
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmyloid engineering – how terminal capping modifies morphology and secondary structure of supramolecular peptide aggregates†
Manuela
Grelich-Mucha
 a,
Thomas
Bachelart
b,
Vladimir
Torbeev
b,
Katarzyna
Ożga
c,
Łukasz
Berlicki
c and
Joanna
Olesiak-Bańska
*a
a,
Thomas
Bachelart
b,
Vladimir
Torbeev
b,
Katarzyna
Ożga
c,
Łukasz
Berlicki
c and
Joanna
Olesiak-Bańska
*a
aInstitute of Advanced Materials, Wroclaw University of Science and Technology, Wybrzeze Wyspianskiego 27, 50-370 Wroclaw, Poland. E-mail: joanna.olesiak@pwr.edu.pl
bÉcole Supérieure de Biotechnologie de Strasbourg (ESBS), CNRS UMR 7242 Biotechnology and Cellular Signalling, University of Strasbourg, 67400 Illkirch, France
cDepartment of Bioorganic Chemistry, Faculty of Chemistry, Wroclaw University of Science and Technology, Wybrzeze Wyspianskiego 27, 50-370 Wroclaw, Poland
First published on 23rd January 2024
Abstract
The effects of peptide N- and C-termini on aggregation behavior have been scarcely studied. Herein, we examine (105–115) peptide fragments of transthyretin (TTR) containing various functional groups at both termini and study their impact on the morphology and the secondary structure. We synthesized TTR(105–115) peptides functionalized with α-amino (H-), N-acetyl-α-amino (Ac-) or N,N-dimethyl-α-amino (DiMe-) groups at the N-terminus, and with amide (–NH2) or carboxyl (–OH) functions at the C-terminus. We also investigated quasi-racemic mixtures by mixing the L-enantiomers with the D-enantiomer capped by H- and –NH2 groups. We observed that fibril formation is promoted by the sufficient number of hydrogen bonds at peptides’ termini. Moreover, the final morphology of the aggregates can be controlled by the functional groups at the N-terminus. Remarkably, all quasi-racemic mixtures resulted in the robust formation of fibrils. Overall, this work illustrates how modifications of peptide termini may help to engineer supramolecular aggregates with a predicted morphology.
Introduction
In all living forms peptides and proteins composed of amino acids give rise to multiple functions regulating the chemistry of life.1 When the amino acids are arranged in a specific sequence, they can result in structures enabling biological catalysis,2–4 fluorescence,5–7 self-assembly,8–11 and many other properties. The self-assembly process of peptides or proteins into amyloid fibrils constitutes an intriguing case. From a biological point of view, amyloid deposits are identified as hallmarks of various neurodegenerative disorders including Alzheimer's and Parkinson's diseases which are still incurable.12,13 Besides their pathological role, amyloid fibrils find various applications in materials science and biotechnology including functional nanodevices,14 biosensors,15,16 hydrogels for cell culture,17 scaffolds for wound healing,18 drug delivery,19,20 tissue engineering,21 or energy conversion materials.22 Amyloids have the potential to be used in biomedical applications thanks to their biocompatibility, biodegradability and synthetic feasibility.18,23,24 All of the mentioned functions and applications arise due to the unique structural features of amyloid fibrils. Typically, these elongated structures are composed of several protofilaments and may reach up to microns in length, and 5–20 nm in width.25,26 A characteristic feature of fibrils is the presence of the secondary β-sheet structure, stabilized by a dense network of hydrogen bonds.27,28 However, apart from these common features, amyloids present rich polymorphism, which influences the mechanical, physical and chemical properties of amyloids.29,30The peptides can possess the standard functional groups such as α-amino or N-acetyl-α-amino at the N-terminus, and carboxyl or amide groups at the C-terminus. Although the modifications of peptides’ termini in vivo play an important role in protein regulation and signal processing,31,32 very few articles explore the influence of peptides’ termini modifications on the aggregation process and the morphology of resulting supramolecular structures. Tao and colleagues33 examined the effect of terminal capping on the self-assembly process of amyloid-β (Aβ) (16–22) peptide fragment. They investigated two variants: uncapped one (KLVFFAE) and peptide capped by N-acetyl-α-amino (Ac-) and amide (–NH2) group at the N- and C-termini (Ac-KLVFFAE-NH2), respectively. The authors noticed different morphology for fibrils formed from these two peptide variants at acidic conditions (pH 2). It was found that the uncapped analogue led to long, straight and unbranched nanofibrils, whereas the capped one formed nanotapes. In another study, Andreasen et al.34 investigated the effect of terminal capping of human islet amyloid (20–29) polypeptide fragment (hIAPP20–29). The authors concluded that the final morphology of amyloid fibrils is governed by the number of hydrogen bonds and electrostatic interactions between the peptide termini. Similar effects were shown for the designed model peptides by Hu et al.35 who reported that electrostatic interactions between the charged peptide termini can tune the transformation from twisted ribbons to flat assemblies. In addition, the role of end capping on morphology and properties of human transthyretin (TTR) fragment 105–115 amyloid fibrils was examined by Lee and Na36 using computational methods. They discovered that the capped TTR(105–115) variant (Ac-TTR-NH2) has twisted and ordered fibrillar conformation, whereas for its uncapped analogue twisted fibers possess disordered terminal regions. Their computational studies indicated that the capping conserves the number of hydrogen bonds and electrostatic interactions. Moreover, it stabilizes the tyrosine residue and therefore increases the overall stability through hydrophobic interactions. The reported results on the stability dictated by end capping are in a good agreement with the computational simulations reported for TTR(105–115) oligomers.37
Biological N-α-methyl modifications of peptides may include mono-, di-, or trimethylation.38,39 Generally it is known that N-α-methyl amino acids can influence the aggregation process of amyloidogenic peptides, for example, among six N-methylated peptide analogues of Aβ(25–35), only one possessing N-α-methyl-amino group at the N-terminus formed fibrils similar to the Aβ(25–35).40 To the best of our knowledge, peptides with the N,N-dimethyl-α-amino group at the N-terminus have not been studied to determine the effect of such N-terminal group on the morphology and the secondary structure.
Herein, we investigated TTR(105–115) peptides which in acidic conditions self-assemble into amyloid fibrils or peptide aggregates.41 Full-length TTR transports thyroxin and retinol throughout the body and brain.42 However, TTR can misfold which leads to TTR amyloidosis.43,44 Full-length TTR, some TTR variants and peptide fragments are capable to form fibrils in vitro.45 Recently, we have reported distinct morphological and optical properties (autofluorescence) for the amyloid fibers of TTR(105–115) prepared in enantiomerically pure forms (L or D) and as racemic mixture (D/L).41 In the current report, we extended our studies and synthesized TTR(105–115) peptides capped by α-amino (H-), N-acetyl-α-amino (Ac-), as well as N,N-dimethyl-α-amino (DiMe–) group at the N-terminus and by carboxyl (–OH) or amide (–NH2) group at the C-terminus. We examined the morphology of the amyloid aggregates formed in the individual L-peptides with atomic force microscopy (AFM). We observed that the functional group at the N-terminus can determine the final morphology of the resulting peptide aggregates. We also studied the morphology of quasi-racemic samples prepared by mixing the D-enantiomer containing H- and –NH2 groups at N- and C-termini, respectively, with each of the synthesized L-enantiomers capped with the above-mentioned functional groups. For all the samples we characterized the secondary structure using attenuated total reflectance Fourier-transform infrared (ATR-FTIR) spectroscopy. Finally, the experimental results were supported with molecular dynamics (MD) simulations providing detailed insights into the relationship between the observed morphology and the secondary structure.
Experimental
Peptide synthesis
All investigated TTR(105–115) fragments (YTIAALLSPYS) were synthesized by the standard 9-fluorenylmethoxycarbonyl (Fmoc) strategy using the Liberty Blue automated microwave peptide synthesizer. Oxyma pure (ethyl 2-cyano-2-(hydroxyimino)acetate) and diisopropylcarbodiimide (DIC) were used as coupling agents and 20% (v/v) piperidine in N,N-dimethylformamide (DMF) as Fmoc-deprotection solution. Peptide cleavage and global deprotection were carried out in the cleavage cocktail composed of trifluoroacetic acid (TFA)/triisopropylsilane (TIPS)/H2O (95/2.5/2.5). The peptides capped by amide group at the C-terminus were prepared using Fmoc-Rink Amide AM resin (100–200 mesh, 0.74 mmol g−1, 1% DVB), whereas to obtain peptides functionalized with carboxyl group, a Wang resin preloaded with L-serine was used, namely Fmoc-L-Ser(t-Bu)-Wang resin (100–200 mesh, 0.7 mmol g−1). To acetylate the N-terminus, after the final Fmoc group deprotection, the following procedure was applied: the reaction vessel was filled with N,N-diisopropylethylamine (DIEA) (200 μL), acetic anhydride (200 μL) and DMF (1.4 mL). The procedure was performed 2 times for 6 minutes each. The absence of α-amino group at the N-terminus was verified by performing a ninhydrin test. All crude peptides were purified by semi-preparative reverse-phase high-performance liquid chromatography (RP-HPLC). Purity of the fractions was determined using analytical RP-HPLC and liquid chromatography–mass spectrometry (LC–MS). Dimethylation of N-termini of peptides was performed according to the procedure adapted from refs. 46–48. To do so, the purified peptides terminated by α-amino group were dissolved in sodium acetate (NaOAc) (0.1 M, pH ∼ 4) at concentration 1 mg mL−1. The mass of the peptide was checked by LC-MS. Then, 37% (aq.) solution of formaldehyde (1000 eq.) and sodium cyanoborohydride (NaBH3CN) (30 eq., 0.2 M in water) were added to the dissolved peptide (ESI Fig. S1†). After a brief mixing, mass of the peptide was checked using LC-MS. After confirmation of a successful modification of the N-terminus (+28 Da), purification was performed using semi-preparative RP-HPLC.Peptide purification
Peptides were purified on a Shimadzu semi-preparative HPLC using a C18 column (Kinetex XB-C18, 250 × 21.2 mm2, 5 μm, 100 Å) at 40 °C. Elution methods were: (1) isocratic step for 10 min at 20% B, then linear gradient from 20% B to 60% B in 50 min for N,N-dimethylated analogue; (2) isocratic step for 10 min at 10% B, then linear gradient 20% B to 53% B in 42 min for other peptides (A = water + 0.1% TFA; B = acetonitrile + 0.1% TFA).Analytical RP-HPLC
Analytical RP-HPLC was performed on a Shimadzu HPLC system equipped with a DAD detector and a Reprosil-XR C18 column (50 × 2 mm2, 3 μm, 120 Å) by using 2 min isocratic step at 10% of B and then linear gradient from 10 to 60% of B over 14 min (A = water + 0.1% TFA; B = acetonitrile + 0.1% TFA).LC-MS analysis
Analytical LC-MS analyses were carried out on a Thermo Ultimate 3000 system using an X-select peptide CSH C18 column (50 × 2.1 mm2, 2.5 μm, 130 Å) connected to a Thermo LCQ Fleet mass-spectrometer. Elution method was: isocratic step at 5% B for 1 min, then linear gradient from 5% to 85% B over 10 min (A = water + 0.1% formic acid + 0.01% TFA; B = acetonitrile + 0.1% formic acid + 0.01% TFA).Preparation of the samples
The peptides were first dissolved in dimethyl sulfoxide (DMSO). Then, a solution of acetonitrile/HCl at pH 1.5 (1/9 (v/v)) was added. The final content of DMSO was 2% (v/v) and the concentration of the peptides was 1 mM. The quasi-racemic mixtures were obtained by mixing in (1/1) molar ratio of the solution of D-enantiomer of TTR(105–115) (D) functionalized with α-amino and amide groups at the N- and C-termini, respectively, and each of the synthesized L-enantiomers capped by the various functional groups described in this manuscript. The samples were incubated at 37 °C in an Eppendorf Thermomixer C for 3 weeks (t1).Morphology analysis
The morphology of the samples was analyzed using an AFM Dimension V Veeco in the tapping mode with NANOSENSORS™ SSS-NCHR tip mounted. For the AFM imaging the samples were diluted (or not) to a concentration enabling analysis of the morphology. Information regarding the samples’ concentration is indicated in the description of the AFM images included in this article and the ESI (Fig. 1, 3 and ESI Fig. S5, S6, S7, S10†). 30 μL aliquots were deposited on mica, and after 30 minutes of the adsorption period the samples were dried. Dimensions of the peptide structures were estimated based on 50 individual profiles using Gwyddion – an open source software for scanning probe microscopy data visualization and analysis.49 | ||
| Fig. 1 Morphology of peptide aggregates formed at t1: (a) H-TTR-NH2, (b) H-TTR-OH, (c) Ac-TTR-NH2, (d) Ac-TTR-OH, (e) DiMe-TTR-NH2, and (f) DiMe-TTR-OH. For the AFM imaging the samples were not diluted. | ||
Absorption spectroscopy
The absorption spectra were acquired using a Jasco V-670 spectrophotometer within the range 240–650 nm. For the measurements, the peptides were diluted to 166 μM.ATR-FTIR spectroscopy
ATR-FTIR spectra were obtained using a Vertex 70v spectrometer (Bruker). Residual TFA was removed from the samples by treatment with 10 mM HCl and lyophilization (3 cycles). For the ATR-FTIR spectra measurements, a droplet (2 μL) of each incubated sample was deposited on the diamond surface. After a background measurement, the ATR signals from samples were collected 64 times and averaged. The spectra were recorded in the range of 4000–400 cm−1 with the resolution equal to 4 cm−1.MD simulations
The starting models of parallel two-layered β-sheet were prepared on the basis of solid state NMR structure of TTR(105–115) (PDB id: 2 m5n) and extended by 8 additional strands to obtain 24-mer bilayered structure of enantiomerically pure peptides (L-TTR peptides). For the quasi-racemic mixtures of L-TTR and D-TTR peptides antiparallel two-layered β-sheet model was prepared by applying transformation on every second strand (180° rotation and reflection) followed by minimization using Discovery Studio Smart Minimizer protocol with distance-dependant dielectric as an implicit water model. Next, the appropriate capping groups on N-terminus (H-, Ac- or DiMe-) and C-terminus (–OH or –NH2) were added to the models. The optimized models (ESI Fig. S13 and S14†) were obtained by energy minimization in explicit water (spc216) using the steepest descent algorithm (GROMACS, Amber03 forcefield). To study the behaviour of the proposed models, up to 100 ns long molecular dynamics simulations was conducted using GROMACS software and Amber03 force field that includes parameters of different terminal caps and in explicit water (spc216). The minimization of the system was performed using the steepest descent algorithm with 5000 maximum number of steps and PME electrostatic method and converged in 700 steps. Equilibration of the system consisted of two phases: 100 ps (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps) under the canonical ensemble (NVT) and 100 ps (50000 steps) under the NPT ensemble for temperature and pressure stabilization followed by 50000 ps (2500000 steps) production run. All the dynamics were performed under periodic boundary conditions using the leapfrog scheme and PME electrostatics with a 1 nm cut-off at a constant temperature of 300 K and a pressure equal to 1 bar.
000 steps) under the canonical ensemble (NVT) and 100 ps (50000 steps) under the NPT ensemble for temperature and pressure stabilization followed by 50000 ps (2500000 steps) production run. All the dynamics were performed under periodic boundary conditions using the leapfrog scheme and PME electrostatics with a 1 nm cut-off at a constant temperature of 300 K and a pressure equal to 1 bar.
Results and discussion
We prepared TTR(105–115) peptides capped by different functional groups at their N- and C-termini (Table 1) via solid phase peptide synthesis, as described in Experimental section. The electrospray ionization mass spectra (ESI MS) (ESI Fig. S2†) confirmed the composition, and analytical RP-HPLC traces evidenced high purity (>98%) (ESI Fig. S3†) of the peptides used in the experiments. In the ESI MS spectra, besides the most abundant peaks assigned to hydrogen ion adducts (M + H+), signals corresponding to ion clusters were also present (i.e., 4M + 3H+ and 3M + 2H+ species (ESI Fig. S2†)). To ensure the same final concentration of the peptides for our experiments, we measured the absorption spectra of the samples (ESI Fig. S4†). Afterwards, we prepared quasi-racemic mixtures for each synthesized L-enantiomeric peptide variant by adding the D-form of TTR(105–115) functionalized with H- and –NH2 groups at its N- and C-termini, respectively (denoted with D) (Table 1).| Name | Composition | Modification at N-terminus | Modification at C-terminus | Amyloid fibrils |
|---|---|---|---|---|
| Enantiomerically pure peptides | ||||
| H-TTR-NH2 | H-L-TTR-NH2 | α-amino group | Amide group | + |
| H-TTR-OH | H-L-TTR-OH | α-amino group | Carboxyl group | + (slow) |
| D | H-D-TTR-NH2 | α-amino group | Amide group | + |
| Ac-TTR-NH2 | Ac-L-TTR-NH2 | N-acetyl-α-amino group | Amide group | + |
| Ac-TTR-OH | Ac-L-TTR-OH | N-acetyl-α-amino group | Carboxyl group | + (slow) |
| DiMe-TTR-NH2 | DiMe-L-TTR-NH2 | N,N-dimethyl-α-amino group | Amide group | − |
| DiMe-TTR-OH | DiMe-L-TTR-OH | N,N-dimethyl-α-amino group | Carboxyl group | − |
| Peptide quasi-racemic mixtures | ||||
| D + H-TTR-NH2 | H-D-TTR-NH2 and H-L-TTR-NH2 | α-amino group | Amide group | + |
| D + H-TTR-OH | H-D-TTR-NH2 and H-L-TTR-OH | α-amino group | Amide group and carboxyl group | + |
| D + Ac-TTR-NH2 | H-D-TTR-NH2 and Ac-L-TTR-NH2 | α-amino group and N-acetyl-α-amino group | Amide group | + |
| D + Ac-TTR-OH | H-D-TTR-NH2 and Ac-L-TTR-OH | α-amino group and N-acetyl-α-amino group | Amide group and carboxyl group | + |
| D + DiMe-TTR-NH2 | H-D-TTR-NH2 and DiMe-L-TTR-NH2 | α-amino group and N,N-dimethyl-α-amino group | Amide group | + |
| D + DiMe-TTR-OH | H-D-TTR-NH2 and DiMe-L-TTR-OH | α-amino group and N,N-dimethyl-α-amino group | Amide group and carboxyl group | + |
We investigated the morphology of the samples after 3 weeks of incubation at 37 °C, referred to as t1 time point, using AFM (Fig. 1 and ESI Fig. S5†) and noticed distinct morphologies for peptide aggregates formed by different peptides (Fig. 1). We observed the formation of fibrils with twisted morphology for the incubated TTR peptide fragments capped by α-amino group at the N-terminus (Fig. 1a, b, and ESI Fig. S5†), regardless of the C-terminus. The AFM images demonstrate that these fibrils differ in handedness and this tendency is chirality-dependent. Namely, H-TTR-NH2 and H-TTR-OH fibrils (Fig. 1a and b) are right-handed, whereas D (ESI Fig. S5†) – left-handed. The handedness of H-TTR-NH2 and D is in agreement with our previous observations.41 According to the analysis of the fibrils’ dimensions (Fig. 2, and ESI Table S1†) in AFM images, the fibrils formed from TTR(105–115) functionalized with the α-amino and the amide groups (H-TTR-NH2) had an average width of 22.1 ± 2.7 nm, average height of 11.7 ± 2.4 nm and average cross-over distance (i.e. corresponding to 180° turn of a helix)50,51 of 98.0 ± 6.5 nm. Fibrils capped by the α-amino and the carboxyl groups (H-TTR-OH) were characterized by an average width of 22.2 ± 3.9 nm, an average height of 8.5 ± 2.7 nm, and an average cross-over distance of 104.4 ± 11.7 nm. Fibrils formed from TTR(105–115) composed of D-amino acids (D) and capped by α-amino and amide groups, had the average width, height and cross-over distance equal to 23.3 ± 2.8 nm, 11.9 ± 2.7 nm, and 96.8 ± 6.9 nm, respectively. Based on the dimension analysis we observed that fibrils formed from the peptide fragment capped by the α-amino group adopt a twisted morphology. However, when a carboxyl group is at the C-terminus, the fibrils are slightly lower and have a longer average cross-over distance compared with the fibrils possessing the amide group at their C-terminus (ESI Table S1†). A graphical representation of width, height and cross-over distance ranges for the samples possessing H- at the N-terminus is included in Fig. 2a.
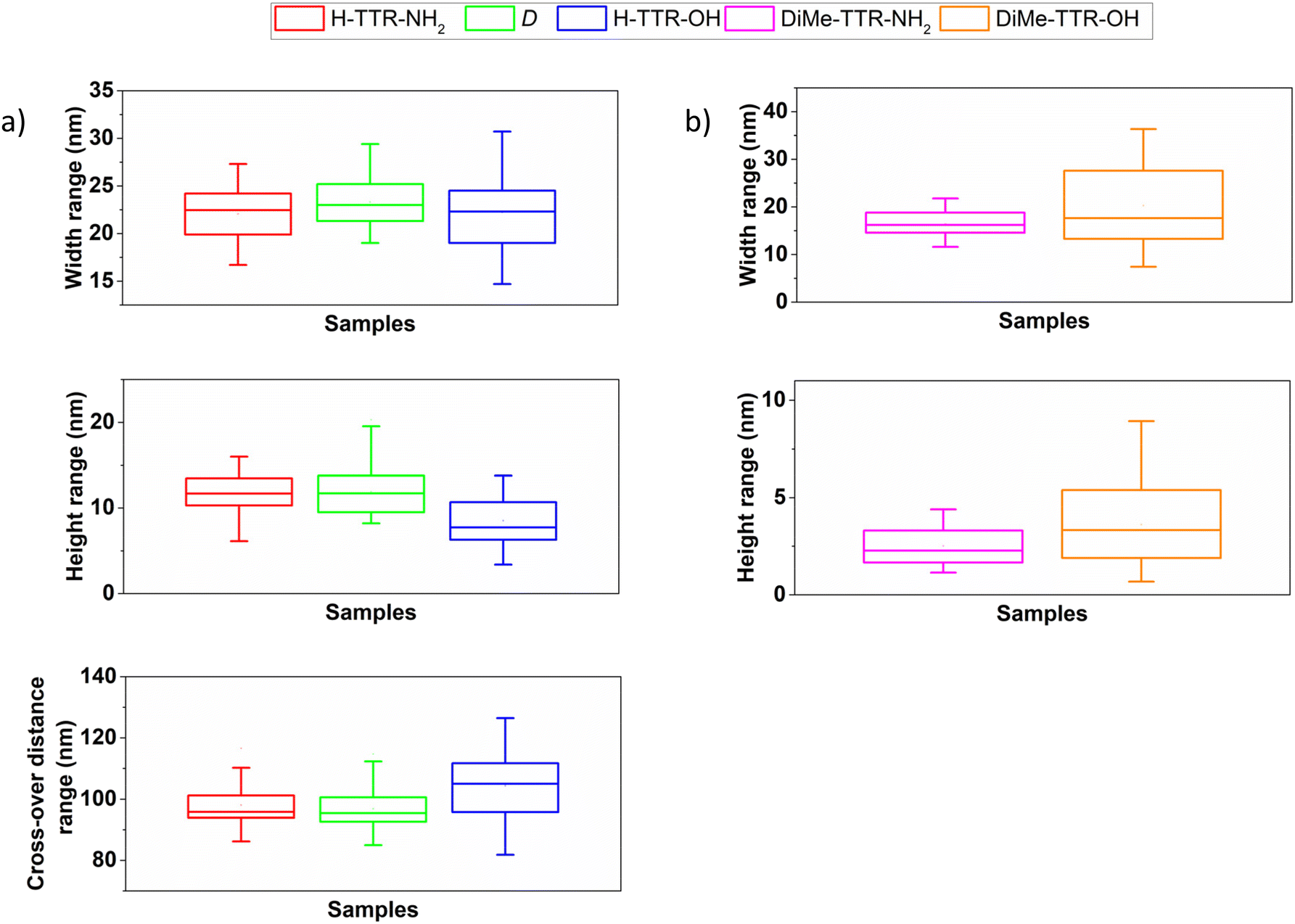 | ||
| Fig. 2 Box charts representing width (nm), height (nm) and cross-over distance ranges (nm) of the peptide supramolecular structures formed upon incubation of the enantiopure peptides capped by (a) H- and (b) DiMe-group at the N-terminus. The dimensions were estimated for fibrils imaged at t1. | ||
The peptides capped by Ac-group (Fig. 1c and d) led to the formation of relatively long amyloid fibrils which tended to stick together instead of being separated. Therefore, it was difficult to find individual amyloid fibrils and calculate their dimensions.
The peptides functionalized by DiMe-group (Fig. 1e and f) did not form any amyloid fibrils. Instead, incubation of these peptides resulted in the formation of globular aggregates. Incubation of DiMe-TTR-NH2 led to the formation of aggregates of an average width of 16.4 ± 2.5 nm and an average height of 2.5 ± 1.0 nm (ESI Table S1†). DiMe-TTR-OH resulted in globular aggregates of an average width and height of 20.3 ± 8.0 nm and 3.6 ± 2.2 nm, respectively (ESI Table S1†). Width and height ranges of those samples are depicted in Fig. 2b.
According to the observations mentioned above we can conclude that the functionalization of N-terminus determines the final morphology of the peptide supramolecular structures:
- the presence of the α-amino group leads to the formation of twisted fibrils (Fig. 1a, b, 2a, ESI Fig. S5, and Table S1†);
- the acetylation of the N-terminus promotes elongated fibrils which tend to associate together (Fig. 1c and d);
- the DiMe-group disrupts the formation of amyloid fibrils with only small peptide aggregates being observed (Fig. 1e, f, 2b, and ESI Table S1†).
We also examined the morphology of the samples directly after their preparation, which we denote as t0 time point. For the individual enantiopure peptides (ESI Fig. S6†) we observed some differences in the fibril formation propensity among the studied samples. Some fibrillar structures were observed at t0 for H-TTR-NH2 (ESI Fig. S6a and b†) and Ac-TTR-NH2 (ESI Fig. S6e and f†). However, in the case of their peptide analogues functionalized by the carboxyl group, no fibrils were detected (ESI Fig. S6c, d, S6g, and h). DiMe-TTR-NH2 as well as DiMe-TTR-OH did not form any fibrils at t0 (ESI Fig. S6i, j and S6k, l, respectively).
Besides the enantiopure individual peptides, we also investigated the morphology of peptide quasi-racemic mixtures at t0 and t1. In this case, fibrillar structures were observed in all samples already at t0 (ESI Fig. S7†), as opposed to the discussed above enantiopure peptides (ESI Fig. S6†). We have already reported an accelerated fibril formation when L- and D-enantiomers of TTR(105–115) capped by α-amino and amide group were mixed in equimolar ratio.41 More rapid fibrillation of racemates than their enantiopure counterparts alone has been reported by other groups as well.52–54 Here, the same response is confirmed regardless of the terminal groups in L-enantiomer. The morphology analysis performed at the later time point, t1, revealed the formation of amyloid fibrils in all quasi-racemic mixtures (Fig. 3). Interestingly, amyloid fibrils were also observed in the case of D + DiMe-TTR-NH2 (Fig. 3e) and D + DiMe-TTR-OH (Fig. 3f), contrary to the suppression of fibrils for the corresponding L-peptides alone, where only aggregates were found at t1 (Fig. 1e and f). These observations demonstrate that the addition of D-enantiomer rescues the formation of amyloid fibrils (Fig. 1e and fvs.Fig. 3e and f). Previously, Dutta et al.52 reported the suppressed oligomer formation in the racemate of Aβ(42) peptide. Their studies evidenced accelerated fibril formation for the racemic mixture, and the kinetic profile was devoid of any lag phase. In contrast, both L- and D-enantiomers of Aβ(42) demonstrated comparable kinetics to each other, starting from a lag phase.52
 | ||
| Fig. 3 Morphology of peptide aggregates formed in quasi-racemic mixtures observed at t1. The samples were prepared by mixing D with (a) H-TTR-NH2, (b) H-TTR-OH, (c) Ac-TTR-NH2, (d) Ac-TTR-OH, (e) DiMe-TTR-NH2, and (f) DiMe-TTR-OH. All of the samples were 10-times diluted except for the mixture of (e) D-form with DiMe-TTR-NH2 which was not diluted. | ||
In our case, for quasi-racemic mixtures the difference in the morphology of fibrils strongly depends on the C-terminal modification. Accordingly, when D-analogue is mixed with peptides capped by the –NH2 group, the resulting quasi-racemic fibrils are wider as compared to their counterpart mixtures composed of peptides functionalized with the –OH group (ESI Table S1†). Fibrils formed upon incubation of D + H-TTR-NH2 had an average width of 103.7 ± 39.8 nm, whereas the fibrils formed from D + H-TTR-OH mixture had an average width of 18.0 ± 3.6 nm. For the mixture of D + Ac-TTR-NH2, the fibrils had a mean width of 104.6 ± 56.3 nm; whereas for D + Ac-TTR-OH mixture it was 21.8 ± 3.4 nm. Fibrils formed upon co-incubation of D and DiMe-TTR-NH2 had a mean width of 63.6 ± 33.5 nm, while for D and DiMe-TTR-OH it was 17.0 ± 3.2 nm. Besides much wider fibrils in the quasi-racemic mixtures of peptides capped by the amide group, these fibrils are characterized by a relatively large standard deviation of their width. The distribution of the width values among these samples is demonstrated in ESI Fig. S8a.† We also included the distribution of the width values of fibrils formed upon mixing the D-enantiomer with peptides functionalized by the –OH group (ESI Fig. S8b†). For the quasi-racemic fibrils we calculated their average height as well (ESI Table S1†). The quasi-racemic fibrils formed from D + H-TTR-NH2 and D + H-TTR-OH had an average height of 5.3 ± 1.3 nm and 6.9 ± 1.7 nm, respectively. In the mixture of D + Ac-TTR-NH2 and D + Ac-TTR-OH were present fibrils of a mean height of 5.1 ± 1.3 nm and 7.4 ± 1.4 nm. Fibrils formed upon co-incubation of D + DiMe-TTR-NH2 and D + DiMe-TTR-OH had an average height of 7.5 ± 2.4 nm and 7.3 ± 1.3 nm.
Our next aim was to correlate the observed morphology with the secondary structure of the studied samples. As predicted by Pauling and Corey, polypeptide chain chirality is a crucial factor in determining the morphology of aggregates. Polypeptide chains composed entirely of L- or D-amino acids arrange into the pleated β-sheet configuration,55–57 whereas the equimolar mixtures of L- and D-polypeptides should assemble into rippled β-sheets wherein the L- and D-peptides are arranged in an alternating L/D pattern.57 Such predictions were confirmed by other groups as well.58–61 Very recent study performed by A. Hazari et al.62 demonstrates that racemic mixtures of amyloid-like peptide assemblies possess rippled sheets and it is their generic property. Additionally, it has been reported that aromatic residues Phe, Tyr, and Trp favor the formation of rippled sheets.62–64 In our case, the investigated TTR(105–115) peptides possess in their sequence 2 Tyr residues and AFM images strongly support the co-assembly of quasi-racemic fibrils. Accordingly, we observed totally different morphologies in enantiopure L- and D-peptide samples (Fig. 1, and ESI Fig. S5†) compared to the corresponding quasi-racemic mixtures (Fig. 3) as these differences are highlighted in ESI Table S2.†
ATR-FTIR spectra recorded for enantiopure peptides at t1 revealed the presence of the secondary β-sheet structure in all of the samples except for the aggregates formed by the peptides with DiMe-group at their N-termini (Fig. 4a). Namely, for H-TTR-NH2 and D-enantiomer the maximum absorption peak was at ∼1630 cm−1, and for Ac-TTR-NH2 it was at ∼1628 cm−1, confirming the presence of the secondary β-sheet structure65,66 and being in good agreement with the observed morphology (Fig. 1a and c and ESI Fig. S5†). The absorption bands at ∼1670 cm−1 recorded for H-TTR-NH2 and D-enantiomer may represent the α-helical structure, β-turns, or a combination of these structures.67 In the case of DiMe-TTR-NH2 the maximum absorption peak was at ∼1639 cm−1 (Fig. 4a) which is on the border between the secondary β-sheet structure (1623–1641 cm−1) and disordered structure (1642–1657 cm−1).68 Taking into account the resolution of the ATR-FTIR measurements which is equal to 4 cm−1 and the observed morphology of the sample evidenced by the AFM (Fig. 1e), we assume that the studied peptide aggregates are composed of the disordered structure. The corresponding ATR-FTIR spectra obtained for the samples termed by the carboxyl group (ESI Fig. S9a†) are analogous to the ones capped by the amide group (Fig. 4a). The only difference was in the case of H-TTR-OH fibrils (ESI Fig. S9a†), as the maximum absorption peak was at ∼1641 cm−1 which could be interpreted as the secondary β-sheet (1623–1641 cm−1) or disordered structure (1642–1657 cm−1).68 The AFM images show amyloid fibrils (Fig. 1b) whose characteristic feature is the presence of the secondary β-sheet structure.12,69 At t2, i.e. after 7 weeks of incubation, we examined again the H-TTR-OH sample. The AFM images (ESI Fig. S10†) demonstrated a more dense network of amyloid fibrils as compared to the lower number of those structures at t1 (Fig. 1b). We calculated the cross-over distance of the fibrils imaged at t1 (Fig. 1b) and t2 (ESI Fig. S10†). The cross-over distance was equal to 104.4 ± 11.7 nm at t1 (ESI Table S1†) and 102.4 ± 10.8 nm at t2. The average width and height of the fibrils were also similar at t1 (22.2 ± 3.9 nm and 8.5 ± 2.7; ESI Table S1†) and at t2 (21.5 ± 3.4 nm and 9.2 ± 1.8 nm). The width, height and cross-over distance ranges of H-TTR-OH fibrils at t1 and t2 are represented in ESI Fig. S11.† These results indicate that dimensions of the fibrils did not significantly change over time, only the number of amyloid fibrils increased. To gain insight into the secondary structure, we compared the ATR-FTIR spectra of the H-TTR-OH samples at both time points, t1 and t2 (ESI Fig. S12†). The IR component at ∼1630 cm−1 (the β-sheet structure) increases its contribution with respect to the absorption peak at ∼1641 cm−1 (the disordered structure) at t2. This indicates the maturation of the secondary β-sheet structure and is in agreement with the increase of amyloid fibrils number observed by AFM (Fig. 1b, ESI Fig. S10 and S12†). Nevertheless, the most intense band with a maximum at ∼1641 cm−1 still appears, indicating that the fibrils were mainly composed of a disordered structure (Fig. 4a and ESI Fig. S12†). In fact, it has been already evidenced, using computational studies, that the uncapped TTR(105–115) fibrils, i.e. termed by α-amino and a carboxyl group, exhibit disordered conformation.36 Moreover, carboxyl termini in proteins are often disordered.32
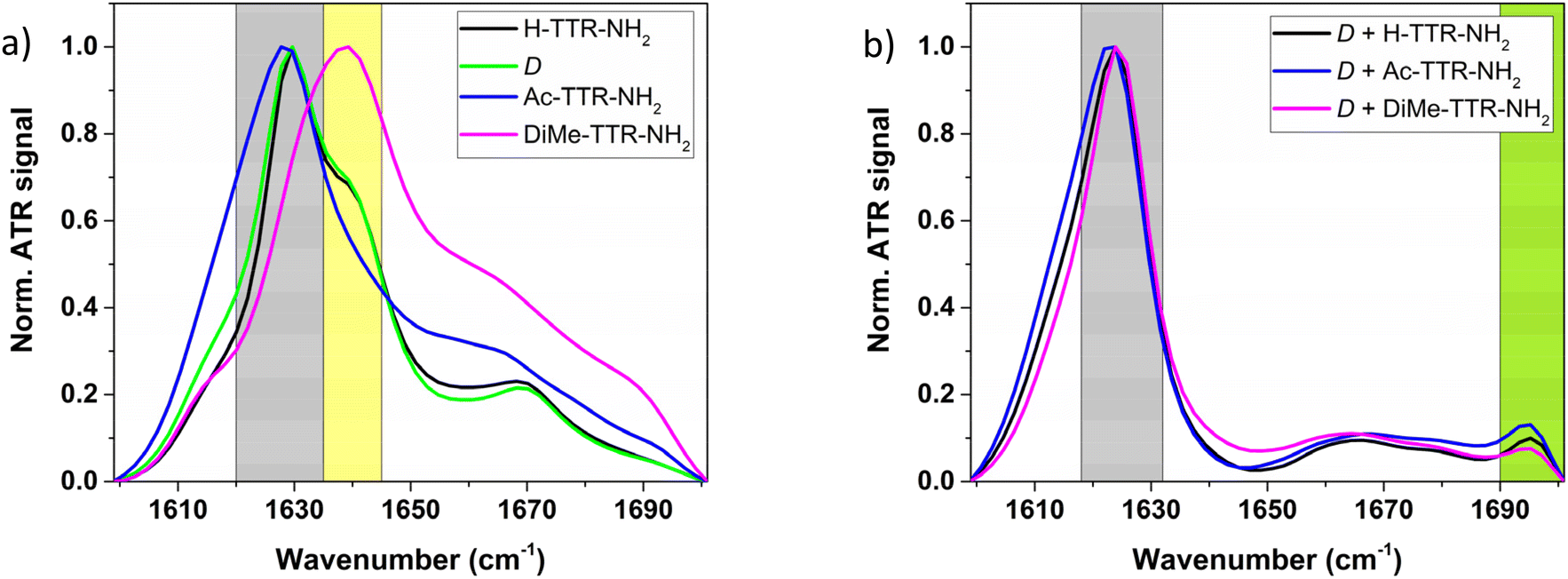 | ||
| Fig. 4 Normalized ATR-FTIR spectra of the studied samples at t1: (a) of the individual peptides C-terminally capped by amide group, (b) of their quasi-racemic mixtures with D-analogue. Grey areas represent the β-sheet structure; yellow area indicates the disordered structure; green area evidences antiparallel β-sheet structure. | ||
We also determined the secondary structure of quasi-racemic mixtures (Fig. 4b, and ESI Fig. S9b†). For all of the samples, regardless of the C-terminus, the β-sheet structure was confirmed with a maximum absorption band centred at ∼1624 cm−1. Among these samples, a quasi-racemic mixture of D-form and Ac-TTR-NH2 had nearly the same intensity at 1622 cm−1 and at 1624 cm−1. The absorption bands at ∼1660 cm−1 may indicate the presence of α-helical structure, disordered structure, β-turn, or a combination of them.67,70 The small absorption band at ∼1695 cm−1 evidences that the β-strands are arranged in an antiparallel orientation.71,72
In order to understand why peptides termed by different functional groups have or do not have the ability to form amyloid fibrils, we performed MD simulations. The fibril formation of peptides H-TTR-NH2, H-TTR-OH, D, Ac-TTR-NH2, and Ac-TTR-OH (Fig. 1a–d, and ESI Fig. S5†) may be explained by the modelled bilayers that show the respective hydrogen bonding formed by the capping groups. N-terminus without capping, i.e. peptides H-TTR-NH2 and H-TTR-OH, may form an interlayer hydrogen bond (ESI Fig. S13a†) between its hydrogen atom and the hydroxyl group oxygen atom of the serine side chain. Acetylated N-terminus (in peptides Ac-TTR-NH2 and Ac-TTR-OH) has the potential to form two hydrogen bonds. The amide oxygen atom may interact with the hydroxyl group hydrogen atom of the serine side chain (interlayer hydrogen bond) and within the layer with the amide group hydrogen of the neighboring strand (ESI Fig. S13b†). Dimethyl-amino group (peptides DiMe-TTR-NH2 and DiMe-TTR-OH) prevents hydrogen bond formation (ESI Fig. S13c†). Amide capping of C-terminus (in peptide Ac-TTR-NH2 but not H-TTR-NH2) allows the formation of stabilizing intralayer hydrogen bond between neighboring strands (ESI Fig. S14a†) which may explain the formation of longer fibrils.
The unstructured aggregates formed by peptides possessing N,N-dimethyl-α-amino group at their N-termini (DiMe-TTR-NH2 and DiMe-TTR-OH) can be explained by disruption of the assembly of the layers due to steric hindrance caused by two methyl groups (ESI Fig. S13c†). To lower the steric hindrance, dimethyl caps of neighbouring strands are rotated relative to each other which excludes most of the amine hydrogen atoms to form a hydrogen bond with a hydroxyl group oxygen atom of the serine side chain, present in the case of peptides without capping. This effect is significantly reduced in the case of an antiparallel β-sheet formed in a 1:1 ratio with D-peptide. The twist of the β-sheet structure of the bilayer formed by peptides H-TTR-NH2, H-TTR-OH and Ac-TTR-OH, can be observed after 5 ns of molecular dynamics simulation and it remains stable until the end of the 100 ns simulation (Fig. 5). The smallest twist is observed for peptide Ac-TTR-NH2 that has charge-neutralizing capping at both termini (acetylated N-terminus and amidated C-terminus). We conclude that the twist is correlated with charge repulsion in parallel β-sheets. The twist cannot be observed in the molecular dynamics of 1:1 mixtures with peptide D which may be consistent with the tape-forming behaviour (Fig. 6). This is also consistent with antiparallel β-sheet where the charge repulsion between the same termini is reduced, as demonstrated in Fig. 6a and b. Taken together, our results suggest that the rippled antiparallel β-sheet structure is thermodynamically more favorable than the pleated parallel one, which was also suggested in other articles.60,73,74
 | ||
| Fig. 5 The results of modelling β-sheet bilayers formed by L-peptides. (a) Representation of two neighbouring β-strands arranged in a parallel orientation to each other, twisting to increase the distance due to the charge repulsion, (b) demonstration of twist formation by peptide chains oriented in a parallel way, (c) twist by different angle due to differences in the distribution of charge in fibrils formed from H-TTR-NH2 (left), H-TTR-OH (middle), and Ac-TTR-NH2 (right). | ||
 | ||
| Fig. 6 The results of modelling β-sheet bilayers formed by peptide quasi-racemic mixtures. (a) Representation of two neighbouring β-strands arranged in an antiparallel orientation to each other to balance the charge repulsion, (b) demonstration of flat fibril formation by peptide chains oriented in an antiparallel way, (c) flat morphology of fibrils formed from quasi-racemic mixtures of D with H-TTR-NH2 (left) and H-TTR-OH (right). | ||
Conclusions
Herein, the influence of different peptide termini on the morphology and the secondary structure of the resulting supramolecular aggregates was examined. We focused on TTR(105–115) peptide fragments which were functionalized by the H-, Ac- or DiMe-groups at the N-terminus, and by –OH or –NH2 groups at the C-terminus. Our studies revealed that the N-terminus has a significant impact on the morphology of the resulting supramolecular aggregates. In order to produce twisted fibrils, the α-amino group at the N-terminus is the best choice; the N-acetyl-α-amino group favours elongated fibrils which tend to stick together instead of being separated, whereas the N,N-dimethyl-α-amino group hinders the formation of amyloid fibrils and leads to small peptide aggregates. In contrast, the C-terminus plays a minor role in morphology, but largely influences the kinetics of the fibril formation. Our studies revealed that the –OH group at C-terminus can enhance a cross-over distance of the resulting twisted amyloid fibrils. By performing the ATR-FTIR measurements, we confirmed the secondary β-sheet structure in the case of the L-peptides, for all of the samples where amyloid fibrils were present. Nevertheless, the H-TTR-OH-derived fibrils were mainly composed of a disordered structure. This conformation was confirmed computationally by another group as well.36 Peptides capped by the DiMe-group led to the formation of peptide aggregates and the ATR-FTIR spectra confirmed that they consist mainly of a disordered structure.Significantly different morphologies were obtained in peptide quasi-racemic mixtures (prepared by mixing the synthesized L-enantiomeric peptides with a D-enantiomer capped by H- and –NH2 groups at the N- and C-termini, respectively, in 1/1 molar ratio). In the case of the quasi-racemic mixtures, amyloid fibrils were present in every case, directly after the preparation. The AFM analysis revealed that all fibrils had a flat morphology, in contrast to the L-enantiomers where the final morphology was governed by the functional group at the N-terminus. Additionally, we observed the formation of wider quasi-racemic fibrils when L-peptides were functionalized by the –NH2 group. In all of the quasi-racemic fibrils antiparallel β-sheet structure was detected.
Finally, we performed MD simulations to better understand the impact of peptides’ termini on the morphology and the secondary structure of the resulting peptide aggregates. Peptides with the H- and Ac- N-terminal groups formed amyloid fibrils due to the additional hydrogen bonds present between the capping groups. Peptides capped by the DiMe-group did not form fibrils because the terminal group prevented the formation of hydrogen bonds. MD simulations provided a molecular explanation of the twisted morphology of amyloid fibrils. For instance, the twist is linked with the charge repulsion in parallel β-sheets. Twisted morphology was not observed for quasi-racemic fibrils because the charge repulsion between the same termini is of less importance in antiparallel β-sheet structure.
Our findings demonstrated that the modulation of peptides’ termini determines the morphology and the secondary structure of the resulting supramolecular peptide aggregates. The knowledge of the role of peptides’ termini may be crucial in the design of novel biomaterials with the desired functional properties. The presented molecular understanding may be used to design peptide nanostructures with a defined morphology. Such nanostructures could be then utilized as supramolecular matrices or scaffolds for nanomaterials and biomaterials, as well as in biocompatible heterostructures which require fibrillar morphology. The role of peptides’ termini may be used to expand the knowledge about novel biomaterials with the desired morphology and functional properties, for applications in controlled cell growth, biosensors, biomimetic catalysts, drug delivery applications, as well as in optoelectronics.
Abbreviations
| TTR(105–115) | (105–115) peptide fragments of transthyretin |
| AFM | Atomic force microscopy |
| ATR-FTIR spectroscopy | Attenuated total reflectance Fourier-transform infrared spectroscopy |
| MD simulations | Molecular dynamics simulations |
| RP-HPLC | Reverse-phase high-performance liquid chromatography |
| LC-MS | Liquid chromatography–mass spectrometry |
| ESI MS | Electrospray ionization mass spectra |
| t 0 | Time point before incubation of the samples |
| t 1 | Time point 3 weeks after incubation of the samples |
| t 2 | Time point 7 weeks after incubation of the samples. |
Author contributions
M.G.-M. – conceptualization, investigation (peptide synthesis, samples’ preparation, absorption spectroscopy, AFM, ATR-FTIR spectroscopy), methodology, visualization, project administration, writing – original draft, writing – review & editing; T.B. – investigation (peptide synthesis), methodology, writing – review & editing; V.T. – methodology, visualization, supervision, resources, writing – review & editing; K.O. – investigation (MD simulations), methodology, writing – review & editing; Ł.B. – investigation (MD simulations), methodology, writing – review & editing; J.O.-B. – conceptualization, methodology, visualization, project administration, supervision, resources, funding, writing – review & editing. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors greatly acknowledge Dr. Mirosława Różycka (Department of Biochemistry, Molecular Biology and Biotechnology; Wroclaw University of Science and Technology) and Klaudia Bielak (Department of Biochemistry, Molecular Biology and Biotechnology; Wroclaw University of Science and Technology) for the possibility to lyophilize the peptides and incubate the samples. Dr. Youssef El Khoury and Prof. Petra Hellwig (University of Strasbourg) are acknowledged for access to FT-IR spectrometer used to acquire preliminary data.This work was supported by NAWA STER Program Internationalisation of Wroclaw University of Science and Technology Doctoral School. M. G.-M. and J. O.-B. acknowledge funding from NONA project performed within the First Team program of the Foundation for Polish Science (First TEAM/2017-3/27) co-financed by the European Union under the European Regional Development Fund. V. T. and T. B. acknowledge the support by the Interdisciplinary Thematic Institute SysChem, via the IdEx Unistra (ANR-10-IDEX-0002) within the French Investments for the Future Program.
References
- F. Sheehan, D. Sementa, A. Jain, M. Kumar, M. Tayarani-Najjaran, D. Kroiss and R. V. Ulijn, Chem. Rev., 2021, 121, 13869–13914 CrossRef CAS PubMed.
- B. Liu, C. G. Pappas, J. Ottelé, G. Schaeffer, C. Jurissek, P. F. Pieters, M. Altay, I. Marić, M. C. A. Stuart and S. Otto, J. Am. Chem. Soc., 2020, 142, 4184–4192 CrossRef CAS PubMed.
- S. N. Semenov, A. S. Y. Wong, R. M. van der Made, S. G. J. Postma, J. Groen, H. W. H. van Roekel, T. F. A. de Greef and W. T. S. Huck, Nat. Chem., 2015, 7, 160–165 CrossRef CAS.
- G. Wei, Z. Su, N. P. Reynolds, P. Arosio, I. W. Hamley, E. Gazit and R. Mezzenga, Chem. Soc. Rev., 2017, 46, 4661–4708 RSC.
- U. N. Morzan, G. Díaz Mirón, L. Grisanti, M. C. González Lebrero, G. S. Kaminski Schierle and A. Hassanali, J. Phys. Chem. B, 2022, 126, 7203–7211 CrossRef CAS PubMed.
- M. Grelich-Mucha, M. Lipok, M. Różycka, M. Samoć and J. Olesiak-Bańska, J. Phys. Chem. Lett., 2022, 13, 4673–4681 CrossRef CAS PubMed.
- P. Obstarczyk, M. Lipok, A. Żak, P. Cwynar and J. Olesiak-Bańska, Biomater. Sci., 2022, 10, 1554–1561 RSC.
- A. Levin, T. A. Hakala, L. Schnaider, G. J. L. Bernardes, E. Gazit and T. P. J. Knowles, Nat. Rev. Chem., 2020, 4, 615–634 CrossRef CAS.
- A. C. Mendes, E. T. Baran, R. L. Reis and H. S. Azevedo, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2013, 5, 582–612 CAS.
- N. J. Sinha, M. G. Langenstein, D. J. Pochan, C. J. Kloxin and J. G. Saven, Chem. Rev., 2021, 121, 13915–13935 CrossRef CAS PubMed.
- X. Yu, Z. Wang, Z. Su and G. Wei, J. Mater. Chem. B, 2017, 5, 1130–1142 RSC.
- P. C. Ke, R. Zhou, L. C. Serpell, R. Riek, T. P. J. Knowles, H. A. Lashuel, E. Gazit, I. W. Hamley, T. P. Davis, M. Fändrich, D. E. Otzen, M. R. Chapman, C. M. Dobson, D. S. Eisenberg and R. Mezzenga, Chem. Soc. Rev., 2020, 49, 5473–5509 RSC.
- M. R. Brown, S. E. Radford and E. W. Hewitt, Front. Mol. Neurosci., 2020, 13, 609073 CrossRef CAS PubMed.
- C. A. E. Hauser, S. Maurer-Stroh and I. C. Martins, Chem. Soc. Rev., 2014, 43, 5326 RSC.
- C. Li, J. Adamcik and R. Mezzenga, Nat. Nanotechnol., 2012, 7, 421–427 CrossRef CAS PubMed.
- L. Sasso, S. Suei, L. Domigan, J. Healy, V. Nock, M. A. K. Williams and J. A. Gerrard, Nanoscale, 2014, 6, 1629–1634 RSC.
- R. S. Jacob, D. Ghosh, P. K. Singh, S. K. Basu, N. N. Jha, S. Das, P. K. Sukul, S. Patil, S. Sathaye, A. Kumar, A. Chowdhury, S. Malik, S. Sen and S. K. Maji, Biomaterials, 2015, 54, 97–105 CrossRef CAS PubMed.
- N. Yadav, M. K. Chauhan and V. S. Chauhan, Biomater. Sci., 2020, 8, 84–100 RSC.
- J. Zhao and P. Yang, Adv. Mater. Interfaces, 2020, 7, 2001060 CrossRef CAS.
- M. Owczarz, T. Casalini, A. C. Motta, M. Morbidelli and P. Arosio, Biomacromolecules, 2015, 16, 3792–3801 CrossRef CAS PubMed.
- S. Das, R. S. Jacob, K. Patel, N. Singh and S. K. Maji, Biomacromolecules, 2018, 19, 1826–1839 CrossRef CAS PubMed.
- J. Ryu, S.-W. Kim, K. Kang and C. B. Park, Adv. Mater., 2010, 22, 5537–5541 CrossRef CAS PubMed.
- Z. Yu, Z. Cai, Q. Chen, M. Liu, L. Ye, J. Ren, W. Liao and S. Liu, Biomater. Sci., 2016, 4, 365–374 RSC.
- S. Catalini, D. R. Perinelli, P. Sassi, L. Comez, G. F. Palmieri, A. Morresi, G. Bonacucina, P. Foggi, S. Pucciarelli and M. Paolantoni, Biomacromolecules, 2021, 22, 1147–1158 CrossRef CAS PubMed.
- W. Close, M. Neumann, A. Schmidt, M. Hora, K. Annamalai, M. Schmidt, B. Reif, V. Schmidt, N. Grigorieff and M. Fändrich, Nat. Commun., 2018, 9, 699 CrossRef PubMed.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2017, 86, 27–68 CrossRef CAS.
- R. S. Harrison, P. C. Sharpe, Y. Singh and D. P. Fairlie, Rev. Physiol., Biochem. Pharmacol., 2007, 159, 1–77 CAS.
- S.-Y. Ow, I. Bekard, A. Blencowe, G. G. Qiao and D. E. Dunstan, J. Mater. Chem. B, 2015, 3, 1350–1359 RSC.
- G. Grasso, M. Rebella, U. Morbiducci, J. A. Tuszynski, A. Danani and M. A. Deriu, Front. Bioeng. Biotechnol., 2019, 7, 83 CrossRef PubMed.
- L. Lutter, L. D. Aubrey and W.-F. Xue, J. Mol. Biol., 2021, 433, 167124 CrossRef CAS PubMed.
- S. Varland, C. Osberg and T. Arnesen, Proteomics, 2015, 15, 2385–2401 CrossRef CAS PubMed.
- S. Sharma and M. R. Schiller, Crit. Rev. Biochem. Mol. Biol., 2019, 54, 85–102 CrossRef CAS PubMed.
- K. Tao, J. Wang, P. Zhou, C. Wang, H. Xu, X. Zhao and J. R. Lu, Langmuir, 2011, 27, 2723–2730 CrossRef CAS PubMed.
- M. Andreasen, K. K. Skeby, S. Zhang, E. H. Nielsen, L. H. Klausen, H. Frahm, G. Christiansen, T. Skrydstrup, M. Dong, B. Schiøtt and D. Otzen, Biochemistry, 2014, 53, 6968–6980 CrossRef CAS PubMed.
- Y. Hu, R. Lin, P. Zhang, J. Fern, A. G. Cheetham, K. Patel, R. Schulman, C. Kan and H. Cui, ACS Nano, 2015, 10, 880–888 CrossRef PubMed.
- M. Lee and S. Na, ChemPhysChem, 2016, 17, 425–432 CrossRef CAS PubMed.
- M. Porrini, U. Zachariae, P. E. Barran and C. E. MacPhee, J. Phys. Chem. Lett., 2013, 4, 1233–1238 CrossRef CAS PubMed.
- A. Stock, S. Clarke, C. Clarke and J. Stock, FEBS Lett., 1987, 220, 8–14 CrossRef CAS.
- K. Diaz, Y. Meng and R. Huang, Curr. Opin. Chem. Biol., 2021, 63, 115–122 CrossRef CAS PubMed.
- E. Hughes, R. M. Burke and A. J. Doig, J. Biol. Chem., 2000, 275, 25109–25115 CrossRef CAS PubMed.
- M. Grelich-Mucha, A. M. Garcia, V. Torbeev, K. Ożga, Ł. Berlicki and J. Olesiak-Bańska, J. Phys. Chem. B, 2021, 125, 5502–5510 CrossRef CAS PubMed.
- J. Magalhães, J. Eira and M. A. Liz, Cell. Mol. Life Sci., 2021, 78, 6105–6117 CrossRef PubMed.
- L. Saelices, L. M. Johnson, W. Y. Liang, M. R. Sawaya, D. Cascio, P. Ruchala, J. Whitelegge, L. Jiang, R. Riek and D. S. Eisenberg, J. Biol. Chem., 2015, 290, 28932–28943 CrossRef CAS PubMed.
- M. Schmidt, S. Wiese, V. Adak, J. Engler, S. Agarwal, G. Fritz, P. Westermark, M. Zacharias and M. Fändrich, Nat. Commun., 2019, 10, 5008 CrossRef PubMed.
- Å. Gustavsson, U. Engström and P. Westermark, Biochem. Biophys. Res. Commun., 1991, 175, 1159–1164 CrossRef PubMed.
- K.-C. Cho, J. W. Kang, Y. Choi, T. W. Kim and K. P. Kim, J. Mass Spectrom., 2016, 51, 105–110 CrossRef CAS PubMed.
- N. Jentoft and D. G. Dearborn, J. Biol. Chem., 1979, 254, 4359–4365 CrossRef CAS PubMed.
- S. Dasgupta, L. M. Castro, A. K. Tashima and L. Fricker, Methods Mol. Biol., 2018, 1719, 161–174 CrossRef CAS PubMed.
- https://gwyddion.net/ .
- H. E. White, J. L. Hodgkinson, T. R. Jahn, S. Cohen-Krausz, W. S. Gosal, S. Müller, E. V. Orlova, S. E. Radford and H. R. Saibil, J. Mol. Biol., 2009, 389, 48–57 CrossRef CAS PubMed.
- M. Fändrich, J. Meinhardt and N. Grigorieff, Prion, 2014, 3, 89–93 CrossRef PubMed.
- S. Dutta, A. R. Foley, C. J. A. Warner, X. Zhang, M. Rolandi, B. Abrams and J. A. Raskatov, Angew. Chem., Int. Ed., 2017, 56, 11506–11510 CrossRef CAS PubMed.
- S. Dutta, A. R. Foley, A. J. Kuhn, B. Abrams, H. W. Lee and J. A. Raskatov, Pept. Sci., 2019, 111, e24139 CrossRef CAS.
- A. M. Garcia, C. Giorgiutti, Y. El Khoury, V. Bauer, C. Spiegelhalter, E. Leize-Wagner, P. Hellwig, N. Potier and V. Torbeev, Chem. – Eur. J., 2020, 26, 9889–9899 CrossRef CAS PubMed.
- L. Pauling and R. B. Corey, Proc. Natl. Acad. Sci. U. S. A., 1951, 37, 251–256 CrossRef CAS PubMed.
- L. Pauling and R. B. Corey, Proc. Natl. Acad. Sci. U. S. A., 1951, 37, 729–740 CrossRef CAS PubMed.
- L. Pauling and R. B. Corey, Proc. Natl. Acad. Sci. U. S. A., 1953, 39, 253–256 CrossRef CAS PubMed.
- K. J. Nagy, M. C. Giano, A. Jin, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2011, 133, 14975–14977 CrossRef CAS PubMed.
- R. J. Swanekamp, J. T. M. DiMaio, C. J. Bowerman and B. L. Nilsson, J. Am. Chem. Soc., 2012, 134, 5556–5559 CrossRef CAS PubMed.
- J. A. Raskatov, J. P. Schneider and B. L. Nilsson, Acc. Chem. Res., 2021, 54, 2488–2501 CrossRef CAS PubMed.
- A. R. Foley and J. A. Raskatov, Curr. Opin. Chem. Biol., 2021, 64, 1–9 CrossRef CAS PubMed.
- A. Hazari, M. R. Sawaya, M. Sajimon, N. Vlahakis, J. Rodriguez, D. Eisenberg and J. A. Raskatov, J. Am. Chem. Soc., 2023, 145, 25917–25926 CrossRef CAS PubMed.
- A. Hazari, M. R. Sawaya, N. Vlahakis, T. C. Johnstone, D. Boyer, J. Rodriguez, D. Eisenberg and J. A. Raskatov, Chem. Sci., 2022, 13, 8947–8952 RSC.
- A. J. Kuhn, B. Ehlke, T. C. Johnstone, S. R. J. Oliver and J. A. Raskatov, Chem. Sci., 2022, 13, 671–680 RSC.
- S. E. Glassford, B. Byrne and S. G. Kazarian, Biochim. Biophys. Acta, Proteins Proteomics, 2013, 1834, 2849–2858 CrossRef CAS PubMed.
- W. K. Surewicz, H. H. Mantsch and D. Chapman, Biochemistry, 2002, 32, 389–394 CrossRef PubMed.
- L. M. Miller, M. W. Bourassa and R. J. Smith, Biochim. Biophys. Acta, Biomembr., 2013, 1828, 2339–2346 CrossRef CAS PubMed.
- A. Barth, Biochim. Biophys. Acta, Bioenerg., 2007, 1767, 1073–1101 CrossRef CAS PubMed.
- J. D. Sipe and A. S. Cohen, J. Struct. Biol., 2000, 130, 88–98 CrossRef CAS PubMed.
- K. A. Conway, J. D. Harper and P. T. Lansbury, Biochemistry, 2000, 39, 2552–2563 CrossRef CAS PubMed.
- S. D. Moran and M. T. Zanni, J. Phys. Chem. Lett., 2014, 5, 1984–1993 CrossRef CAS PubMed.
- E. Cerf, R. Sarroukh, S. Tamamizu-Kato, L. Breydo, S. Derclaye, Y. F. Dufrêne, V. Narayanaswami, E. Goormaghtigh, J.-M. Ruysschaert and V. Raussens, Biochem. J., 2009, 421, 415–423 CrossRef CAS PubMed.
- J. A. Raskatov, A. R. Foley, J. M. Louis, W.-M. Yau and R. Tycko, J. Am. Chem. Soc., 2021, 143, 13299–13313 CrossRef CAS PubMed.
- J. A. Raskatov, Chem. – Eur. J., 2017, 23, 16920–16923 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Chemical steps to obtain N,N-dimethylated peptides, ESI MS spectra and analytical RP-HPLC traces, absorption spectra, AFM images and morphology analysis, ATR-FTIR spectra, molecular models of bilayers formed by proper samples. See DOI: https://doi.org/10.1039/d3bm01641b |
| This journal is © The Royal Society of Chemistry 2024 |