
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA quaternary ammonium-based nanosystem enables delivery of CRISPR/Cas9 for cancer therapy†
Mengzhu
Zhang‡
a,
Siyu
Sun‡
a,
Xiao
Liang
a,
Zengguang
Liu
a,
Jiaxin
Yin
a,
Quanshun
Li
 *ab and
Shengcai
Yang
*a
*ab and
Shengcai
Yang
*a
aKey Laboratory for Molecular Enzymology and Engineering of Ministry of Education, School of Life Sciences, Jilin University, Changchun 130012, China. E-mail: yang_shengcai@jlu.edu.cn; quanshun@jlu.edu.cn; Fax: +86-431-85155200; Tel: +86-431-85155200
bCenter for Supramolecular Chemical Biology, Jilin University, Changchun 130012, China
First published on 11th January 2024
Abstract
Genome editing mediated by CRISPR/Cas9 is an attractive weapon for cancer therapy. However, in vivo delivery of CRISPR/Cas9 components to achieve therapeutic efficiency is still challenging. Herein, a quaternary ammonium-functionalized poly(L-lysine) and a cholesterol-modified PEG (QNP) were self-assembled with a negatively charged CRISPR Cas9/sgRNA ribonucleoprotein (RNP) to form a ternary complex (QNP/RNP). Such a delivery system of QNP exhibited multiplex genome editing capabilities in vitro (e.g., the GFP gene and the PLK1 gene). In addition, QNP/RNPPLK1 containing PLK1 sgRNA led to 30.99% of genome editing efficiency in MCF-7 cells with negligible cytotoxicity of the carrier. QNP/RNPPLK1, which was capable of simultaneously inhibiting cell proliferation, mediating cell cycle arrest and downregulating expression of PLK1, held great in vitro therapeutic efficiency. Moreover, QNP/RNPPLK1 exhibited outstanding accumulation in tumors and high biocompatibility in vivo. In an MCF-7 xenograft animal model, QNP/RNPPLK1 showed excellent anti-tumor efficacy and achieved 17.75% indels ratio. This work showcases the successful delivery of CRISPR Cas9/sgRNA RNP with enhanced genome editing efficiency and provides a potential on-demand strategy for cancer therapy.
Introduction
Cancer is one of the leading causes of death globally, but common treatments including surgery, radiotherapy or chemotherapy have some limitations for cancer therapy.1 In recent years, gene therapy strategies, such as messenger RNA (mRNA), small interfering RNA (siRNA), DNAzyme or clustered regularly interspaced short palindromic repeats (CRISPR)-associated protein (CRISPR/Cas)-based technologies, have attracted great attention for cancer therapy in clinical applications.2–4 CRISPR/Cas9 is the most commonly used CRISPR/Cas, which has acted as the groundbreaking genetic engineering tool and is composed of a Cas9 nuclease and single guide RNA (sgRNA).5 The CRISPR/Cas9 genome editing system can be delivered in three different modes: plasmid DNA (“all-in-one” plasmid), mRNA (Cas9 mRNA with sgRNA), or protein (Cas9/sgRNA ribonucleoprotein (RNP) complex).6–8 However, the “all-in-one” plasmid, expressing both Cas9 protein and sgRNA, has high off-target efficacy and must process transcription and translation for targeted deletion or insertion in the genomic DNA. The mRNA of Cas9 and sgRNA is easily degraded by nucleases, causing low stability and unsatisfactory efficacy for in vivo delivery.9 In contrast, CRISPR/Cas9 RNP complexes could be directly delivered into cells for rapid and highly efficient genome editing without transcription and translation.10 The preformed RNP also reduces the possibility of off-target events and minimizes immunogenicity.11 Nevertheless, the delivery of the RNP also faces many difficulties: RNP is easily denatured and has negligible intracellular delivery activity due to the high molecular weight of Cas9 (160 kDa) and sgRNA (∼31 kDa, ∼130 bases).12–14 Thus, there is an urgent need to develop nanovectors to simultaneously protect RNP from undesired degradation and enhance the efficiency of its intracellular delivery.To date, a lot of nanovectors have been developed to overcome the limitations and increase the genome editing efficiency of RNPs.15 For example, inorganic nanoparticles, polymeric micelles, DNA-based nanocarriers, lipid nanoparticles or extracellular vesicles could efficiently deliver the RNP to the nucleus for genome editing.16–18 These delivery systems not only protect the RNP from unnecessary denaturation, but also contribute to the intracellular delivery of the RNP. In addition, the immunogenicity, cytotoxicity and poor stability of these nanocarriers urge us to exploit viable delivery systems. As CRISPR/Cas9 RNP is a negatively charged complex, it is possible for cationic polymers to self-assemble with the RNP through electrostatic interaction.19 For instance, poly(L-lysine)-based polymers, cell-penetrating peptides, arginine-based nanoparticles or other polycation polymers have been rationally developed and investigated to deliver RNPs to targeted cells or tissues for genome editing.20,21 However, the charged proteins or polypeptides in blood could attack the nanoparticles with weak interaction, which would lead to poor stability of the nanoparticles and their easy dissociation during circulation in the body. To this end, our previous study indicated that quaternary ammonium-functionalized polypeptides could self-assemble with negatively charged polymers to obtain more stable nanoparticles in comparison with amine-functionalized polymers.22
Inspired by these results, the quaternary ammonium-functionalized polymer may be a potential material to interact with the negative RNP to construct stable nanoparticles. Meanwhile, a previous report has demonstrated that a hydrophobic moiety (such as cholesterol) could facilitate the formation of π–π stacking interactions and hydrogen bonds with proteins.23 PEGylated nanoparticles could decrease the generation of protein corona and avoid the interaction with charged proteins or polypeptides in the blood.24,25 Thus, we hypothesized that the combination of the quaternary ammonium-functionalized polymer with cholesterol-functionalized PEG may effectively stabilize the RNP during its circulation in the body.
Polo-like kinase 1 (PLK1) is a mitotic protein kinase that is overexpressed in many cancers, including breast cancer, ovarian cancer, non-small cell lung cancer, colorectal cancer and others.26 PLK1 plays a critical role in carcinogenesis, and the inhibition of its expression can induce apoptosis of tumor cells and lead to G2/M phase cell cycle arrest.27,28 Therefore, downregulating its expression level has been considered a promising approach in cancer treatment, e.g., using siRNA to knockdown the expression29–31 and the CRISPR/Cas9 technique to achieve its knockout.32,33 In this study, a CRISPR/Cas9 delivery system was developed for the delivery of Cas9/PLK1 sgRNA RNP complexes (denoted as RNPPLK1) to improve genome editing efficiency (Scheme 1). The delivery system consisted of a quaternary ammonium-functionalized poly(L-lysine) (Qua-PLL) and a cholesterol-modified PEG (Chol-PEG), which were self-assembled with negatively charged RNPPLK1 to form a ternary complex (Qua-PLL/Chol-PEG/RNPPLK1, abbreviated as QNP/RNPPLK1). The QNP delivery system was firstly optimized and multiplex genome editing (e.g., GFP gene and PLK1 gene) was evaluated in vitro. Finally, the in vivo therapeutic efficiency and genome editing of QNP/RNPPLK1 were investigated in MCF-7 tumor xenograft nude mice.
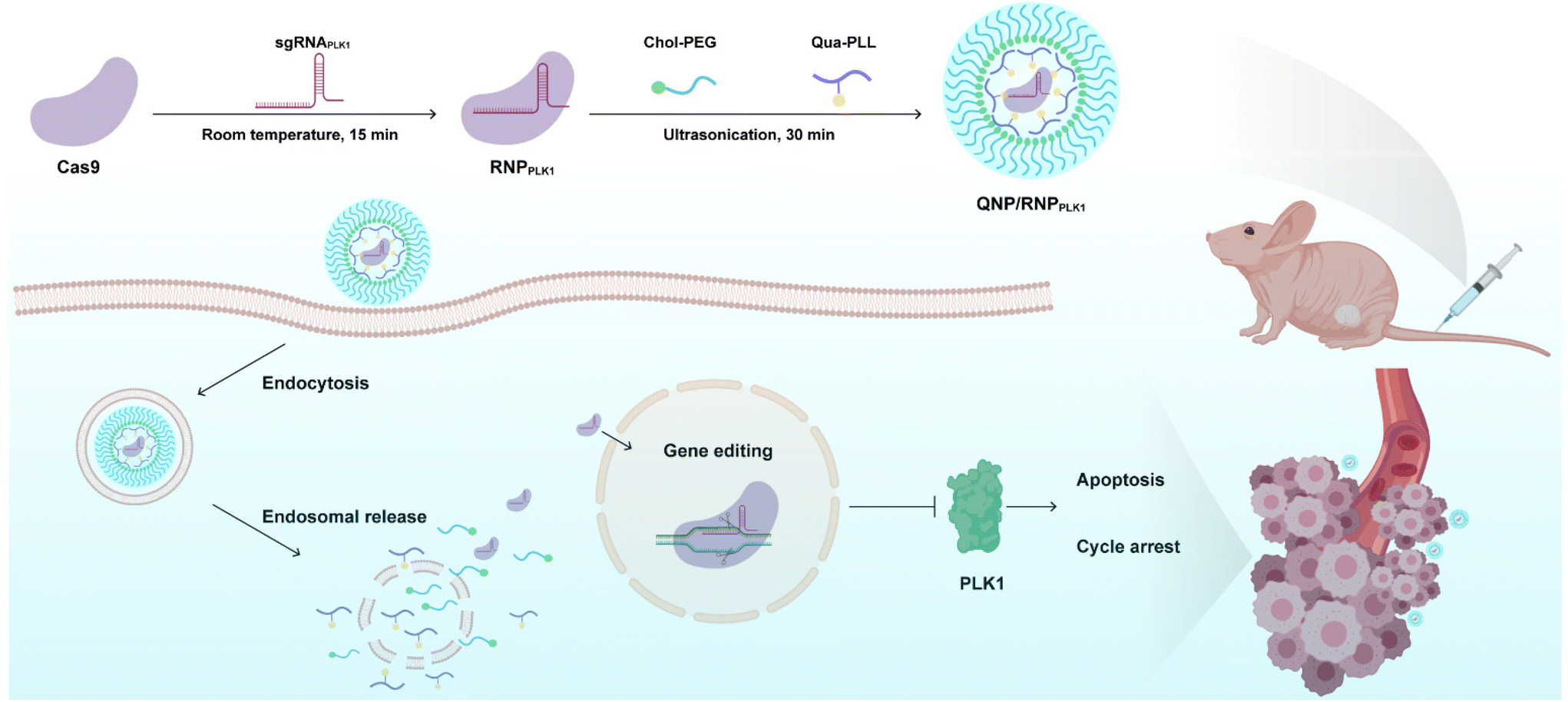 | ||
| Scheme 1 Schematic illustration of the preparation of QNP/RNPPLK1 and the genome editing efficiency in vivo for cancer therapy. | ||
Experimental section
Materials
Cholesterol hydrogen succinate (Chol-COOH) was obtained from TCI Development Co., Ltd (Shanghai, China). Methoxypoly(ethylene glycol) amine (mPEG-NH2, Mn = 2000 g mol−1) was purchased from Macklin Biochemical Technology Co., Ltd (Shanghai, China). 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS) were purchased from Energy Chemical Co., Ltd (Shanghai, China). Isopropyl β-D-thiogalactopyranoside (IPTG) and Ni Sepharose 6FF were purchased from Gentihold (Beijing, China) and Solarbio (Beijing, China), respectively. Q5 DNA polymerase and T7 endonuclease I (T7E1) were purchased from New England Biolabs, Inc. (Ipswich, MA, USA). A TUNEL staining kit was acquired from Beyotime (Shanghai, China). Anti-PLK1 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000, ab17057), anti-pro caspase 9 (1:5000, ab138412), anti-cyclin B1 (1:2000, ab181593) and anti-Ki67 (1:250, ab16667) antibodies were purchased from Abcam (Cambridge, UK). Dulbecco's modified Eagle's medium (DMEM) and Roswell Park Memorial Institute (RPMI) 1640 medium were obtained from Gibco (Grand Island, NY, USA). Fetal bovine serum (FBS) was obtained from Kangyuan (Beijing, China). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and Lipofectamine 2000 (Lipo2000) were purchased from Amersco (Solon, OH, USA) and Invitrogen (CarIsbad, CA), respectively. An Annexin V-fluoresceine isothiocyanate (FITC)/propidium iodide (PI) apoptosis detection kit and a PI-based cell-cycle detection kit were purchased from Bestbio (Shanghai, China). Rhodamine B isothiocyanate (RBITC) was obtained from Sangon Biotech. (Shanghai, China). The kits for detecting alanine aminotransferase (ALT), aspartate aminotransferase (AST), urea (UREA) and creatinine (CREA) were obtained from Rayto Life Sciences Co. (Shenzhen, China). The sgRNAs and PCR primers were synthesized by GenScript Biotech Co. (Nanjing, China) as follows:
:1000, ab17057), anti-pro caspase 9 (1:5000, ab138412), anti-cyclin B1 (1:2000, ab181593) and anti-Ki67 (1:250, ab16667) antibodies were purchased from Abcam (Cambridge, UK). Dulbecco's modified Eagle's medium (DMEM) and Roswell Park Memorial Institute (RPMI) 1640 medium were obtained from Gibco (Grand Island, NY, USA). Fetal bovine serum (FBS) was obtained from Kangyuan (Beijing, China). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and Lipofectamine 2000 (Lipo2000) were purchased from Amersco (Solon, OH, USA) and Invitrogen (CarIsbad, CA), respectively. An Annexin V-fluoresceine isothiocyanate (FITC)/propidium iodide (PI) apoptosis detection kit and a PI-based cell-cycle detection kit were purchased from Bestbio (Shanghai, China). Rhodamine B isothiocyanate (RBITC) was obtained from Sangon Biotech. (Shanghai, China). The kits for detecting alanine aminotransferase (ALT), aspartate aminotransferase (AST), urea (UREA) and creatinine (CREA) were obtained from Rayto Life Sciences Co. (Shenzhen, China). The sgRNAs and PCR primers were synthesized by GenScript Biotech Co. (Nanjing, China) as follows:
GFP target: GCGGTTGCCGTACATGAAGG
Sense primer: CCAAACACAGCCTGAGCAAGGAGAT
Anti-sense primer: TCGGTTAGGTGCCACTTCTGGTTCT
PLK1 target: TACCTACGGCAAATTGTGCT
Sense primer: GAGAAGGGGTGCTGCGAATG
Anti-sense primer: GACTTGTGGGTTGTCTCCTTCCT.
Synthesis of quaternary ammonium-functionalized poly(L-lysine) (Qua-PLL)
Poly(L-lysine) (PLL) with a polymerization degree of 90 was synthesized according to our previous protocol.34 PLL (50.0 mg) was firstly dissolved in H2O and dimethyl sulfoxide (DMSO) (v/v = 1/1, 6.0 mL), and then triethylamine (Et3N, 0.5 mL) was added. 0.2 mL of methyl iodide (CH3I) was added to the above solution and stirred overnight at room temperature (RT). The reaction solution was dialyzed (MWCO = 1000 Da) against an NaCl solution (0.1 M) for 1 day and deionized water for 2 days. Finally, the sample was lyophilized to obtain white powder Qua-PLL.Synthesis of cholesterol-modified PEG (Chol-PEG)
Firstly, Chol-COOH (146.0 mg) was dissolved in 6.0 mL of DMSO. EDC (171.9 mg) and NHS (103.5 mg) were then added and stirred at RT to activate the carboxyl group of Chol-COOH. After 4 h, mPEG-NH2 (200.0 mg) was added to the above solution and stirred overnight at RT. The reaction solution was dialyzed against deionized water for 3 days (MWCO = 1000 Da) and lyophilized to obtain white solid Chol-PEG.Preparation and characterization of Qua-PLL/Chol-PEG/BSA nanocomplexes
The Qua-PLL and Chol-PEG polymers were mixed with BSA at a mass ratio of 5:5:1 and sonicated at RT for 30 min to obtain the Qua-PLL/Chol-PEG/BSA nanocomplexes. Similarly, the PLL/BSA and Qua-PLL/BSA nanocomplexes without Chol-PEG were prepared at a mass ratio of 5:1. In addition, the Qua-PLL/Chol-PEG/BSA nanocomplexes were prepared with different mass ratios (5:2.5:1 and 5:10:1). The hydrodynamic diameter and zeta potential of the nanocomplexes were measured using a Malvern Nano ZS90 zetasizer (Malvern, UK) at RT. Besides, the nanocomplexes (mass ratio: 5/5/1) were incubated in phosphate-buffered saline (PBS, 10 mM, pH = 7.4) at 4 °C for 7 days, and their hydrodynamic diameter and zeta potential values were monitored at predetermined times to evaluate their stability.
Cell culture
Human cervical cell line HeLa–GFP cells were purchased from FuHeng Biology (Shanghai, China), and human breast cancer cell line MCF-7, murine cancer cell line CT-26 and human embryonic kidney cell line HEK-293T cells were obtained from the Shanghai Institute Cell Bank (Shanghai, China). HeLa–GFP cells and other cells (MCF-7, CT-26 and HEK-293T) were cultured in RPMI 1640 medium and DMEM, respectively, and supplemented with 10% (v/v) FBS and 1% penicillin/streptomycin at 37 °C in a humidified environment with 5% CO2.In vitro cellular internalization of Qua-PLL/Chol-PEG/FITC-BSA nanocomplexes
To study the cellular internalization of Qua-PLL/Chol-PEG/BSA nanocomplexes, BSA was labelled with a fluorescent dye, FITC. In brief, BSA and FITC were dissolved in PBS and DMSO, respectively. Then the above solutions were mixed at a molar ratio of 1:3 and stirred overnight at RT in the dark. Afterward, the reaction solution was dialyzed against deionized water for 3 days (MWCO = 8000–14000 Da) and lyophilized to obtain FITC-labelled BSA (FITC-BSA). Similarly, the Qua-PLL/Chol-PEG/FITC-BSA nanocomplexes were prepared according to the protocol described above. MCF-7 cells were plated into a 6-well plate (2 × 105 cells per well) and incubated for 12 h prior to ensure 70% confluence at the time of transfection. Then Qua-PLL/Chol-PEG/FITC-BSA and other nanocomplexes at a final FITC-BSA concentration of 1 μg mL−1 were added and incubated at 37 °C for 6 h. The cells were washed and collected in 1 mL of PBS for detection using CytoFLEX flow cytometry (Beckman Coulter, IN, USA), and the data were analyzed by CytExpert 2.5.
MTT assay
To investigate the cytotoxicity of the QNP, MCF-7 cells were seeded into a 96-well plate (5 × 103 cells per well) and incubated overnight at 37 °C. The QNP with different concentrations (10, 5, 2.5 and 1.25 μg mL−1) was added into the wells and incubated for 72 h. Then the medium was replaced with 100 μL of MTT (1.0 mg mL−1 in DMEM) and incubated for another 4 h. Subsequently, the medium containing MTT was removed, and 150 μL of DMSO was added. Finally, the absorbance at 520 nm was measured using a GF-M3000 microplate reader (Shandong, China), and the cell viability was calculated as follows:| Cell viability (%) = (Aexperimental/Acontrol) × 100 |
Similarly, the cell killing ability of QNP/RNPPLK1 was determined using the MTT assay, with MCF-7, CT-26 and HEK-293T cells as models. The final Cas9 concentration of free RNPPLK1, QNP/RNPPLK1 and Lipo2000/RNPPLK1 was 2 μg mL−1.
Expression and purification of the Cas9 protein
A pET-NLS-Cas9-6 × His plasmid sequence was obtained from Addgene (#62934) and synthesized by GenScript Biotech Co. (Nanjing, China). The Cas9 protein was expressed in chemically competent One Shot™ BL21 Star™ (DE3) (Thermo Fisher, C601003) by induction with 0.5 mM IPTG at 20 °C overnight. The cells were lysed by sonication in buffer A (50 mM Tris–HCl, pH 8.0, 1 M NaCl, 20% glycerol), and the lysate was centrifuged at 6000g for 15 min. The supernatant was subjected to Ni Sepharose 6FF resin and eluted with buffer B (50 mM Tris–HCl, pH 8.0, 300 mM imidazole, 0.1 M NaCl, 20% glycerol). The collected Cas9 protein was dialyzed at 4 °C for 12 h, and concentrated by centrifugation (3500g, 15 min) in a 100 kDa ultrafiltration tube. The supernatant was collected and analyzed using 8% SDS-PAGE.Preparation and characterization of QNP/RNP nanocomplexes
First, Cas9 protein and GFP sgRNA were mixed at a molar ratio of 1:1 and incubated at RT for 15 min to obtain RNPGFP nanocomplexes. Then, Qua-PLL, Chol-PEG and RNPGFP at a Qua-PLL/Chol-PEG/Cas9 mass ratio of 5:5:1 were sonicated and self-assembled at RT for 30 min to prepare Qua-PLL/Chol-PEG/RNPGFP nanocomplexes (denoted as QNP/RNPGFP). Similarly, Lipo2000/RNPGFP nanocomplexes were prepared and acted as a positive control. When GFP sgRNA was replaced with PLK1 sgRNA, the RNPPLK1, QNP/RNPPLK1, and Lipo2000/RNPPLK1 nanocomplexes were synthesised. The hydrodynamic diameter and zeta potential of these nanocomplexes were also measured using a Malvern Nano ZS90 Zetasizer (Malvern, UK), and the stability of the nanocomplexes (mass ratio: 5/5/1) was evaluated by monitoring the changes of their hydrodynamic diameter and zeta potential values in PBS at 4 °C for 7 days.
In vitro GFP disruption by QNP/RNPGFP nanoparticles
HeLa–GFP cells were seeded into 6-well plates (2 × 105 cells per well) and incubated overnight, and then free RNPGFP, QNP/RNPGFP and Lipo2000/RNPGFP nanocomplexes with a final Cas9 concentration of 2 μg mL−1 were added. After treatment at 37 °C for 6 h, the cells were treated with a fresh RPMI 1640 medium and incubated for additional 72 h. Finally, the cells were washed with PBS three times and visualized using an IX73P1F fluorescence microscope (Olympus, Tokyo, Japan).Similarly, the HeLa–GFP cells were washed and collected after the 72 h incubation, and then subjected to analysis on a CytoFLEX flow cytometry instrument (Beckman Coulter, IN, USA).
Cell apoptosis
MCF-7 cells were seeded into 6-well plates (2 × 105 cells per well) and incubated at 37 °C for 12 h. Then the cells were treated with free RNPPLK1, QNP/RNPPLK1, Lipo2000/RNPPLK1 or QNP with a final Cas9 concentration of 2 μg mL−1 in DMEM (200 μL) for 72 h. The cells were washed with cold PBS three times and resuspended with 100 μL of 1× binding buffer provided in the detection kit. Then 5 μL of Annexin V-FITC and 5 μL of PI were added to the samples according to the manufacturer's instructions. Finally, the apoptotic cells were detected using the CytoFLEX flow cytometry instrument (Beckman Coulter, IN, USA), and the data were analyzed by CytExpert 2.5.Cell cycle arrest
Cell culture and transfection were conducted as described in the section “Cell apoptosis”. The cells were collected, washed with cold PBS and resuspended with 100 μL of PBS. Then 300 μL of cold anhydrous ethanol was added followed by cell incubation at −20 °C for 1 h. Afterward, the cells were washed and resuspended with PBS (100 μL), and 20 μL of RNase A and 300 μL of PI dye solution were added. Finally, the cells were detected by the CytoFLEX flow cytometry system after incubation in the dark at 4 °C for 1 h.Western blotting
Cell culture and transfection were conducted as described in the section “Cell apoptosis”. Cellular lysates were obtained by the treatment of RIPA lysis buffer and collected by centrifugation at 14000g for 15 min. The proteins were separated by 10% SDS-PAGE and transferred into a polyvinylidene fluoride (PVDF) membrane. The membrane was blocked with 5% non-fat milk at RT for 2 h and incubated with primary antibodies overnight at 4 °C. Then, the membrane was incubated with a horseradish peroxidase (HRP)-labelled secondary antibody for 1 h. Finally, the bands were detected using an enhanced chemiluminescence (ECL) reagent and visualized using a Tanon 2500 imaging system (Shanghai, China).
T7E1 assay
Cell culture and transfection were conducted as described in the section “Cell apoptosis”. Genomic DNA of the samples was extracted using the genomic DNA extraction kit according to the manufacturer's instructions, and the flanking region of the PLK1 gene was amplified by PCR. First, a purified PCR product (200 ng) was diluted in 20 μL of NEBuffer 2 and denatured at 95 °C for 5 min. Then, the solution was reannealed at 95–85 °C (ramp rate: −2 °C s−1) and 85–25 °C (ramp rate: −0.1 °C s−1). While cooling down to 4 °C, 1 μL of T7E1 was added and the solution was incubated at 37 °C for 15 min. Finally, 1.5 μL of 0.25 M EDTA was added to stop the reaction. The digested PCR products were analyzed by electrophoresis with 2% (w/v) agarose gels (120 V, 20 min). The disrupted fragments were imaged using a Universal Hood II system (Bio-Rad, USA) and analyzed by ImageJ software. The gene knockout efficiency was calculated using the following formula: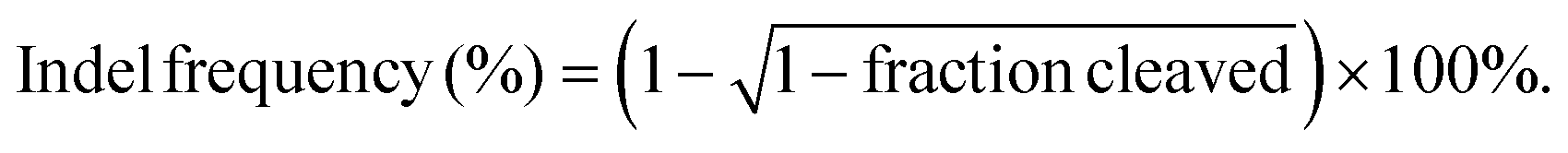
Similarly, the genomic DNA of tumors was extracted, and the T7E1 assay was conducted as described above.
Sanger sequencing assay
The DNA fragment (500 bp) containing the target sequence was ligated into the p-EASY-Blunt vector (TransGen Biotech., Beijing, China) and transformed into E. coli Trans5α. Then, monoclonal colonies were randomly selected for the extraction of plasmids. Using universal primers M13F (5′-GTAAAACGACGGCCAGT-3′) and M13R (5′-CAGGAAACAGCTATGAC-3′), Sanger sequencing of the target fragment was performed using an Applied Biosystems 3730XL DNA analyzer (Thermo Fisher, Waltman, MA, USA) to clarify the mutant sequence, which was conducted at Sangon Biotech. (Shanghai, China).Animals
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Jilin University and approved by the Animal Ethics Committee of Jilin University (approval no. YNPZSY2023001). Female BALB/c nude mice (6 weeks old) were purchased from Vital River Laboratory Animal Technology Co., Ltd (Beijing, China). All mice were raised in a specific pathogen free (SPF) animal laboratory and provided with a standard diet. An MCF-7 cell suspension (5 × 106 cells in 100 μL PBS) was injected into the back of each mouse to obtain MCF-7 bearing nude mice for further studies.Biodistribution analysis
Firstly, RBITC was added to the Cas9 sample at a molar ratio of 3:1 and the mixture was stirred overnight at RT in the dark. The above solution was dialyzed against deionized water for 3 days (MWCO = 3500 Da) and lyophilized to obtain RBITC-labelled Cas9 (RBITC-Cas9). Then RBITC-Cas9 was loaded by QNP to obtain QNP/RBITC-Cas9 nanoparticles. The RBITC-Cas9 (10 mg kg−1, 125 μL) and QNP/RBITC-Cas9 (10 mg kg−1 on RBITC-Cas9 basis, 125 μL) were intravenously administered via the tail vein into the MCF-7 bearing mice. After the administration at 1, 4 or 24 h, the mice were sacrificed. Tumors and major organs (heart, liver, spleen, lungs and kidneys) were collected and ex vivo fluorescence (λex: 579 nm and λem: 595 nm) was observed using the Caliper IVIS Lumina III In Vivo Imaging System (PerkinElmer, USA).
In vivo anti-tumor studies
When the tumor volume reached about 50 mm3, the BALB/c nude mice bearing MCF-7 tumors were randomly divided into 5 groups (n = 5) and intravenously administered with 100 μL of saline, free RNPPLK1 (50 μg on Cas9 basis), QNP/RNPPLK1 (50 μg on Cas9 basis), QNP/RNPScr (50 μg on Cas9 basis) and QNP (125 μg) on days 0, 3, 6, 9, and 12. Body weight and tumor volume were recorded every other day. When the tumor volume of the saline group reached 1200 mm3, all the mice were sacrificed, and tumors and major organs were excised for further analysis. Blood samples were collected for serum biochemistry analysis. The tumor volume was calculated using the following equation:| Tumor volume (Vt, mm3) = a × b2/2 |
H&E staining and TUNEL assay
The harvested tumors and major organs were fixed in formalin and embedded in paraffin. The paraffin-embedded tumors and organs were cut into 5 μm-thick slices that were stained with hematoxylin and eosin (H&E). Similarly, the TUNEL assay was performed using a TUNEL apoptosis assay kit. Images were obtained using an IX73P1F fluorescence microscope (Olympus, Tokyo, Japan).Immunofluorescence analysis
The 5 μm-thick slices were also used for immunofluorescence analysis according to a previous report.35 Significantly, the sections were incubated with an anti-PLK1 (or anti-Ki67) primary antibody overnight at 4 °C and with a Cy5 (or FITC)-labelled secondary antibody for 1 h at RT, whereas the cell nuclei were stained with DAPI. Images were obtained using an IX73P1F fluorescence microscope (Olympus, Tokyo, Japan).Statistical analysis
All experiments were performed at least three times and expressed as mean value ± standard deviation (SD). Data were analyzed for statistical significance using the Student's t-test. *p < 0.05 was considered statistically significant, whereas **p < 0.01 and ***p < 0.001 were considered highly and extremely significant. The abbreviation “ns” means no significance.Results and discussion
Preparation and characterization of Qua-PLL and Chol-PEG
The cationic polymer Qua-PLL was synthesized through the methylation of PLL according to our previous report (Fig. S1A†).22 The appearance of characteristic 1H NMR peaks d (–CH3 of the quaternary ammonium group) and e (–CH2–CH2–C(CH3)) indicated the successful synthesis of Qua-PLL, where the m and n values were measured to be 55 and 35, respectively (Fig. S2†). The amphiphilic Chol-PEG was prepared through the amidation reaction of Chol-COOH and mPEG-NH2 (Fig. S1B†). A 1H NMR spectrum of Chol-PEG showed the characteristic peak of PEG (peak a, –CH2–CH2–O–) and Chol-COOH (peak b, –CH3), demonstrating the successful preparation of Chol-PEG (Fig. S3†).Preparation and characterization of Qua-PLL/Chol-PEG/BSA nanoparticles
The model protein BSA was used to optimize the feeding ratio of Qua-PLL and Chol-PEG. As shown in Fig. 1A, both PLL and Qua-PLL could self-assemble with BSA through electrostatic interaction at a mass ratio of 5:1 to obtain uniform PLL/BSA and Qua-PLL/BSA nanoparticles, respectively. The addition of Chol-PEG did not affect the self-assembly of ternary complexes PLL/Chol-PEG/BSA or Qua-PLL/Chol-PEG/BSA at a mass ratio of 5:5:1. The hydrodynamic diameters of PLL/Chol-PEG/BSA and Qua-PLL/Chol-PEG/BSA were 110.9 ± 16.2 nm and 117.5 ± 9.0 nm, respectively, which were larger than those without Chol-PEG. As shown in Fig. 1B, all four of these nanoparticles showed a net positive charge due to the introduction of positively charged PLL or Qua-PLL. Meanwhile, there were almost no obvious changes in the hydrodynamic diameter and zeta potential of Qua-PLL/Chol-PEG/BSA during the incubation in PBS at 4 °C for 7 days, demonstrating the favourable stability of the nanocomplex (Fig. S4†).
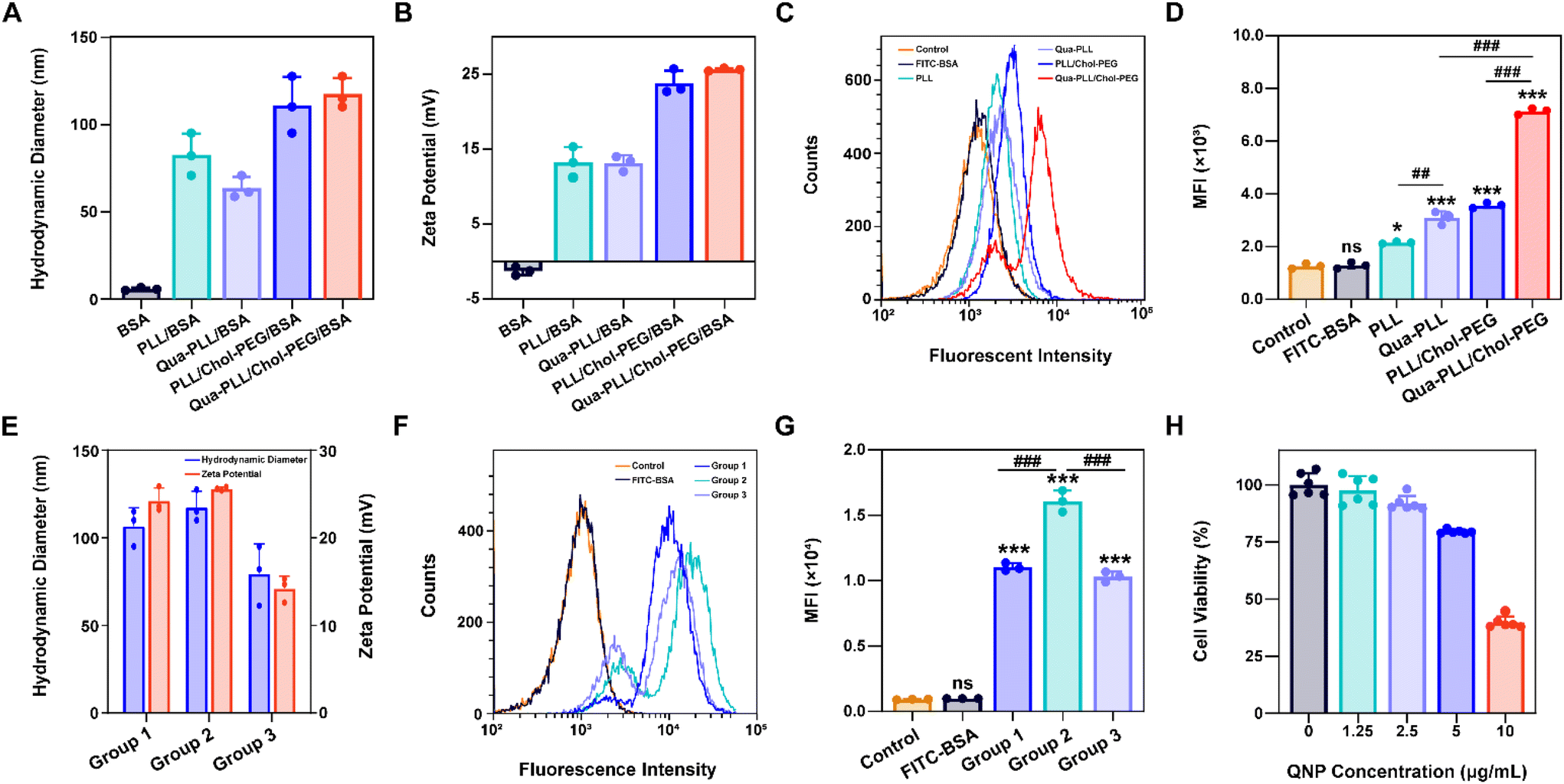 | ||
| Fig. 1 Preparation and characterization of the Qua-PLL/Chol-PEG/BSA nanoparticles. (A) Hydrodynamic diameter and (B) zeta potential of BSA, PLL/BSA, Qua-PLL/BSA, PLL/Chol-PEG/BSA and Qua-PLL/Chol-PEG/BSA. (C) The cellular uptake and (D) mean fluorescence intensity (MFI) of MCF-7 after the treatment with different formulations for 72 h. (E) Hydrodynamic diameter and zeta potential of Qua-PLL/Chol-PEG/BSA at different mass ratios (group 1: 5/2.5/1, group 2: 5/5/1 and group 3: 5/10/1). (F) The cellular uptake and (G) MFI of MCF-7 after the treatment with Qua-PLL/Chol-PEG/BSA at different mass ratios for 72 h. (H) The cell viability of MCF-7 after the treatment with Qua-PLL/Chol-PEG (abbreviated as QNP) for 72 h. Data are presented as mean value ± SD (*p < 0.05, ***p < 0.001 vs. the control group; ##p < 0.01, ###p < 0.001, ns: no significance). | ||
To investigate the in vitro intracellular delivery efficacy, BSA was labelled with FITC to obtain FITC-BSA. After 72 h of treatment, the FITC fluorescence of the FITC-BSA group showed no significant differences compared with the control group, indicating the biomacromolecules could not be delivered into the cells without a delivery system (Fig. 1C and D). As expected, all the components contributed to the intracellular delivery of BSA. However, the fluorescence intensity of the Qua-PLL/BSA or Qua-PLL/Chol-PEG/BSA group was significantly higher than that of PLL/BSA or PLL/Chol-PEG/BSA, and these results demonstrated that the quaternary ammonium-functionalized Qua-PLL was more effective in the intracellular delivery of proteins. Compared with the Qua-PLL/BSA group, the fluorescence intensity of Qua-PLL/Chol-PEG/BSA increased by 2.30 times, signifying the importance of hydrophobic cholesterol in the formation of ternary complexes. The Qua-PLL and Chol-PEG may self-assemble with BSA through electrostatic interaction, π–π stacking and hydrogen bonding, and these multiple interactions were beneficial for the stabilization of the nanoparticles, especially in the in vivo environment. Based on these results, the Qua-PLL/Chol-PEG carrier was selected as the protein delivery system for further studies.
Then the optimal formula of the delivery system was studied by changing the feeding ratio of Qua-PLL to that of Chol-PEG. As shown in Fig. 1E, the ternary complexes of Qua-PLL/Chol-PEG/BSA at a mass ratio of 5:2.5:1 (group 1), 5:5:1 (group 2), and 5:10:1 (group 3) all showed uniform size and net positive charge. However, the fluorescence intensity of group 2 was 1.46-fold and 1.56-fold higher than that of group 1 and group 3, respectively (Fig. 1F and G). For all subsequent studies, the mass ratio of Qua-PLL:Chol-PEG:protein was determined to be 5:5:1. In addition, the biosafety of the Qua-PLL/Chol-PEG delivery system (abbreviated as QNP) was investigated by MTT assay using MCF-7 cells. As shown in Fig. 1H, after their incubation for 72 h, the cell viability was above 75% at a concentration of 5 μg mL−1, which was selected as the maximal concentration in further in vitro experiments.
In vitro GFP disruption by QNP/RNPGFP nanoparticles
Cas9 protein was firstly extracted and purified according to the previously reported protocols,9,36 and the SDS-PAGE image indicated the successful purification of Cas9 from bacterial lysates (Fig. S5†). The reporter gene GFP was chosen, and GFP sgRNA was mixed with Cas9 at a molar ratio of 1:1 to obtain RNPGFP nanocomplexes. The RNPGFP was firstly loaded into a commercial delivery system, Lipo2000, to prepare Lipo2000/RNPGFP nanoparticles to explore genome editing in HeLa–GFP cells, which contained the GFP reporter gene and expressed unstable GFP protein. The in vitro GFP disruption could be detected by fluorescence microscopy and flow cytometry. As shown in Fig. 2A–D, the GFP fluorescence of the Lipo2000/RNPGFP group was significantly lower in comparison with that of the control group after 72 h of treatment, while free RNPGFP could not downregulate the expression of GFP. As ultrasonication was employed to construct Qua-PLL/Chol-PEG/RNPGFP, the effect of ultrasonication on the activity of Cas9 was first studied (Fig. S6†). Clearly, there were no differences between the fluorescence intensity of Lipo2000/RNPGFP groups with or without ultrasonication, implying the ultrasonication did not influence the activity of Cas9.
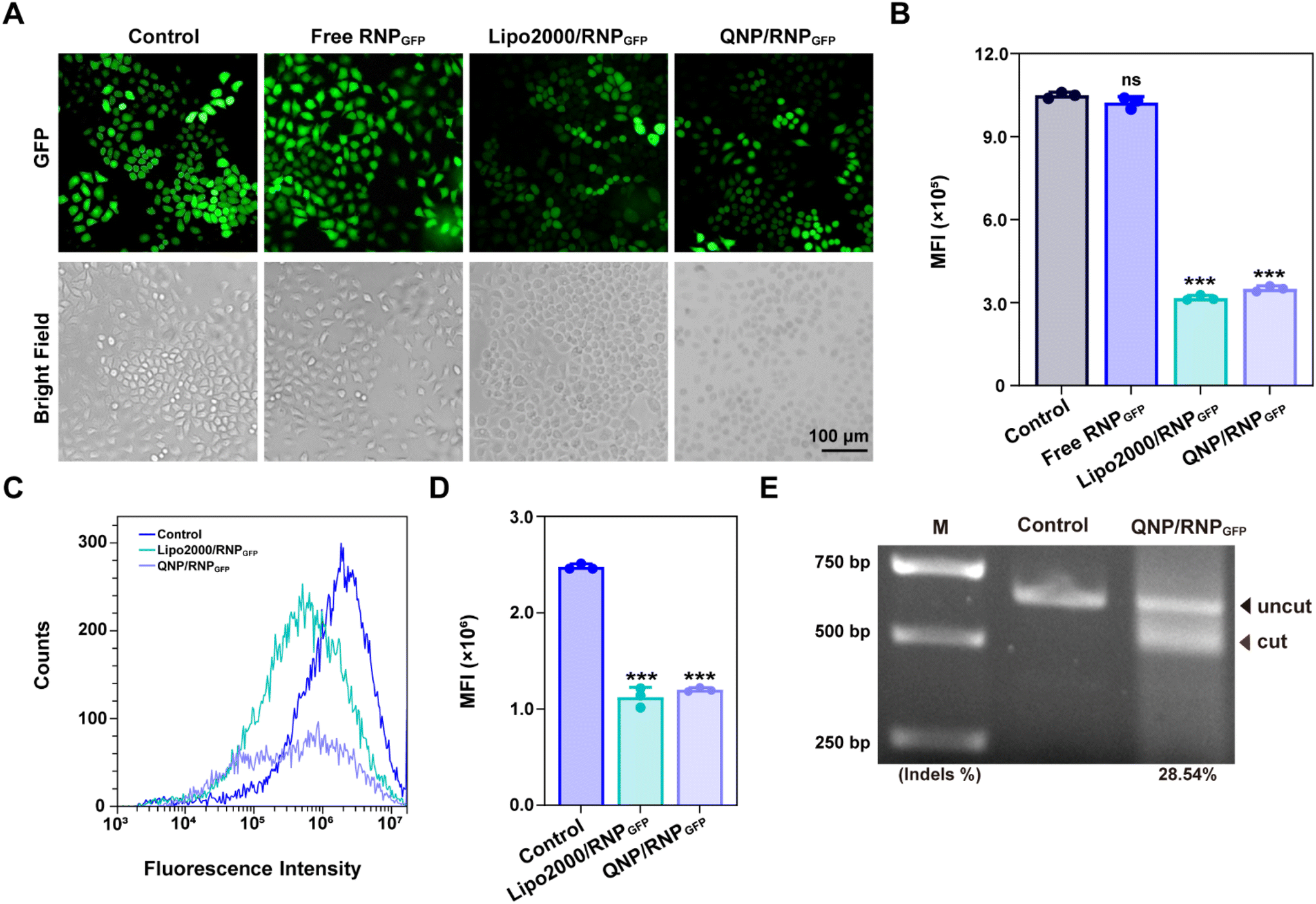 | ||
| Fig. 2 In vitro GFP disruption of HeLa–GFP cells with QNP/RNPGFP nanoparticles. (A) Fluorescence microscopy analysis and (B) quantification of GFP expression in HeLa–GFP cells treated with PBS, free QNP/RNPGFP, Lipo2000/RNPGFP and QNP/RNPGFP. (C) Flow cytometry analysis and (D) quantification of GFP expression in HeLa–GFP cells after the treatment with different formulations for 72 h. (E) T7E1 assay of indels around the PLK1 locus in HeLa–GFP cells treated with PBS and QNP/RNPGFP. Data are presented as mean value ± SD (***p < 0.001 vs. the control group; ns: no significance). | ||
Subsequently, the in vitro genome editing of our QNP delivery system was evaluated in HeLa–GFP cells. As expected, QNP loaded the preformed RNPGFP to form a ternary complex (Qua-PLL/Chol-PEG/RNPGFP, denoted as QNP/RNPGFP). As shown in Fig. 2C and D, QNP/RNPGFP downregulated 51.56% of the GFP protein expression after the incubation for 72 h, which was consistent with the results of fluorescence microscopy (Fig. 2A and B), signifying the importance of the delivery system to increase the cellular uptake of Cas9/sgRNA RNP. In addition, the QNP/RNPGFP complex produced similar genome editing efficiency in comparison with Lipo2000/RNPGFP, further suggesting the advantage of the quaternary ammonium-mediated delivery system. In addition, the frequency of mutation after the treatment with QNP/RNPGFP was investigated using the T7E1 assay. As shown in Fig. 2E, the frequency of insertion–deletion (indels, %) at the targeted GFP locus was 28.54% for QNP/RNPGFP. All these results indicated that QNP/RNPGFP was able to achieve CRISPR/Cas9 genome editing in vitro and avoid off-target effects.
QNP/RNPPLK1 induces efficient therapeutic genome editing in vitro
To explore the potential of therapeutic genome editing, QNP/RNPPLK1 containing PLK1 sgRNA was prepared. PLK1 plays a critical role in carcinogenesis, which has been found to be overexpressed in various tumors.26 Inhibition or depletion of its expression leads to cell cycle arrest and apoptosis of tumor cells, providing a promising strategy for cancer therapy.27,28 Therefore, the PLK1 gene in the MCF-7 cells was selected for targeting, and PLK1 sgRNA was synthesized. The commercial formulation of Lipo2000 was used as a positive control to deliver RNPPLK1. After self-assembly with free RNPPLK1, the hydrodynamic diameter increased from 95.7 ± 14.0 nm (QNP) to 170.9 ± 23.1 nm (QNP/RNPPLK1) (Fig. 3A), whereas the zeta potential of QNP/RNPPLK1 was lower than that of QNP (7.04 ± 2.92 mV vs. 16.73 ± 1.89 mV) due to the net positive charge of free RNPPLK1 (Fig. 3B). Meanwhile, the hydrodynamic diameter and zeta potential values of QNP/RNPPLK1 did not change during the incubation in PBS at 4 °C for 7 days, indicating its good stability (Fig. S4†).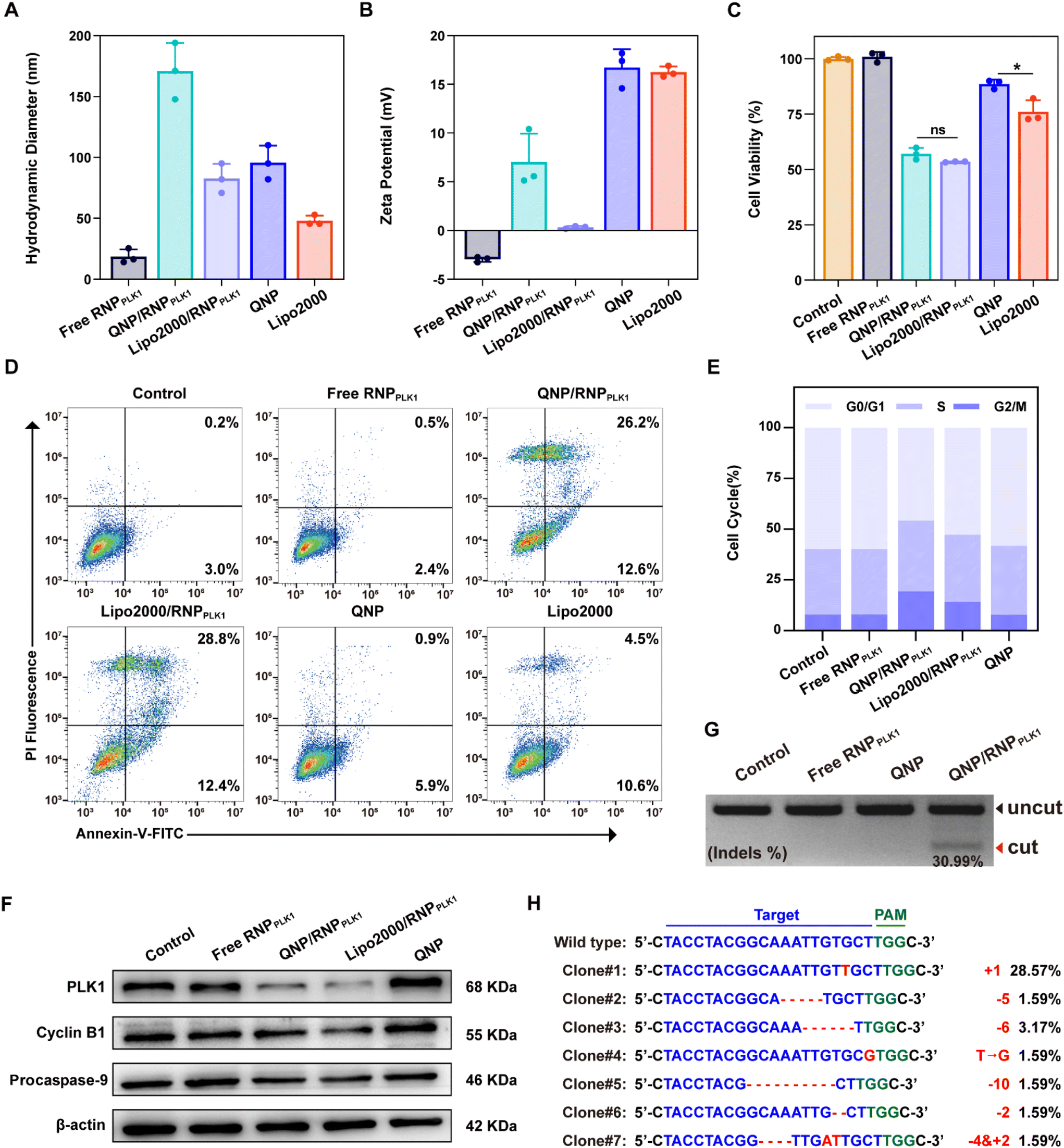 | ||
| Fig. 3 In vitro therapeutic genome editing efficiency of QNP/RNPPLK1. (A) Hydrodynamic diameter and (B) zeta potential of free RNPPLK1, QNP/RNPPLK1, Lipo2000/RNPPLK1, QNP or Lipo2000. (C) Cell viability and (D) apoptosis of MCF-7 cells after the treatment with free RNPPLK1, QNP/RNPPLK1, Lipo2000/RNPPLK1, QNP or Lipo2000. (E) Cell cycle arrest and (F) western blotting analysis of PLK1, cyclin B1 and procaspase-9 in the MCF-7 cells after the treatment with free RNPPLK1, QNP/RNPPLK1, Lipo2000/RNPPLK1, or QNP. (G) T7E1 indels analysis of the PLK1 locus in the MCF-7 cells after the treatment with free RNPPLK1, QNP and QNP/RNPPLK1 for 72 h. (H) Sanger sequencing of the on-target site of the MCF-7 cells treated with QNP/RNPPLK1. Data are presented as mean value ± SD (*p < 0.05; ns: no significance). | ||
The potential of therapeutic genome editing was investigated using the MTT, cell apoptosis, cell cycle arrest and western blotting assays. First, the MTT assay was conducted to evaluate the cell viability of the MCF-7 cells treated with different formulations. As shown in Fig. 3C, no inhibition of cell viability has been observed for the free RNPPLK1 group after the treatment for 72 h, validating that RNPPLK1 could not be delivered into cells to enable the genome editing of the PLK1 gene. Notably, QNP/RNPPLK1 inhibited the proliferation of the MCF-7 cells by 57.05 ± 2.62%, which was comparable to the cell viability of the Lipo2000/RNPPLK1 group. Additionally, the cell viability of QNP was significantly higher than that of Lipo2000 (88.61 ± 2.04% vs. 76.03 ± 5.23%), further suggesting the low cytotoxicity of our QNP delivery system. Moreover, the treatment with QNP/RNPPLK1 in CT26 cells (PLK1 overexpression) could inhibit the cell viability in a concentration-dependent manner, while QNP/RNPPLK1 had no significant influences on the cell viability of HEK-293T owing to its lower expression of the PLK1 gene (Fig. S7†). Furthermore, the cell apoptosis was detected by flow cytometry using Annexin V-FITC/PI staining. As shown in Fig. 3D, QNP/RNPPLK1 and Lipo2000/RNPPLK1 induced 38.8% and 41.2% of the apoptotic rate, respectively, while free RNPPLK1 could not induce the apoptosis as evidenced by inappreciable apoptotic rate. The apoptosis-inducing ability of RNPPLK1 delivery was identified to be related to the activation of the mitochondrial dependent signalling pathway, as evidenced by the decreased level of procaspase 9 (Fig. 3F). Besides, the commercial carrier Lipo2000 showed higher ability to induce apoptosis than QNP. As PLK1 is a key regulator of the cell cycle and its blockade could achieve G2/M phase arrest, the cell cycle arrest was detected for the MCF-7 cells after the treatment with different formulations using flow cytometry (Fig. 3E). After 72 h of treatment, there was no significant G2/M phase cell cycle arrest of the free RNPPLK1 and QNP groups. In contrast, the G2/M phase ratios of QNP/RNPPLK1 and Lipo2000/RNPPLK1 were found to be 19.28% and 14.10%, respectively. The typical G2/M cycle arrest was elucidated to be associated with the decreased expression level of cyclin B1 (Fig. 3F). All these results demonstrated that the QNP delivery system was biocompatible and contributed to the intracellular delivery of RNPPLK1 for cancer therapy, based on the induction of cell apoptosis and cell cycle arrest.
To investigate whether the genome editing occurred at the PLK1 locus, a T7E1 assay was conducted. As shown in Fig. 3G, there was an obvious digestion band in the PLK1 locus of the QNP/RNPPLK1 group (indels %: 30.99%), whereas no cleavage bands were observed with the free RNPPLK1 and QNP groups. Then PLK1 expression was detected using western blotting analysis to verify the on-targeted genome editing (Fig. 3F). Both QNP/RNPPLK1 and Lipo2000/RNPPLK1 significantly downregulated the expression of PLK1 in comparison with the control group. However, free RNPPLK1 and delivery system QNP were unable to inhibit PLK1 expression, which was also evidenced by the inappreciable indels ratio. The highly on-targeted genome editing of QNP/RNPPLK1 was further confirmed by DNA Sanger sequencing (Fig. 3H and S8†). Compared with the control group, there were two or more peaks at the same targeted site of PCR fragments, and the indels ratio was around 40.90%. Notably, QNP/RNPPLK1 induced several mutation sequences, including substitution, insertion and base deletion around PAMs (TGG). Collectively, QNP/RNPPLK1 was capable of inducing efficient therapeutic genome editing in vitro.
In vivo biodistribution analysis
To investigate the in vivo biodistribution of the QNP/protein nanoparticles, BALB/c nude mice bearing MCF-7 tumors were subjected to an ex vivo optical imaging study. First, Cas9 was labelled with fluorescent dye, RBITC, to obtain RBITC-Cas9, and QNP/RBITC-Cas9 was intravenously administered via the tail vein. At predetermined time points (1, 4 and 24 h), the mice were euthanized, and the tumors and normal tissues (heart, liver, spleen, lungs and kidneys) were excised. As shown in Fig. 4A and B, both RBITC-Cas9 and QNP/RBITC-Cas9 showed obvious fluorescence at the tumor sites after 1 h of injection, with no significant differences. However, the fluorescence intensity of QNP/RBITC-Cas9 was 2.04-fold higher than that of RBITC-Cas9 at 4 h (Fig. 4A and C). The fluorescence of QNP/RBITC-Cas9 was weakened over time, but the fluorescence signal of the tumors was still significantly stronger than that of RBITC-Cas9 after 24 h (Fig. 4A and D). In addition, after the intravenous administration of RBITC-Cas9 and QNP/RBITC-Cas9, there was no obviously negligible fluorescence in the liver and kidneys, which was similar to other nano-formulations.37 Owing to the hepatic reticuloendothelial system, it has been reported that most of the administered nanoparticles would be accumulated and sequestered in the liver after the administration.38,39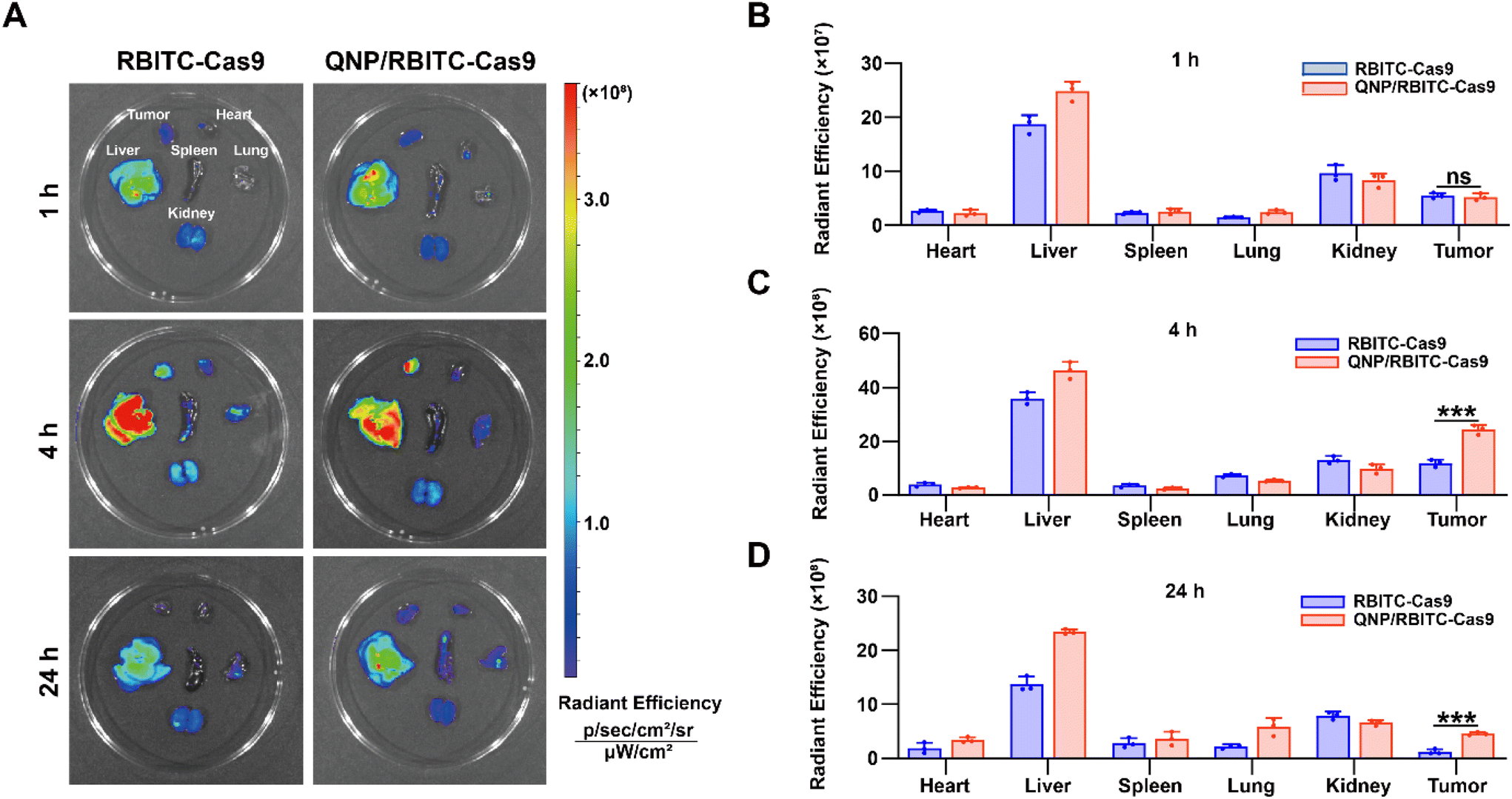 | ||
| Fig. 4 In vivo biodistribution of QNP/RBITC-Cas9. (A) The tumors and major organs were visualized on a Caliper IVIS Lumina III In Vivo Imaging System. The radiant efficiency of the tumors and major organs at (B) 1 h, (C) 4 h, and (D) 24 h after the intravenous injection. Data are presented as mean value ± SD (***p < 0.001; ns: no significance). | ||
QNP/RNPPLK1 induces efficient genome editing in vivo
The efficient in vitro genome editing of QNP/RNPPLK1 and outstanding accumulation at tumor sites encouraged us to study the anti-tumor efficacy in vivo. When the tumor volume reached about 50 mm3, BALB/c nude mice bearing MCF-7 tumors were randomly divided into 5 groups (n = 5) and intravenously administered with saline, free RNPPLK1, QNP/RNPPLK1, QNP/RNPScr and QNP on days 0, 3, 6, 9, and 12 (Fig. 5A). Free RNPPLK1, QNP/RNPScr and the QNP delivery system could not significantly inhibit tumor growth in comparison with the saline group (Fig. 5B). Notably, QNP/RNPPLK1 showed the strongest tumor suppression and the best therapeutic efficacy. On day 28, the mice were euthanized and their tumors were excised, photographed and weighed (Fig. 5C and D). The tumors of the mice in the QNP/RNPPLK1 group were the smallest and the tumor weight was only 0.12 ± 0.03 g. In addition, the tumor weights of free RNPPLK1, QNP/RNPScr and delivery system QNP mice were significantly larger than that of QNP/RNPPLK1 mice. All these results indicated that our QNP delivery system protected RNPPLK1 from degradation and enhanced its anti-tumor efficacy in vivo.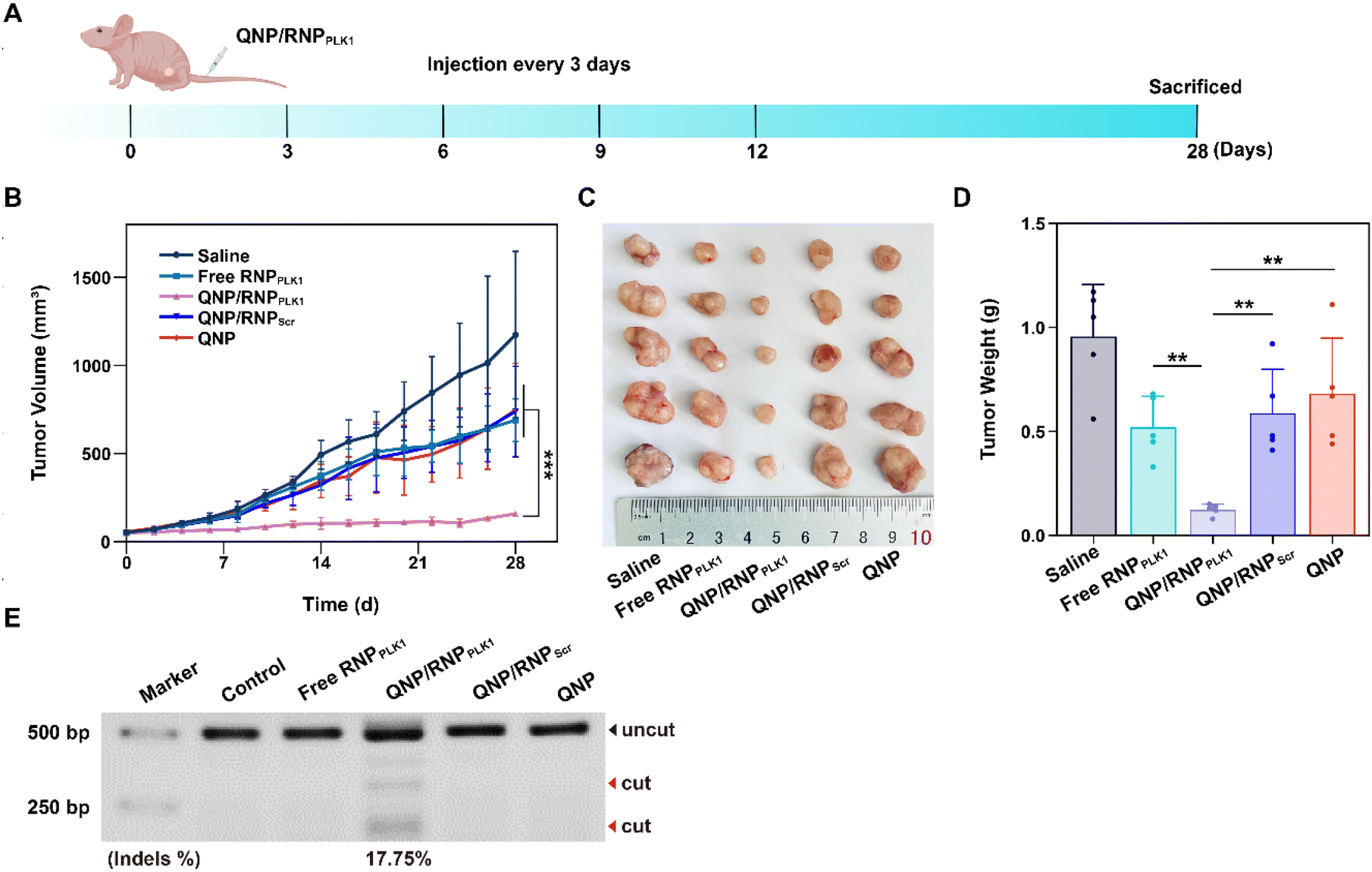 | ||
| Fig. 5 Evaluation of the therapeutic efficiency of QNP/RNPPLK1. (A) MCF-7 tumor-bearing mice were injected with saline, free RNP, QNP/RNPPLK1, QNP/RNPScr and QNP via the tail vein on days 0, 3, 6, 9 and 12. (B) The changes of the tumor volume within 28 days after the intravenous administration. (C) The images and (D) average weight of the tumors excised from the mice with different treatments. (E) T7E1 indels analysis of the PLK1 locus in the tumors with different treatments. Data are presented as mean value ± SD (**p < 0.01 and ***p < 0.001). | ||
To further assess the genome editing efficiency of QNP/RNPPLK1, a T7E1 assay of tumor tissues was performed. As shown in Fig. 5E, free RNPPLK1, QNP/RNPScr and QNP could not achieve obvious genome editing efficiency with no digestion bands. In contrast, there were two cleavage bands after the QNP/RNPPLK1 treatment, and the indels ratio was calculated to be 17.75%. Meanwhile, the expression of PLK1 was visualized using immunofluorescence staining (Fig. 6). As expected, the saline group showed strong red fluorescence owing to the overexpression of PLK1, which was consistent with other reports.36,40 The fluorescence intensity of PLK1 significantly decreased in the QNP/RNPPLK1 group due to the successful genome editing of the PLK1 locus. As the overexpression of PLK1 was related to the tumorigenesis, the remarkably lower expression of PLK1 in the QNP/RNPPLK1 group was a key reason for obtaining excellent anti-tumor efficacy.
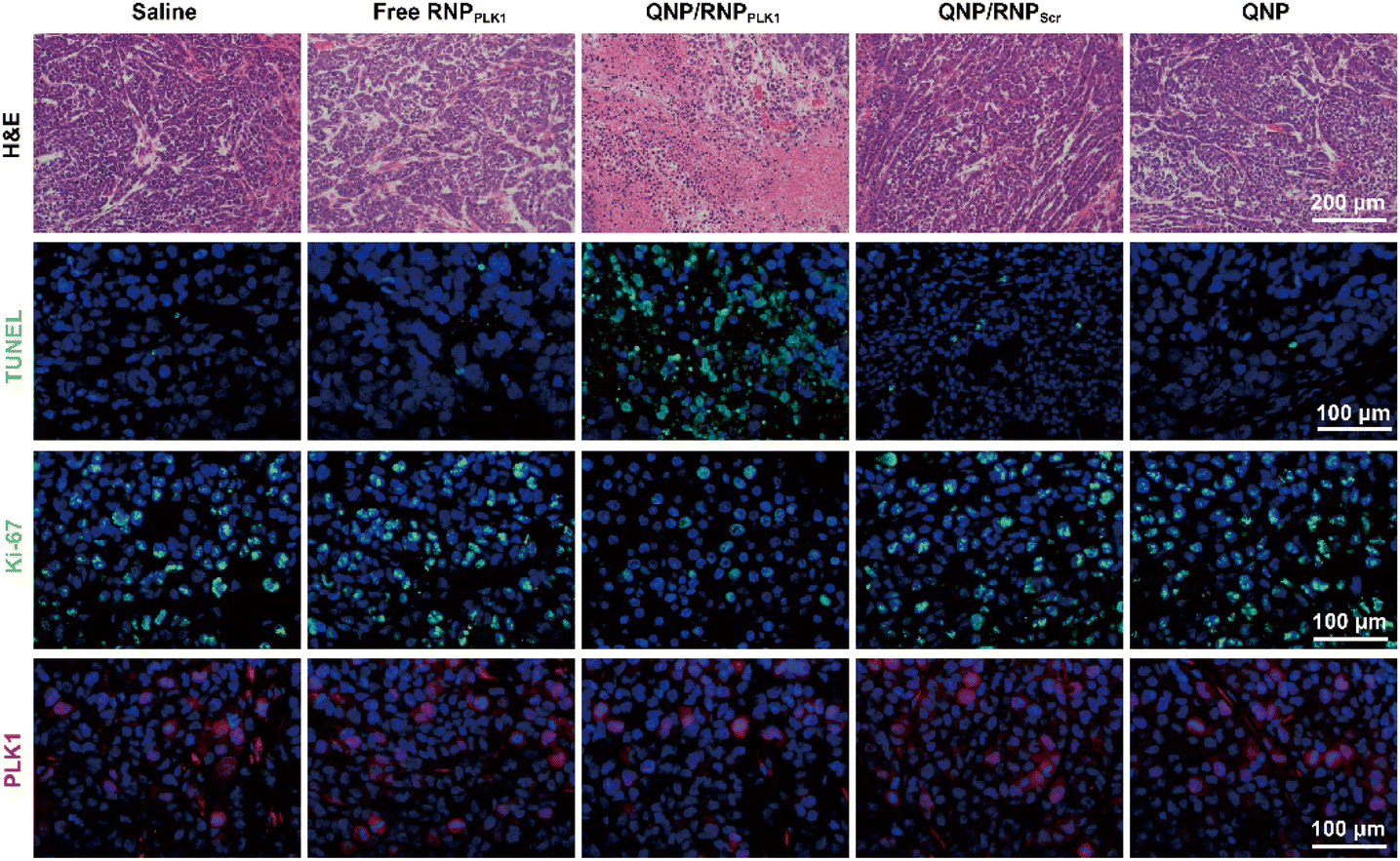 | ||
| Fig. 6 H&E, TUNEL, Ki-67 and PLK1 immunofluorescence staining of the tumor sections after the treatment of the different groups. Scale bar: 100 μm. | ||
On day 28, the tumors were excised and subjected to H&E staining (Fig. 6). As expected, free RNPPLK1, QNP/RNPScr and QNP could not induce obvious necrosis in tumor tissues. However, there was significant necrosis in the QNP/RNPPLK1 group compared with the saline group, which was consistent with the results of tumor weight. Additionally, the tumor tissues were processed with TUNEL staining to visualize the apoptotic cells, which were stained with green fluorescence. As shown in Fig. 6, the QNP/RNPPLK1 group showed strong green fluorescence, while the fluorescence intensities of the other groups were significantly lower. The tumors were also employed for Ki-67 immunofluorescence staining, in which the proliferative cells were stained green (Fig. 6). Compared with other groups, the QNP/RNPPLK1 group exhibited the lowest green fluorescence. These results indicated that QNP/RNPPLK1 could induce the apoptosis of tumor cells and inhibit the proliferation of tumor cells, thereby achieving favourable in vivo anti-tumor efficacy.
Biocompatibility analysis of QNP/RNPPLK1
Finally, the biocompatibility of QNP/RNPPLK1 was evaluated. As shown in Fig. S9,† there were no significant changes of body weight after the treatment with different formulations. Furthermore, the H&E staining of normal tissues (heart, liver, spleen, lungs and kidneys) was conducted on day 28, and no obvious abnormality could be observed (Fig. 7A). In addition, on day 28, the serum was collected for biochemical analysis. As shown in Fig. 7B, the levels of ALT, AST, CREA and BUN were all within the normal ranges, indicating that there was no liver and kidney damage for these groups. These results suggest that our QNP delivery system and genome editing tool QNP/RNPPLK1 are of favourable biocompatibility and thus hold great potential in in vivo therapeutic applications.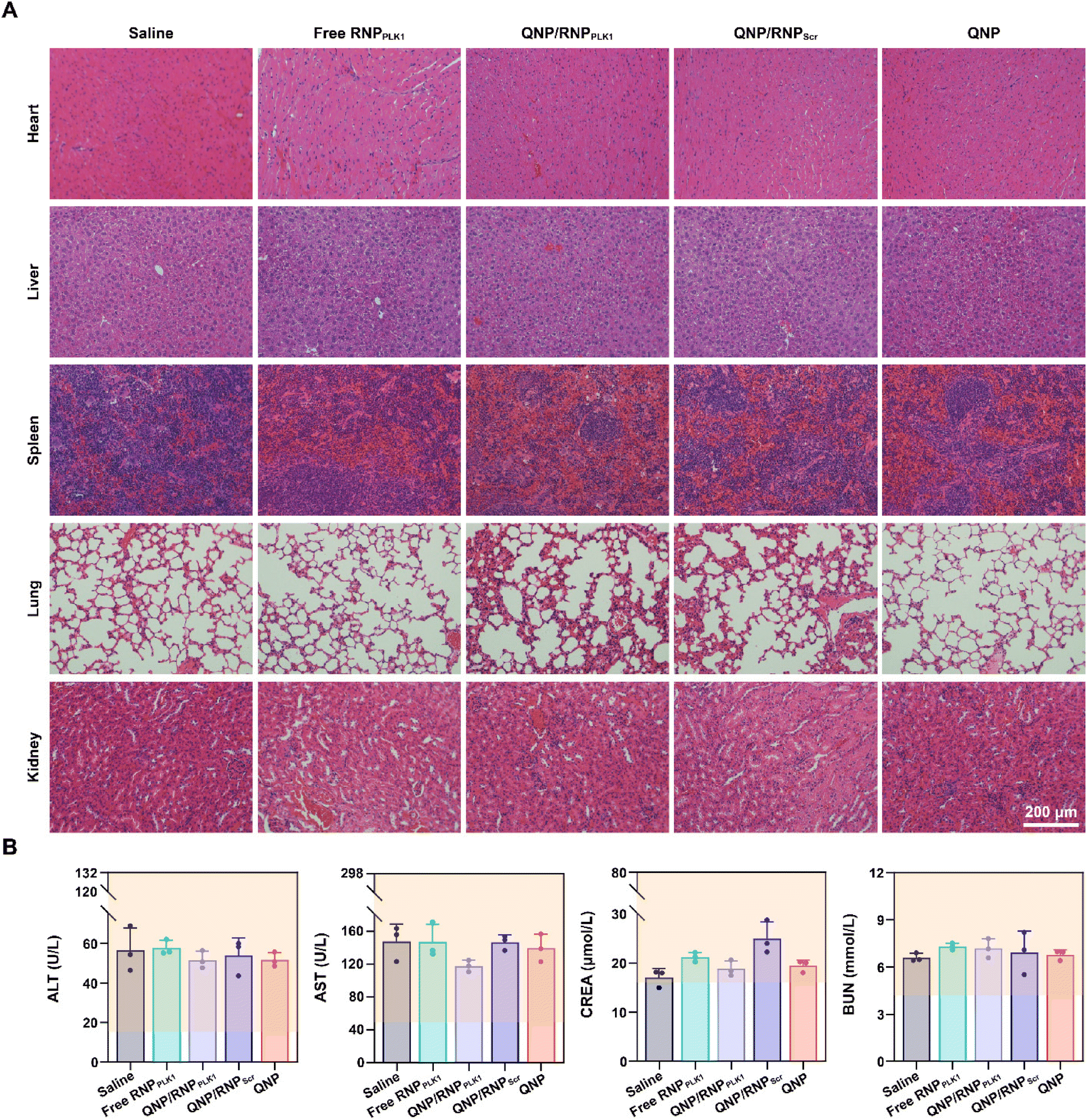 | ||
| Fig. 7 Biocompatibility analysis of QNP/RNPPLK1. (A) H&E staining of the heart, liver, spleen, lungs and kidneys. (B) Blood biochemical analysis of the mice with different treatments. | ||
Conclusions
In summary, the QNP carrier was developed for the delivery of the CRISPR/Cas9 RNP in tumor therapy. The RNP was prepared from the Cas9 ribonucleoprotein and required sgRNA (target GFP or PLK1 gene), and the QNP carrier endowed the nanocomplex QNP/RNP with efficient genome editing efficiency. Compared with free RNPPLK1, QNP/RNPPLK1 induced significant inhibition of cell proliferation, cell apoptosis and cell cycle arrest, attributed to the remarkable downregulation of PLK1 expression through efficient genome editing. Furthermore, QNP/RNPPLK1 effectively inhibited tumor progression and exerted high therapeutic potential in MCF-7 xenograft mouse models. Benefiting from its outstanding genome editing efficiency and biocompatibility, the delivery carrier provided a promising alternative tool for cancer therapy based on CRISPR/Cas9 genome editing.Author contributions
Mengzhu Zhang: data curation, conceptualization, investigation, formal analysis, methodology, project administration, resources, and validation; Siyu Sun: data curation, formal analysis, methodology, resources, software, validation, visualization, and writing – original draft; Xiao Liang: formal analysis, investigation, methodology, software, and project administration; Zengguang Liu: methodology, software, and validation; Jiaxin Yin: methodology, project administration, and visualization; Quanshun Li: conceptualization, funding acquisition, project administration, supervision, and writing – review & editing; Shengcai Yang: conceptualization, funding acquisition, investigation, project administration, supervision, writing – original draft, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (52203168, 32271319 and 32071267), the Development and Reform Commission of Jilin Province (2023C015), the Science and Technology Department of Jilin Province (YDZJ202301ZYTS537), the “Medicine + X” Cross-Innovation Team of Bethune Medical Department of Jilin University “Leading the Charge with Open Competition” Construction Project (2022JBGS04) and the Fundamental Special Research Funds for the Central Universities (2023-JCXK-25).References
- R. L. Siegel, K. D. Miller, N. S. Wagle and A. Jemal, CA Cancer J. Clin., 2023, 73, 17–48 CrossRef PubMed.
- D. L. Puhl, D. Mohanraj, D. W. Nelson and R. J. Gilbert, Adv. Drug Delivery Rev., 2022, 183, 114161 CrossRef CAS PubMed.
- Y. Chen and Y. Zhang, Adv. Sci., 2018, 5, 1700964 CrossRef PubMed.
- M. Chen, A. Mao, M. Xu, Q. Weng, J. Mao and J. Ji, Cancer Lett., 2019, 447, 48–55 CrossRef CAS PubMed.
- H. Bloomer, J. Khirallah, Y. Li and Q. Xu, Adv. Drug Delivery Rev., 2022, 181, 114087 CrossRef CAS PubMed.
- P. Yang, A. Y.-T. Lee, J. Xue, S.-J. Chou, C. Lee, P. Tseng, T. X. Zhang, Y. Zhu, J. Lee, S.-H. Chiou and H.-R. Tseng, Nano Today, 2022, 44, 101482 CrossRef CAS.
- Z. Iqbal, K. Rehman, J. Xia, M. Shabbir, M. Zaman, Y. Liang and L. Duan, Biomater. Sci., 2023, 11, 3762–3783 RSC.
- Q. Cheng, T. Wei, L. Farbiak, L. T. Johnson, S. A. Dilliard and D. J. Siegwart, Nat. Nanotechnol., 2020, 15, 313–320 CrossRef CAS PubMed.
- S. Y. Chae, E. Jeong, S. Kang, Y. Yim, J. S. Kim and D. H. Min, J. Controlled Release, 2022, 345, 108–119 CrossRef CAS PubMed.
- Y. Liang, W. Han, C. Xu, J. Wang, J. Zhang, J. An, W. Liu, J. Liu, Z. Zhang, J. Shi and K. Zhang, Adv. Funct. Mater., 2023, 33, 2301256 CrossRef CAS.
- K. Yi, H. Kong, Y. H. Lao, D. Li, R. L. Mintz, T. Fang, G. Chen, Y. Tao, M. Li and J. Ding, Adv. Mater., 2023, e2300665 CrossRef PubMed.
- Y. Li, J. Bolinger, Y. Yu, Z. Glass, N. Shi, L. Yang, M. Wang and Q. Xu, Biomater. Sci., 2019, 7, 596–606 RSC.
- W. Cai, T. Luo, L. Mao and M. Wang, Angew. Chem., Int. Ed., 2021, 60, 8596–8606 CrossRef CAS PubMed.
- T. Taharabaru, T. Kihara, A. Obata, R. Onodera, Y. Wen, J. Li, K. Motoyama and T. Higashi, Carbohydr. Polym., 2024, 323, 121443 CrossRef CAS PubMed.
- S. Zhang, J. Shen, D. Li and Y. Cheng, Theranostics, 2021, 11, 614–648 CrossRef CAS PubMed.
- T. Wu, Y. Cao, Q. Liu, X. Wu, Y. Shang, J. Piao, Y. Li, Y. Dong, D. Liu, H. Wang, J. Liu and B. Ding, J. Am. Chem. Soc., 2022, 144, 6575–6582 CrossRef CAS PubMed.
- T. Wei, Q. Cheng, Y. L. Min, E. N. Olson and D. J. Siegwart, Nat. Commun., 2020, 11, 3232 CrossRef CAS PubMed.
- Y. Lu, F. Wu, Y. Xu, C. He, S. Luo and X. Sun, J. Controlled Release, 2023, 354, 57–68 CrossRef CAS PubMed.
- M. Zhu, X. Wang, R. Xie, Y. Wang, X. Xu, J. Burger and S. Gong, ACS Appl. Mater. Interfaces, 2023, 15, 10464–10476 CrossRef CAS PubMed.
- X. Xu, C. Liu, Y. Wang, O. Koivisto, J. Zhou, Y. Shu and H. Zhang, Adv. Drug Delivery Rev., 2021, 176, 113891 CrossRef CAS PubMed.
- Q. Liu, J. Cai, Y. Zheng, Y. Tan, Y. Wang, Z. Zhang, C. Zheng, Y. Zhao, C. Liu, Y. An, C. Jiang, L. Shi, C. Kang and Y. Liu, Nano Lett., 2019, 19, 7662–7672 CrossRef CAS PubMed.
- S. Yang, J. Leong, Y. Wang, R. Sim, K. H. Tan, Y. H. Chua, N. Tan, A. L. Z. Lee, J. Tay and Y. Y. Yang, J. Controlled Release, 2022, 345, 464–474 CrossRef CAS PubMed.
- S. Asayama, K. Nagashima, Y. Negishi and H. Kawakami, Bioconjugate Chem., 2018, 29, 67–73 CrossRef CAS PubMed.
- H. Park, A. Otte and K. Park, J. Controlled Release, 2022, 342, 53–65 CrossRef CAS PubMed.
- P. C. Ke, S. Lin, W. J. Parak, T. P. Davis and F. Caruso, ACS Nano, 2017, 11, 11773–11776 CrossRef CAS PubMed.
- Y. Degenhardt and T. Lampkin, Clin. Cancer Res., 2010, 16, 384–389 CrossRef CAS PubMed.
- S. Su, G. Chhabra, C. K. Singh, M. A. Ndiaye and N. Ahmad, Transl. Oncol., 2022, 16, 101332 CrossRef CAS PubMed.
- J. Li, S. Ohmura, A. Marchetto, M. F. Orth, R. Imle, M. Dallmayer, J. Musa, M. M. L. Knott, T. L. B. Holting, S. Stein, C. M. Funk, A. Sastre, J. Alonso, F. Bestvater, M. Kasan, L. Romero-Perez, W. Hartmann, A. Ranft, A. Banito, U. Dirksen, T. Kirchner, F. Cidre-Aranaz and T. G. P. Grunewald, Nat. Commun., 2021, 12, 5356 CrossRef CAS PubMed.
- M. Shahidi, O. Abazari, P. Dayati, J. Z. Reza, M. H. Modarressi, D. Tofighi, B. F. Haghiralsadat and F. Oroojalian, Nanomedicine, 2023, 18, 259–277 CrossRef CAS PubMed.
- D. Jain and A. K. Yadav, Drug Delivery Transl. Res., 2023, 13, 1730–1744 CrossRef CAS PubMed.
- Y. Yang, H. Ning, T. Xia, J. Du, W. Sun, J. Fan and X. Peng, Adv. Mater., 2023, 35, e2301409 CrossRef PubMed.
- P. Wang, L. Zhang, Y. Xie, N. Wang, R. Tang, W. Zheng and X. Jiang, Adv. Sci., 2017, 4, 1700175 CrossRef PubMed.
- P. Pan, X. Liu, M. Fang, S. Yang, Y. Zhang, M. Li and Y. Liu, Pharmaceutics, 2023, 15, 2756 CrossRef PubMed.
- S. Yang, Y. Wang, J. Tan, J. Y. Teo, K. H. Tan and Y. Y. Yang, Adv. Healthcare Mater., 2022, 11, 2101770 CrossRef CAS PubMed.
- S. Yang, Z. Tang, C. Hu, D. Zhang, N. Shen, H. Yu and X. Chen, Adv. Mater., 2019, 31, e1805955 CrossRef PubMed.
- F. Li, N. Song, Y. Dong, S. Li, L. Li, Y. Liu, Z. Li and D. Yang, Angew. Chem., Int. Ed., 2022, 61, e202116569 CrossRef CAS PubMed.
- L. He, W. Xu, X. Wang, C. Wang, J. Ding and X. Chen, Biomater. Sci., 2018, 6, 1433–1444 RSC.
- Y. N. Zhang, W. Poon, A. J. Tavares, I. D. McGilvray and W. C. W. Chan, J. Controlled Release, 2016, 240, 332–348 CrossRef CAS PubMed.
- R. Pattipeiluhu, G. Arias-Alpizar, G. Basha, K. Y. T. Chan, J. Bussmann, T. H. Sharp, M. A. Moradi, N. Sommerdijk, E. N. Harris, P. R. Cullis, A. Kros, D. Witzigmann and F. Campbell, Adv. Mater., 2022, e2201095 CrossRef PubMed.
- C. Li, T. Yang, Y. Weng, M. Zhang, D. Zhao, S. Guo, B. Hu, W. Shao, X. Wang, A. Hussain, X. J. Liang and Y. Huang, Bioact. Mater., 2022, 9, 590–601 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3bm01629c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |