, which combine the advantages of injectability and microscale porosity.
 | ||
| Fig. 4 Granular hydrogel injectability assessed by (a) shear-thinning behavior of all formulations through rheological characterization, (b) injectability and direct writing of “MERLN” representing the Institute for Technology-Inspired Regenerative Medicine, 1.2 mm tip and syringe of 15% w/v αCD GPR. (c) Bright field image of printed fluorescent 15% w/v αCD GPR with 1% alginate solution. (d) Fluorescent image of printed gels showing microgels shape retained after injection. (c and d) scale bars: 500 μm. | ||
Further assessment of injectability was performed using a conical injector (inner diameter: 1.2 mm), shedding light on the hydrogels’ adaptability for precise deposition and bioprinting capabilities. The inherent stickiness of the hydrated microgel mass and the subsequent pressure buildup from GPR clogging at the injector tip highlighted challenges in maintaining uniform GPR distribution. To mitigate this, a 1% alginate carrier was incorporated as a lubricant, facilitating smoother transit through the injector tip. The alginate effectively enabled the microgels to pass smoothly through tip without obstruction which was confirmed through visual analysis. Without the alginate, the microgels could pass through but tended to clump. Bright-field and fluorescent imaging of the injected fluorescent microgels with the alginate carrier demonstrated that the microgels maintained their structure post-injection. This indicates that the microgels not only exhibit desirable flow properties under shear conditions but also preserve their structural integrity during and after the injection process, affirming their practicality for biomedical applications.
Determination of interparticle interaction
Our study further explored the effect of interparticle crosslinking in granular hydrogels (Fig. 5a and b) on their mechanical strength. Leveraging insights from research on microgel fabrication through extrusion fragmentation, which enhances yield strength and maintains porosity,29 we adopted a novel heat-based annealing method aimed at intensifying microgel interactions, diverging from traditional jamming techniques. This approach was predicated on the assumption that heat annealing would enhance interparticle crosslinking through increased CD interactions (Fig. 5a and b). To begin, rheological analysis was conducted on purified GPR microgels both before (Fig. 5c–e) and after heat treatment (Fig. 5f and g). Amplitude sweeps (Fig. 5c) were employed to evaluate the yield behavior of the GPR hydrogels. We observed an increase in yield stress but a decrease in yield strain for the GPR hydrogels in comparison to the PEG microgels (Fig. 5d). The presence of the polyrotaxane structure leads to a higher crosslinking density, thereby reinforcing the network and necessitating higher stress for flow. Despite the noted increase in strength, the reduction in yield strain suggests that these stronger interactions might be more susceptible to disassembly.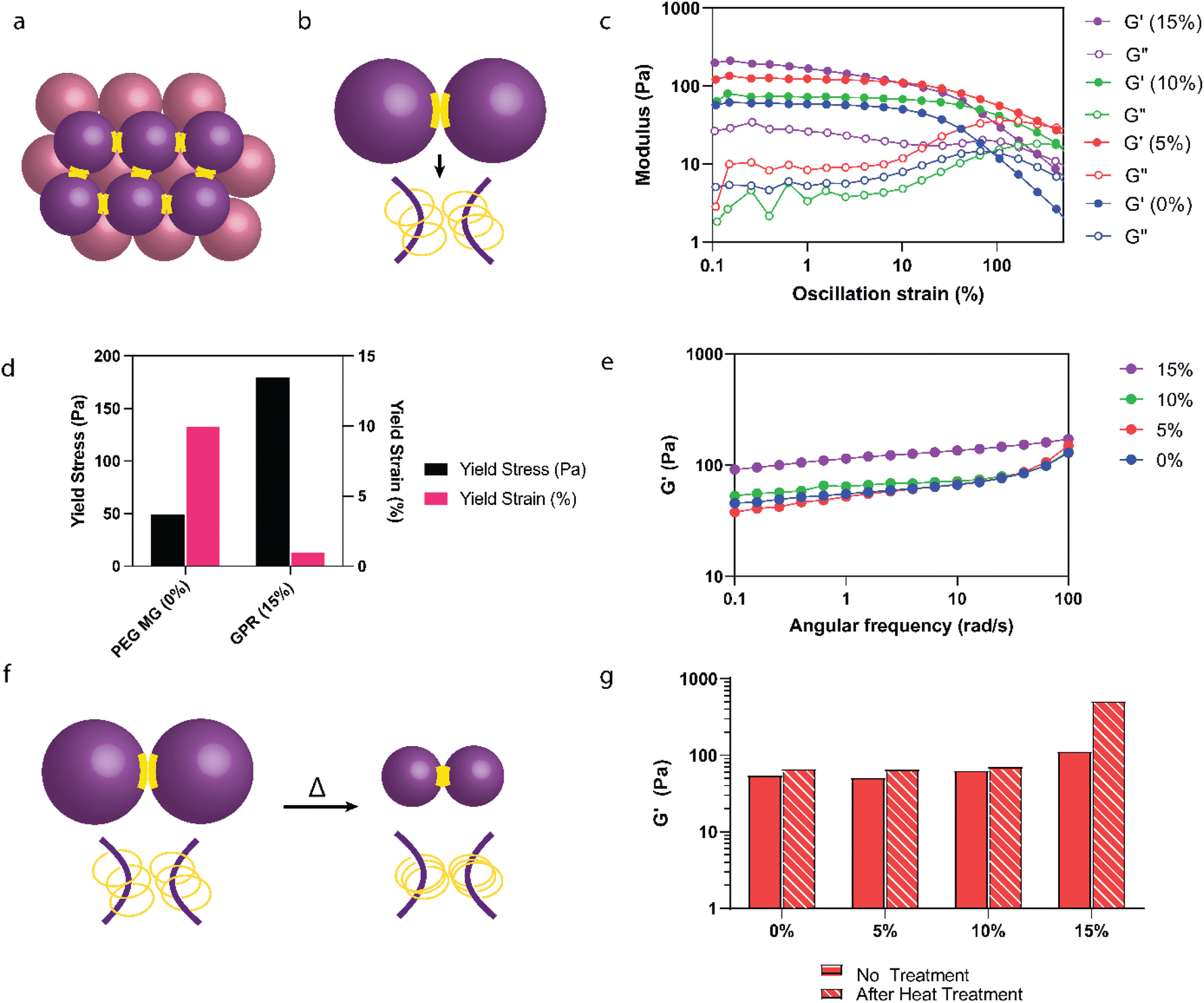 | ||
| Fig. 5 Exploring heat annealing to improve interparticle adhesion. (a) Schematic representation of jammed interparticle interactions. (b) Schematic illustrating the proposed interparticle polyrotaxane interactions among individual microgels. (c) Amplitude sweeps of GPR post purification without heat annealing. (d) Yield stress and yield strain comparisons taken from amplitude sweeps of 0% and 15% w/v αCD GPR indicating an increase in yield stress and decrease in yield strain without heat annealling. (e) Frequency sweeps (G′ shown for clarification) of GPR post purification without heat annealing. (f) Proposed schematic demonstrating the heat annealing (50 °C for 15 minutes) process of polyrotaxanes. Particle size decreases but interactions between CDs increase. (g) Stiffness increased significantly (4.5×) only for the GPR with a 15% αCD concentration. | ||
For 0–10% w/v αCD, the stiffness of the microgels remained stable between 55–64 Pa at 1 rad s−1 (Fig. 5e). The stiffness of the 15% w/v αCD GPR doubled to 115 Pa due to the increased polyrotaxane interactions at these higher αCD concentrations compared to 0–10% w/v αCD GPR. Upon heating (Fig. 5f), only the granular hydrogels containing 15% w/v αCD exhibited superior stiffness (an increase of 4.5× compared to the GPR without heat treatment, Fig. 5g). Enhanced moduli shown here can improve the handling, surgical manipulation, and suturing of the gel during application.61 Additionally, a higher modulus can provide temporary mechanical support to the cornea during the healing process. Extracellular matrix (ECM) stiffness has been shown to influence keratocyte phenotype, with cells displaying characteristics closer to their native state on substrates that mimic physiological corneal stiffness. While these improvements are outside the scope of our study, there remains potential in optimizing mechanical properties for these beneficial effects.
Previously, we observed that microgels containing 15% w/v αCD undergo a reduction in diameter upon heat treatment in water, a change attributed to stronger interactions between the CDs on the polyrotaxane chains. This thermal response leads to a decrease in the GPR microgels’ surface area while potentially expanding the volume of the spaces between particles. This expansion could reduce the contact area between individual microgels. This reduction in contact area necessitates the use of a higher αCD concentration, such as 15% w/v, to promote and maintain the increased interparticle interactions essential for the desired properties of the hydrogel network. This observation highlights the potential of heat annealing as a valuable technique for enhancing the mechanical properties of granular hydrogels, yet also sheds light on the intricate interplay between microgel composition, interparticle crosslinking, and resulting stiffness.
In vitro tissue culture and cellular invasion of GPR
Lab-grown tissues mark a major step forward in regenerative medicine, with successful applications such as creating arteries for human implants showing that it's possible to produce high-quality tissues in large quantities.62 Building on this success, our study investigates the use of injectable GPR hydrogels for developing corneal tissue analogs, aiming to extend the benefits of lab grown tissue to Opthamology. Our approach employs a dynamic, biocompatible hydrogel matrix that supports the organized growth and proliferation of corneal stromal keratocytes. We must, therefore develop tissue that is transparent and contains tissue-like structures that can be integrated into the existing corneal tissue, offering new treatments for corneal disorders and injuries.We initially evaluated the cytotoxicty of the GPR hydrogels. The LIVE/DEAD cellular assay provided compelling evidence of cellular viability, with 91% of corneal keratocytes remaining viable when exposed to the GPR supernatant (Fig. 6a), compared to 87% viability in the control. However, it is worth noting that morphological alterations were discernible after a 24-hour incubation period (Fig. S12†); the cells appeared rounder and less spread out. Despite these morphological changes, our study does not anticipate toxicity attributable to the microgels themselves as they are developed from materials of proven biocompatibility, including PEG and αCD-both widely used in the biomedical domain. Additionally, the possible presence of unbound PEG and αCD in the supernatant may cause the cells to become less adherent, as these highly hydrophilic compounds have been used for antifouling coatings.63,64
 | ||
| Fig. 6 Assessment of annealed GPR hydrogels for corneal tissue formation. (a) Live/dead assay of immortalized human corneal keratocytes with no treatment and in the presence of 15% w/v microgel supernatant. No significant difference between two groups according to unpaired t-test analysis. (b) Annealed GPR hydrogels molded in the shape of a cornea held on a glove. (c) Bright-field images of cells after 3 days in expansion media (c) forming cell aggregates on TCP and (d) infiltrating GPR. Aggregate formation after 2 weeks in culture on (e and f) TCP (non-adherent) compared to the development of complex tissue-like structures on annealed GPR (g and h). | ||
To evaluate GPR's ability for cellular infiltration and tissue formation, we cultured immortalized human corneal keratocytes on GPR (15% w/v αCD). Prior to cell culture, an annealing process was executed using a corneal-lens shaped mold (15-minute incubation period at 50 °C) to impart the hydrogels with corneal-like shape (Fig. 6b). We seeded keratocytes onto the materials in expansion medium for 3 days followed by culture in serum-free differentiation medium to induce keratocyte differentiation. After 2 weeks of culture, cell morphology was assessed by fluorescence microscopy, Fig. 6.
In our experiments, cells cultured on bulk polyrotaxane hydrogels for two weeks demonstrated minimal adhesion or infiltration into the hydrogel matrix (Fig. S13†). In first trials with the gels, after two weeks of culture we observed no cells. To rule out any problems with the culture method, we decided to use adherent cell culture plates which provided a surface that cells could adhere to. Upon imaging the hydrogels from above, we primarily observed that cells were attached to the perimeter of the hydrogel material. In contrast, cells that were in direct contact with the TCP surfaces displayed a more spread morphology, indicating better attachment on the TCP compared to the hydrogel surface.
GPR hydrogels, on the other hand, displayed a significantly enhanced capacity to support cell adhesion, facilitating the spreading of keratocytes across their surfaces. Already apparent after 3 days, we observed very different cell behaviors (Fig. 6c and d), including aggregate formation on non-adherent TCP while on GPR we observed cell infiltration into the matrix. Furthermore, in these hydrogels keratocytes were observed to migrate into the material, forming intricate, multilayered structures (Fig. 6g and h) after 2 weeks. For the non-adherent TCP control wells, we observed that keratocytes tended to form only cell aggregates and did not form multilayers (Fig. 6e and f). The observed differences in cell attachment and cell infiltration indicates that granular hydrogels, such as the GPR hydrogels examined in our investigation, could potentially offer a more conducive environment for cell adhesion, infiltration, and organization compared to their bulk hydrogel counterparts of similar composition.
These granular hydrogels provide unique advantages over existing corneal scaffolds. Firstly, due to their intrinsic nature as a granular material, cells actively infiltrate the network, as shown in Fig. 6g and h. Additionally, the corneal shape can be annealed into the scaffold using the GPR's thermoresponsiveness and subsequent crystallization between adjacent polyrotaxane CDs. This interaction enhances not only the mechanical properties but also enables handleability of the microgels into corneal shapes (Fig. 6b). Current corneal scaffolds made of collagen, GelMA, and alginate, although capable of being implanted, can face challenges post-implantation, such as corneal blurring and extended periods before the scaffold is repopulated with corneal cells.65 In our GPR, cellular infiltration is observed already after 3 days, and cells continue to grow and form structures after 2 weeks. Developing cultured transparent tissue in vitro could be a method to maintain optical clarity after implantation.
In this vein, we evaluated two critical properties of our scaffolds to facilitate corneal tissue production: the GPR biodegradability (achieved through basic hydrolysis, which has been used as a method to decellularize corneal tissue66) and the transparency of the tissue-like structures they formed. After adjusting the pH level from neutral (pH 7) to highly alkaline (pH 13), the scaffolds disintegrated due to the basic hydrolysis process (Fig. S14†). While in vitro basic hydrolysis does not exactly replicate in vivo conditions, these results provide a preliminary indication that the GPR microgels could degrade gradually in a biological setting. In an actual physiological environment, we anticipate that the GPR scaffold could, due to its material properties and response to alkaline conditions, support tissue development and, over time, degrade without leaving residual foreign materials. Additionally, the tissue-like constructs that were formed after a two-week culture period exhibited transparency that was comparable to that of natural corneal tissue, across the entire spectrum of visible light (Fig. S15†). While differences are noted between the native cornea's thickness and that of the cell layers formed through GPR infiltration, the development of a GPR scaffold that supports tissue growth is a significant stride forward. The GPR developed here opens avenues for future innovations in creating more effective and biocompatible scaffolds for medical applications, but also shows potential for the upscaling and manufacturing of corneal tissue. Further in vitro studies with extended culture observation and proto-tissue characterization are necessary to better understand the long-term effects on cell morphology, cell viability and the biological, chemical, and mechanical properties of the formed proto-tissue. Following these studies, in vivo validation will be required to comprehensively assess the biocompatibility and safety of our granular polyrotaxane hydrogel. These additional studies will ensure that our material is both safe and effective for corneal tissue regeneration.
Conclusion
In this study, we explored the design, fabrication, and characterization of granular hydrogels composed of polyrotaxane capped networks stabilized by thiol-norbornene chemistry. These innovative hydrogels offer a unique combination of structural features, including micron-sized porosity, enhanced mechanical properties, and interconnectivity of pores, which are pivotal for creating biomimetic environments conducive to cellular adhesion and infiltration.In GPR microgel synthesis, the use of an 8 arm PEG norbornene crosslinker, combined with thiol-RGD peptide incorporation, yielded hydrogels with excellent shape fidelity, resistance to dissolution, and rapid gelation kinetics. A flow-focusing microfluidic device was successfully used to fabricate GPR microgels with superior stability and shape retention in water and after injection.
The GPR developed in this study offer a multifaceted platform for tissue engineering. Their microscale porosity, enhanced mechanical properties, and cellular compatibility make them an attractive candidate for generating exogenous tissues. The adjustable nature and enhancement of hydrogel properties, along with their inherent ability to be injected and printed, make these constructs versatile tools in regenerative medicine. Our initial findings on creating corneal tissue-like structures offer a positive indication that our approach could contribute to evolving tissue engineering techniques in various biomedical fields.
Data availability
Data for this article, including NMR, rheology, microscopy images, FTIR, XRD, immunofluorescence and stained cell images, transmittance and SEM images are available at DataVerse. The DOI has been generated and is https://doi.org/10.34894/LG3KAU.Conflicts of interest
Authors declare no conflicts of interest.Acknowledgements
AF and MB would like to thank the InSciTe funding agency under the “EyeSciTe” consortium for funding and the feedback of all the consortium members. All the authors would like to thank the Province of Limburg for the LINK project and the gracious funding. YAS and ZNTB thank Dr Hoon Suk Rhu for the generous help with fabricating the SU-8 molds for the microfluidic chip. YAS and PH acknowledge the financial support of the Talent Program Vidi (project ‘Bone microfactory’; No. 15604) of the Dutch Research Council (Nederlandse Organisatie voor Wetenschappelijk Onderzoek; NWO). A portion of this research was financially supported by the Gravitation Program (project ‘Materials-driven regeneration: Regenerating tissue and organ function with intelligent, life-like materials’; no. 024.003.013) of the Dutch Research Council (NWO).References
- W. Armitage, C. Goodchild, M. D. Griffin, D. J. Gunn, J. Hjortdal, P. Lohan, C. C. Murphy, U. Pleyer, T. Ritter, D. M. Tole and B. Vabres, Transplantation, 2019, 2468–2478 CrossRef CAS PubMed.
- H. Kim, M. N. Park, J. Kim, J. Jang, H. K. Kim and D. W. Cho, J. Tissue Eng., 2019, 10, 2041731418823382 Search PubMed.
- S. Ulag, E. Ilhan, A. Sahin, B. K. Yilmaz, D. M. Kalaskar, N. Ekren, O. Kilic, F. N. Oktar and O. Gunduz, Eur. Polym. J., 2020, 133, 109744 CrossRef CAS.
- S. Ulag, E. Uysal, T. Bedir, M. Sengor, N. Ekren, C. B. Ustundag, S. Midha, D. M. Kalaskar and O. Gunduz, Polym. Adv. Technol., 2021, 32(8), 3287–3296 CrossRef CAS.
- M. Rafat, M. Jabbarvand, N. Sharma, M. Xeroudaki, S. Tabe, R. Omrani, M. Thangavelu, A. Mukwaya, P. Fagerholm, A. Lennikov, F. Askarizadeh and N. Lagali, Nat. Biotechnol., 2022, 41(1), 70–81 CrossRef PubMed.
- C. D. McTiernan, F. C. Simpson, M. Haagdorens, C. Samarawickrama, D. Hunter, O. Buznyk, P. Fagerholm, M. K. Ljunggren, P. Lewis, I. Pintelon, D. Olsen, E. Edin, M. Groleau, B. D. Allan and M. Griffith, Sci. Adv., 2020, 6(25), 2187–2204 CrossRef PubMed.
- A. J. Feliciano, C. van Blitterswijk, L. Moroni and M. B. Baker, Acta Biomater., 2021, 1–14 CrossRef CAS.
- C. Samarawickrama, A. Samanta, A. Liszka, P. Fagerholm, O. Buznyk, M. Griffith and B. Allan, Cornea, 2018, 37(5), 609–616 CrossRef PubMed.
- Y. A. Seo, K. Chen, G. Fernandes-Cunha, S. H. Jung, G. H. Lee, S. K. Hahn, A. R. Djalilian, S. Jabbehdari and D. Myung, Invest. Ophthalmol. Visual Sci., 2020, 61(7), 162 Search PubMed.
- Y. Li, C. Zhu, Y. Dong and D. Liu, Polymer, 2020, 210, 122993 CrossRef CAS.
- C. M. Madl and S. C. Heilshorn, Adv. Funct. Mater., 2018, 28(11), 1706046 CrossRef PubMed.
- S. Uman, A. Dhand and J. A. Burdick, J. Appl. Polym. Sci., 2020, 137(25), 48668 CrossRef CAS.
- A. C. Daly, L. Riley, T. Segura and J. A. Burdick, Nat. Rev. Mater., 2020, 20–43 CAS.
- D. B. Emiroglu, A. Bekcic, D. Dranseikiene, X. Zhang, T. Zambelli, A. J. DeMello and M. W. Tibbitt, Sci. Adv., 2022, 8(50), eadd8570 CrossRef CAS PubMed.
- M. Behra, N. Azzouz, S. Schmidt, D. V. Volodkin, S. Mosca, M. Chanana, P. H. Seeberger and L. Hartmann, Biomacromolecules, 2013, 14(6), 1927–1935 CrossRef CAS.
- S. Xin, O. M. Wyman and D. L. Alge, Adv. Healthcare Mater., 2018, 7(11), e1800160 CrossRef.
- S. Xin, D. Chimene, J. E. Garza, A. K. Gaharwar and D. L. Alge, Biomater. Sci., 2019, 7(3), 1179–1187 RSC.
- Z. Jiang, B. Xia, R. McBride and J. A. Oakey, J. Mater. Chem. B, 2017, 5(1), 173–180 RSC.
- L. P. B. Guerzoni, J. Bohl, A. Jans, J. C. Rose, J. Koehler, A. J. C. Kuehne and L. De Laporte, Biomater. Sci., 2017, 5(8), 1549–1557 RSC.
- F. Shao, L. Yu, Y. Zhang, C. An, H. Zhang, Y. Zhang, Y. Xiong and H. Wang, Front. Bioeng. Biotechnol., 2020, 8, 1181 RSC.
- S. H. Ahn, M. Rath, C. Y. Tsao, W. E. Bentley and S. R. Raghavan, ACS Appl. Mater. Interfaces, 2021, 13(16), 18432–18442 CrossRef CAS PubMed.
- S. Utech, R. Prodanovic, A. S. Mao, R. Ostafe, D. J. Mooney and D. A. Weitz, Adv. Healthcare Mater., 2015, 4(11), 1628–1633 CrossRef CAS.
- C. B. Highley, K. H. Song, A. C. Daly and J. A. Burdick, Adv. Sci., 2019, 6(1), 1801076 CrossRef PubMed.
- E. Jooybar, M. J. Abdekhodaie, M. Karperien, A. Mousavi, M. Alvi and P. J. Dijkstra, Int. J. Biol. Macromol., 2020, 144, 837–846 CrossRef CAS PubMed.
- X. Jia, Y. Yeo, R. J. Clifton, T. Jiao, D. S. Kohane, J. B. Kobler, S. M. Zeitels and R. Langer, Biomacromolecules, 2006, 7(12), 3336–3344 CrossRef CAS PubMed.
- A. Charlet, F. Bono and E. Amstad, Chem. Sci., 2022, 13(11), 3082–3093 RSC.
- K. Krutkramelis, B. Xia and J. Oakey, Lab Chip, 2016, 16(8), 1457–1465 RSC.
- A. E. Widener, M. Bhatta, T. E. Angelini and E. A. Phelps, Biomater. Sci., 2021, 9(7), 2480–2493 RSC.
- V. G. Muir, T. H. Qazi, S. Weintraub, B. O. Torres Maldonado, P. E. Arratia and J. A. Burdick, Small, 2022, 18(36), 2201115 CrossRef CAS PubMed.
- A. M. D'Elia, O. L. Jones, G. Canziani, B. Sarkar, I. Chaiken and C. B. Rodell, ACS Biomater. Sci. Eng., 2024, 10(3), 1577–1588 CrossRef PubMed.
- A. K. Rajendan, Y. Arisaka, N. Yui and S. Iseki, Inflammation Regener., 2020, 1–8 Search PubMed.
- F. Huang and H. W. Gibson, Prog. Polym. Sci., 2005, 982–1018 CrossRef CAS.
- J. Wankar, N. G. Kotla, S. Gera, S. Rasala, A. Pandit and Y. A. Rochev, Adv. Funct. Mater., 2020, 1909049 CrossRef CAS.
- Q. Lin, L. Li, M. Tang, S. Uenuma, J. Samanta, S. Li, X. Jiang, L. Zou, K. Ito and C. Ke, Chem, 2021, 7(9), 2442–2459 CAS.
- Q. Lin, M. Tang and C. Ke, Polym. Chem., 2020, 11(2), 304–308 RSC.
- J. H. Seo, S. Kakinoki, T. Yamaoka and N. Yui, Adv. Healthcare Mater., 2015, 4(2), 215–222 CrossRef CAS PubMed.
- R. Sekiya-Aoyama, Y. Arisaka, M. Hakariya, H. Masuda, T. Iwata, T. Yoda and N. Yui, Biomater. Sci., 2021, 9(3), 675–684 RSC.
- Y. Arisaka and N. Yui, Biomater. Sci., 2021, 9(6), 2271–2278 RSC.
- S. Kakinoki, J. H. Seo, Y. Inoue, K. Ishihara, N. Yui and T. Yamaoka, Acta Biomater., 2015, 13, 42–51 CrossRef CAS PubMed.
- Y. Arisaka and N. Yui, J. Biomed. Mater. Res., Part A, 2019, 107(5), 1080–1085 CrossRef CAS PubMed.
- T. Ooya, T. Ichi, T. Furubayashi, M. Katoh and N. Yui, React. Funct. Polym., 2007, 67, 1408–1417 CrossRef CAS.
- M. Terauchi, G. Ikeda, K. Nishida, A. Tamura, S. Yamaguchi, K. Harada and N. Yui, Macromol. Biosci., 2015, 15(7), 953–964 CrossRef CAS PubMed.
- X. Lei, Y. G. Jia, W. Song, D. Qi, J. Jin, J. Liu and L. Ren, ACS Appl. Bio Mater., 2019, 2(9), 3861–3869 CrossRef CAS PubMed.
- X. Zhao, W. Song, W. Li, S. Liu, L. Wang and L. Ren, RSC Adv., 2017, 7(46), 28865–28875 RSC.
- A. A. Gavrilov and I. I. Potemkin, Soft Matter, 2018, 14(24), 5098–5105 RSC.
- S. Zheng, K. Liu, P. Chen, C. Song, J. Yan and A. Zhang, Macromolecules, 2022, 55(16), 7127–7135 CrossRef CAS.
- O. Bekers, E. V. Uijtendaal, J. H. Beijnen, A. Bult and W. J. M. Underberg, Drug Dev. Ind. Pharm., 1991, 17(11), 1503–1549 CrossRef CAS.
- H. W. Ooi, C. Mota, A. Tessa ten Cate, A. Calore, L. Moroni and M. B. Baker, Biomacromolecules, 2018, 19(8), 3390–3400 CrossRef CAS PubMed.
- M. M. Perera and N. Ayres, Polym. Chem., 2017, 8(44), 6741–6749 RSC.
- C. C. Lin, C. S. Ki and H. Shih, J. Appl. Polym. Sci., 2015, 132(8), 41563 CrossRef PubMed.
- S. Hafeez, A. A. Aldana, H. Duimel, F. A. A. Ruiter, M. C. Decarli, V. Lapointe, C. van Blitterswijk, L. Moroni and M. B. Baker, Adv. Mater., 2023, 35(24), 2207053 CrossRef CAS PubMed.
- A. Harada, J. Li and M. Kamachi, Macromolecules, 1993, 26(21), 5698–5703 CrossRef CAS.
- Q. Lin, X. Hou and C. Ke, Angew. Chem., Int. Ed., 2017, 56(16), 4452–4457 CrossRef CAS PubMed.
- S. Uenuma, R. Maeda, H. Yokoyama and K. Ito, Macromolecules, 2019, 52(10), 3881–3887 CrossRef CAS.
- J. Li, X. Yu, Y. Zhao, H. Zhang, M. H. Li and J. Hu, Polym. Chem., 2020, 11(40), 6492–6498 RSC.
- L. Riley, L. Schirmer and T. Segura, Curr. Opin. Biotechnol., 2019, 1–8 CrossRef CAS PubMed.
- J. Wu, S. Yadavali, D. A. Issadore and D. Lee, Adv. Mater. Technol., 2022, 7(5), 1–6 Search PubMed.
- A. K. Zhang, J. Ling, K. Li, G. D. Fu, T. Nakajima, T. Nonoyama, T. Kurokawa and J. P. Gong, J. Polym. Sci., Part B: Polym. Phys., 2016, 54(13), 1227–1236 CrossRef CAS.
- A. C. Daly, Adv. Healthcare Mater., 2023, 2301388, 1–19 Search PubMed.
- W. Cheng, J. Zhang, J. Liu and Z. Yu, View, 2020, 1(3), 1–8 CrossRef.
- N. Formisano, C. van der Putten, R. Grant, G. Sahin, R. K. Truckenmüller, C. V. C. Bouten, N. A. Kurniawan and S. Giselbrecht, Adv. Healthcare Mater., 2021, 2100972 CrossRef CAS PubMed.
- L. E. Niklason and J. H. Lawson, Science, 2020, 370(6513), eaaw8682 CrossRef CAS PubMed.
- S. Lowe, N. M. O'Brien-Simpson and L. A. Connal, Polym. Chem., 2015, 198–212 RSC.
- S. Xu, P. Wang, Z. Sun, C. Liu, D. Lu, J. Qi and J. Ma, J. Membr. Sci., 2019, 569, 124–136 CrossRef CAS.
- J. F. Jameson, M. O. Pacheco, H. H. Nguyen, E. A. Phelps and W. L. Stoppel, Bioengineering, 2021, 8(11), 1–28 CrossRef PubMed.
- J. S. Choi, J. K. Williams, M. Greven, K. A. Walter, P. W. Laber, G. Khang and S. Soker, Biomaterials, 2010, 31(26), 6738–6745 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4bm00409d |
| This journal is © The Royal Society of Chemistry 2024 |