
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Engineered human H-chain ferritin with reversed charge of the internal cavity exhibits RNA-mediated spongelike effect for loading RNA/DNA-binding molecules†
Marketa
Charousova‡
a,
Marie
Kudlickova Peskova‡
 b,
Paulina
Takacsova
a,
Katerina
Kapolkova
a,
Yazan
Haddad
a,
Jan
Bilek
a,
Ladislav
Sivak
a,
Tomas
Bartejs
b,
Zbynek
Heger
*a and
Vladimir
Pekarik
*ab
b,
Paulina
Takacsova
a,
Katerina
Kapolkova
a,
Yazan
Haddad
a,
Jan
Bilek
a,
Ladislav
Sivak
a,
Tomas
Bartejs
b,
Zbynek
Heger
*a and
Vladimir
Pekarik
*ab
aDepartment of Chemistry and Biochemistry, Mendel University in Brno, Zemedelska 1, Brno CZ-613 00, Czechia. E-mail: vladimir.pekarik@mendelu.cz
bCentral European Institute of Technology, Masaryk University, Kamenice 5, Brno CZ-625 00, Czechia. E-mail: zbynek.heger@mendelu.cz; Fax: +420-5-4513-3285; Tel: +420-5-4513-3350
First published on 5th January 2024
Abstract
Ferritins are globular proteins with an internal cavity that enables the encapsulation of a plethora of low-mass compounds. Unfortunately, the overall negative surface charge of ferritin's internal cavity hampers efficient loading of negatively charged molecules. Therefore, we produced a genetically engineered human H-chain ferritin containing a cationic RKRK domain, reversing the natural net charge of the cavity to positive, thus allowing for efficient encapsulation of negatively charged siRNA. Due to the reversed, positive charge mediated by RKRK domains, the recombinant ferritin produced in E. coli inherently carries a load of bacterial RNA inside its cavity, turning the protein into an effective sponge possessing high affinity for DNA/RNA-binding substances that can be loaded with markedly higher efficiency compared to the wildtype protein. Using doxorubicin as payload, we show that due to its loading through the RNA sponge, doxorubicin is released in a sustained manner, with a cytotoxicity profile similar to the free drug. In summary, this is the first report demonstrating a ferritin/nucleic acid hybrid delivery vehicle with a broad spectrum of properties exploitable in various fields of biomedical applications.
1. Introduction
Ferritins are iron-sequestering proteins widely spread throughout nature and expressed in nearly all living organisms, from archaea and bacteria and to plants and mammals. Ferritins possess a unique hollow globular structure adapted for storing mineralized iron deposits, sequestering the iron ions from the cellular environment and protecting biointegrity by suppressing the detrimental Fenton reaction, which can damage the cell by triggering oxidative stress.1It has been shown over the years that the hollow internal cavity of ferritins can be utilized to encapsulate therapeutic agents, dyes or nanoparticles, which can be then effectively delivered to a variety of cell types. To date, many examples of ferritin-mediated delivery of anticancer drugs such as doxorubicin (Dox),2 cisplatin,3 gefitinib,4 and many others have been published. Moreover, ferritins can be also employed for delivery of cell-protecting compounds, such as Prussian blue,5 reactive oxygen species–scavenging nanoceria,6 or curcumin.7 In addition, dye-loaded ferritins have been successfully utilized for multimodal imaging8 or encapsulation of gadolinium chelate to develop an efficient magnetic resonance imaging contrast agent.9 In combination with Dox, ferritins’ ability to accumulate large quantities of iron has been exploited to trigger cellular ferroptosis, a type of cell death caused by selective iron poisoning.10 Last but not least, a broad spectrum of metal species has been encapsulated inside the ferritin nanocage. These include gold,11–13 silver,14 and palladium15,16 nanoparticles or bimetallic complexes, such as those formed by palladium and iridium,17 to name few.
Despite the efficient entrapment of small molecules or ions into the ferritin cavity, the encapsulation of macromolecules remains problematic. Of particular interest might be the encapsulation of small noncoding RNAs that could be thus selectively delivered to desired locations, mainly tumours or even to the brain, due to H-chain ferritins’ ability to cross the blood–brain barrier.18 To the best of our knowledge, only a few reports suggested that ferritin might be used for such purposes. Among them, Li et al. exploited a reversible pH-driven disassembly-reassembly procedure to encapsulate anti-insulin receptor siRNA into the ferritin cavity.19 Another published approach utilized genetically modified ferritin with an inverted, positively charged, amino acid-rich C-terminus containing a peptide derived from protamine, linked to the ferritin core with a cathepsin cleavage site and directed toward the external surface of ferritin.20 Recently, Yuan and coworkers designed ferritin with mutated arginine residues on the luminal surface to allow active loading with siRNA and confirmed the ability of H-ferritin to traverse the blood–brain barrier in vitro.21 In another interesting work, a mutated ferritin was used to deliver TLR-activating nucleic acids, which increased immune activation.22 An alternative approach to protein engineering is chemical surface modification. Pediconi et al. modified the ferritin surface with piperazine moieties to improve the affinity of humanized AfuFt for nucleic acids.23 The system was effective for the delivery of siRNAs to various cell lines. Despite these efforts, the efficient encapsulation and intracellular delivery of siRNA by ferritins remains a challenge.
To address this challenge, we present a genetically engineered human H-chain ferritin (hereinafter referred to as HsaFtH-RK). The HsaFtH engineering was based on the knowledge that each of the 24 monomeric units forming the ferritin supramolecular complex is composed of four aligned α-helices (A–D) and an E-helix directed toward the internal cavity terminated by nine unstructured amino acids.24 The E-helix has been previously utilized as a site for the insertion of an immunogenic epitope (Epstein–Barr nuclear antigen 1) without compromising the folding properties of ferritin.25 Therefore, in this work, we replaced the six terminal predominantly negatively charged amino acids with a positively charged RKRK amino acid cluster. We were surprised to find that during the self-assembly process in E. coli, the engineered HsaFtH-RK is capable of loading bacterial RNA. We show that the loaded RNA molecules can be subsequently utilized as an affinity matrix to bind positively charged or nucleic acid–binding compounds. In addition, we demonstrate that the bacterial RNA can be replaced with other exogenously added small nucleic acids (siRNA, LNA or DNA oligonucleotides) during the pH-mediated reversible disassembly-reassembly process. Taken together, HsaFtH-RK presents a novel versatile nanodelivery biomaterial tool with promising properties towards improved delivery of nucleic acids and anticancer compounds that can benefit from the intrinsic spongelike effect of the RNA affinity matrix due to the reversed cationic charge of the engineered E-helix terminal amino acid cluster.
2. Materials and methods
2.1. Chemicals and material
The recombinant wildtype (wt) HsaFtH expression plasmid was a kind gift from Yi Yuan, Beijing, China. If not stated otherwise, all reagents were purchased from Merck (Kenilworth, NJ, USA). Solvents were from Penta Chemicals (Prague, Czech Republic).2.2. Cell cultures
Cell-culture experiments were performed using HEK-293 (human embryonic kidney) and MDA-MB-468 (triple-negative breast cancer) cells grown in a complete medium [DMEM/F12 or DMEM (Sigma-Aldrich, St Louis, MO, USA) supplemented with 10% fetal bovine serum, 2 mM L-glutamine or 1× GlutaMAX™ and 1× Penicillin/Streptomycin] at 37 °C in a humidified 5% CO2 atmosphere. For experiments with anti-EGFP siRNA, we prepared HEK-293 cells stably expressing destabilized EGFP (d2EGFP). For this purpose, HEK-293 cells were transfected with pd2EGFP-N1 plasmid (Addgene, Watertown, MA, USA) using polyethyleneimine (PEI, Polysciences, Warrington, PA, USA). The transfection was carried out with 1.25 μg of DNA and 2 μg of PEI applied in one well of a 24-well-plate in OptiMEM medium (Thermo-Fisher Scientific, Waltham, MA, USA) and cultured overnight, followed by medium exchange. After 48 h, the confluent cells were trypsinized (1× Trypsin-EDTA solution containing 0.5 g porcine trypsin and 0.2 g EDTA) (Sigma-Aldrich) and seeded as a single-cell suspension to ∼10% confluency in HEK-293 conditioned media (conditioning was done for at least 2 days). Then, the cells were allowed to form colonies with around 100 cells, and d2EGFP-positive colonies were picked using a 10 μL tip under an Olympus IX53 fluorescence microscope (Olympus, Tokyo, Japan), then reseeded at one colony per well in a 24-well plate. After reaching ∼60–70% confluency, the colony with the highest distribution of d2EGFP+ cells was trypsinized and seeded as a single-cell suspension to approximately ∼10% confluency in HEK-293 conditioned media. The production and picking of d2EGFP+ colonies continued as described for 3 months until a population of HEK-293 cells stably expressing d2EGFP was obtained. Subsequently, d2EGFP+ HEK-293 cells were grown in the presence of 200 μg mL−1 of G418 (Sigma-Aldrich) to prevent the exclusion of d2EGFP expression.2.3. Molecular cloning
The ferritin open reading frame (ORF) was amplified from a plasmid containing HsaFtH13 with the following primers: forward primer, atatccatgGGCacgaccgcgtccacctcgcaggtgc, which is complementary to the beginning of the ORF and contains the NcoI site preceding the start codon. The reverse primer sequence was atatggatccttaTTTGCGTTTACGtcccagggtgtgcttgtcaaagag, introducing the coding sequence for the RKRK domain, followed by a stop codon, and the BamHI restriction site. The ORF was amplified by polymerase chain reaction (PCR) with proofreading Phusion DNA polymerase (New England Biolabs, Ipswich, MA, USA). Purified PCR product was digested with NcoI and BamHI and cloned into pET-28a(+) plasmid (Addgene) cleaved by the same enzymes. The plasmid was subsequently propagated in XL10-Gold E. coli, and the sequence integrity was verified by sequencing of the entire ORF. The design of the forward primer introduces an additional codon following the translational start and coding for an extra glycine.2.4. Recombinant ferritin production
The HsaFtH-RK was produced in the BL21-CodonPlus (DE3)-RIL E. coli strain (Agilent, Santa Clara, CA, USA). Bacteria were incubated in standard LB medium (10 g L−1 bacto-tryptone, 10 g L−1 NaCl, 5 g L−1 yeast extract) supplemented with kanamycin and chloramphenicol (50 μg mL−1 and 34 μg mL−1, respectively) overnight at 37 °C. Then, bacteria were collected by centrifugation and inoculated in fresh LB medium with kanamycin and chloramphenicol (50 μg mL−1 and 34 μg mL−1, respectively). After an hour of incubation, the bacteria were induced with 0.5 mM isopropyl β-D1-thiogalactopyranoside (Sigma-Aldrich) and cultured for an additional 4 h. Induced bacteria were collected by centrifugation and resuspended in ferritin lysis buffer (FLB; 25 mM ½ Na HEPES, 150 mM NaCl ). Bacteria were dispersed by sonication with Q700 sonicator (QSonica, Newtown, CT, USA), centrifuged, and the cleared lysate was transferred to a new tube. Mesophilic proteins were denatured with 10 min heating at 70 °C. The lysate was cooled on ice, and denatured proteins were removed by centrifugation. The clear supernatant was transferred to a new tube and treated with DNaseI (10 μg mL−1) at 37 °C overnight to remove bacterial DNA. DNaseI and residual proteins were removed from the sample by a second heat denaturation at 85 °C per 10 min, followed by cooling and centrifugation. Ammonium sulfate precipitation of ferritins was done in two steps. At the first step, a suboptimal amount of (NH4)2SO4 was used to precipitate undesired proteins that were subsequently removed by centrifugation. The cleared supernatant was mixed with additional (NH4)2SO4 to precipitate ferritin at a final concentration of ammonium sulfate of 400 mg mL−1. The precipitate was collected by centrifugation. The isolated protein precipitate was washed three times with ammonium sulfate (400 mg mL−1 in FLB), dissolved in FLB, and finally desalted by size-exclusion chromatography (SEC) using PD MiniTrap G-25 Sephadex columns (GE Healthcare, Wauwatosa, WI, USA). Importantly, the washing step using ammonium sulphate was found to substantially reduce the amount of endotoxins from recombinant production. Finally, protein concentration was quantified by the standard Bradford protein assay (Bio-Rad, Hercules, CA, USA) using bovine serum albumin as standard. The morphology and size distribution of recombinant ferritins were investigated using transmission electron microscopy (TEM, FEI Tecnai F20, FEI, Eindhoven, Netherlands) and dynamic light scattering (DLS, Zeta Sizer Nano, Malvern Instruments, Malvern, UK) according to our previously published study.162.5. Di-tert-butyl dicarbonate surface modification
HsaFtH-RK was dissolved in 100 mM sodium bicarbonate buffer (pH 9.0), and di-tert-butyl dicarbonate (BOC) in acetone was added to the desired concentration (1 and 5 mM). The amount of acetone did not exceed 10%. The reaction mix was incubated for 1 h at ambient temperature. The reaction was stopped by adding Tris base at a final concentration of 100 mM, and the modified HsaFtH-RK was purified through SEC with PD MiniTrap G-25 columns (GE Healthcare) against FLB buffer.2.6. Prussian blue staining
To evaluate the ability of HsaFtH-RK to accumulate Fe3+, we performed Prussian blue staining and compared the ferroxidase activity of HsaFtH-RK with the activity of HsaFtH. For this purpose, 10 μg ferritin was added to 10 mM Tris (pH 6.8) with 150 mM NaCl. Samples were incubated at room temperature, and aliquots of Fe2+ stock solution (20 mM HCl, 40 mM FeCl2·4H2O) were added in 10 min intervals to reach a final Fe2+ concentration of 3.0 mM. After the last addition of Fe2+, the samples were incubated for an additional 15 min. To sequester free Fe2+/Fe3+ from the ferritin surface, 1.3 mM Na citrate was added, followed by incubation for 5 min. Then, loading buffer containing bromophenol blue was added to the samples, and proteins were separated in 5% native polyacrylamide gel electrophoresis (PAGE). After that, gels were quickly rinsed with distilled water, soaked in 0.75% HCl, and incubated on a rocking table until the bromophenol blue from the loading buffer turned yellow, indicating acidification of the whole gel. Prussian blue Fe3+ staining solution was prepared by mixing solutions of 2% K4[Fe(CN)6]·3H2O and 0.75% HCl (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, v/v). The gels were placed in the freshly prepared staining solution until blue bands developed. The proteins were subsequently stained with Coomassie Brilliant Blue R-250 (CBB, Bio-Rad) in fixation solution (H2O:MeOH:HAc, 50:40:10, v/v/v).
:1 ratio, v/v). The gels were placed in the freshly prepared staining solution until blue bands developed. The proteins were subsequently stained with Coomassie Brilliant Blue R-250 (CBB, Bio-Rad) in fixation solution (H2O:MeOH:HAc, 50:40:10, v/v/v).
2.7. Measurement of ferroxidase activity
The ferroxidase catalytic activity of ferritins was measured as a change in absorbance at 315 nm due to the catalytic oxidation of Fe2+ from Fe3+. For the assay, ferritins were dissolved in 50 mM NaCl, as we have noticed that HEPES, which is commonly used as the storage buffer, greatly enhances the oxidation of Fe2+ by air oxygen. The assay was carried out in 200 μL of 10 mM Tris·HCl (pH 6.8) containing 25 μg of ferritin and 1 mM freshly prepared ferrous ammonium sulphate. The absorbance at 315 nm was recorded in 1-minute intervals in triplicate. As negative control, we used Fer8, an L-chain ferritin lacking the ferroxidase catalytic site.2.8. Stability of HsaFtH-RK in organic solvents
HsaFtH-RK (20 μg) was dispersed in four different organic solvents: dimethylsulfoxide (DMSO), EtOH, dimethylformamide (DMF) and acetonitrile (ACN). The concentrations of solvents used were 10%, 20% and 30% in the final volume. After mixing, samples were incubated at ambient temperature for 30 min. Evaluation and visualization of samples was done by 5% native PAGE electrophoresis. For detection, staining with ethidium bromide (EtBr, 0.5 mg mL−1) was performed, and the gel was visualized under a UV transilluminator (FireReader XS D, Uvitec). The proteins were also visualized after CBB staining (Bio-Rad) as described above.2.9. Loading of HsaFtH-RK with fluorescent compounds
HsaFtH or HsaFtH-RK (25 μg) was incubated (3 h) at room temperature with EtBr, propidium iodide (PropI), acridine orange (AO), Dox, 3,3′-dihexyloxacarbocyanine (DiOC6) and azocarmine (AzoC) at a final concentration of 1 mM in 10 μL of reaction mixture. After incubation to enable passive loading of ferritins with fluorescent dyes, the samples were resolved on 5% native PAGE, stained with CBB (Bio-Rad), and visualized under a UV transilluminator (FireReader XS D, Uvitec).2.10. Digestion of HsaFtH-RK by trypsin and proteinase K
Bovine serum albumin (BSA) or HsaFtH-RK (10 μg) was digested for 1 h at 37 °C with respective amounts of trypsin or proteinase K in 100 mM Tris·HCl at pH 6.8 for trypsin and pH 7 for proteinase K. After incubation, the samples were resolved in 5% native PAGE, stained with CBB (Bio-Rad), and visualized under a UV transilluminator (FireReader XS D, Uvitec).2.11. Fluorescent labeling and evaluation of cellular uptake
HsaFtH-RK and HsaFtH-RK-BOC were labeled with 5-carboxytetramethylrhodamine succinimidyl ester (TAMRA, Thermo Fisher Scientific) in a carbonate buffer (90 mM NaHCO3, 10 mM Na2CO3). The unbound TAMRA was removed by purification with Amicon® Ultra Centrifugal Filters Ultracel®-50 K (exclusion limit 50000 NMWL; EMD Millipore, Danvers, MA, USA). Validation of labelling was done by protein separation on 1% agarose gel in TAE buffer [40 mM Tris-acetate, 1 mM EDTA]. Protein concentration was determined by the Bradford protein assay (Bio-Rad). For cellular uptake experiments, HEK-293 cells were cultured in a 24-well plate to ∼60% confluence and treated with the TAMRA-HsaFtH-RK or TAMRA-HsaFtH-RK-BOC (100 μg mL−1) in complete medium and incubated overnight. Three hours prior to imaging, 4 μg mL−1 of Hoechst 33342 and 2.5 μM quinacrine (QC, Sigma-Aldrich) for lysosomal staining were added to the culture. At the end of incubation, the cells were washed with fresh complete medium. Then, 100 μM chloroquine (CHQ) was added to the cell culture to disrupt endolysosomes. Micrographs of cells were captured using an Olympus IX53 fluorescence microscope (Olympus).
2.12. Encapsulation of siRNA into HsaFtH-RK
HsaFtH (100 μg) dissolved in 100 μL of water was mixed with 10 μL of 1 M NaOH to achieve alkaline disassembly (at pH 13.0) and incubated for 5 min at ambient temperature. Then, 10 μL of 10 μM universal noncoding control carboxyfluorescein (FAM)-labeled siRNA (Sigma-Aldrich) was added, followed by immediate addition of 10 μL of 1 M HCl to neutralize the pH to 7.5 and reassemble the HsaFtH-RK quaternary structure. Then, the samples were incubated for another 10 min at ambient temperature, followed by the addition of RNaseI (5 μg mL−1) to degrade free unencapsulated siRNA. Then, the samples were incubated for 2 h at 37 °C. The siRNA encapsulation was validated on 1% agarose gel without EtBr staining and visualized using Azure c600 (Azure Biosystems, Dublin, CA, USA).2.13. Evaluation of d2EGFP RNA interference
HsaFtH-RK loaded with mission esiRNA targeting EGFP (anti-GFP-siRNA, Sigma Aldrich) or universal noncoding control Cy3-labeled siRNA (Sigma Aldrich), encapsulated according to the optimized procedure, were mixed with complete medium and added to ∼40% confluent d2EGFP+ HEK-293 cells in a 24-well plate. All samples were diluted to the same siRNA concentration (45 nM). The cells were cultured overnight, and one hour prior to medium exchange, 100 μM CHQ was added. After one hour, the CHQ-containing medium was replaced with fresh complete medium. The silencing effect of siRNA was observed at four different timepoints (16, 24, 26, and 48 h). In all experiments, Metafectene SI+ (Biontex, Munchen, Germany) was used as positive control. Such prepared samples were observed under an Olympus IX53 fluorescence microscope (Olympus). In addition, we evaluated RNA interference using fluorescence-activated cell sorter (FACS). For this purpose, cells treated as described above were washed (2×) with phosphate-buffered saline (PBS) and trypsinized with 1× trypsin-EDTA solution (Sigma-Aldrich). Then, trypsin activity was quenched by the addition of serum-containing culture medium, and cells were collected by centrifugation, after which the supernatant was discarded and cells were resuspended in PBS. The FACS analyses were performed using a BD FACSVerse Flow Cytometer (BD Biosciences, Franklin Lakes, NJ, USA).2.14. Passive loading of Dox into wt HsaFtH and HsaFtH-RK
HsaFtH-RK or HsaFtH were diluted in 100 mM glycine buffer at pH 8.5 to the final volume of 500 μL and final concentration of 5 mg mL−1. Dox was added to the HsaFtH-RK or HsaFtH solution at the final concentration of 2 mM. In addition, a reaction mixture containing free Dox without ferritins was prepared to evaluate the differential fluorescence behavior between free Dox and Dox-loaded ferritins. Immediately after that, photographs of reaction mixtures were captured under ambient light and UV excitation using a UV transilluminator (FireReader XS D, Uvitec) at the exposure time of 200 ms. Then, the samples were incubated for 1 h at 60 °C, and the reaction mixtures were photographed as described above. After that, the samples were purified with Amicon® Ultra Centrifugal Filters Ultracel®-50 K (exclusion limit 50000 NMWL; EMD Millipore) five times against sterile FLB. After the last purification, the samples were diluted with sterile FLB to a final volume of 500 μL. Samples were then purified using size exclusion chromatography (SEC) on PD MiniTrap G-25 Sephadex columns (GE Healthcare, Wauwatosa, WI, USA), and the resulting encapsulates were again photographed under ambient light using a UV transilluminator. After purification, protein concentration was quantified by Bradford protein assay (Bio-Rad). The concentrations of loaded Dox were determined by plotting the fluorescence of loaded samples against the calibration curve of Dox. The analyses were done in DMSO to ensure complete dissociation of the protein and release of the loaded drugs. The fluorescence of the samples was analyzed with SPARK 10M spectrophotometer (Tecan, Maennedorf, Switzerland) at 485/595 nm. HsaFtH-Dox, HsaFtH-RK-Dox, and Dox-free ferritins were also analyzed using 5% native PAGE. After separation, Dox was visualized colorimetrically through its absorbance observable by naked eye under ambient conditions and also through its fluorescence, visualized under the UV transilluminator. The proteins were subsequently stained with CBB (Bio-Rad) in fixation solution (H2O:MeOH:HAc, 50:40:10, v/v/v).
2.15. Cellular uptake of Dox-loaded HsaFtH and HsaFtH-RK
For uptake experiments, the cells were grown to 50% confluence in complete medium on a 24-well plate and treated with 5 μg of Dox-loaded ferritins. Final concentrations of loaded Dox were 7.0 μM for HsaFtH-RK and 5.3 μM for HsaFtH, reflecting the improved ability of HsaFtH-RK to encapsulate nucleic acid-interacting compounds (i.e. Dox). The concentration of free Dox used as a control was 7.0 μM. Treated cells were incubated overnight. After that, nuclei were counterstained with Hoechst 33342; endolysosomal compartments were stained with 0.5 μM QC; and the cells were observed under an Olympus IX53 fluorescence microscope (Olympus). We also performed cotreatment with QC (0.5 μM) to visualize the lysosomes.2.16. Cytotoxicity of Dox-loaded HsaFtH and HsaFtH-RK
The tested concentrations of Dox ranged from 0 μM to 10 μM. As a positive control, free Dox was administered. The analyses were performed in triplicate in 96-well plates. MDA-MB-468 cells were seeded to ∼40% confluence and allowed to attach for 24 h, while HEK-293 cells were seeded to ∼40% confluence and allowed to attach for 7 h. After that, cells were treated with the indicated concentrations of free Dox, HsaFtH-Dox and HsaFtH-RK-Dox for 72 h. Then, 4.5 μg of resazurin sodium salt dissolved in DMSO in 50 μL of fresh medium was added, and the cells were incubated for an additional 2 h. The weakly fluorescent resazurin was then reduced via mitochondrial reductase of viable cells to a highly fluorescent resorufin. The intensity of resorufin fluorescence was analyzed using a SPARK 10 M spectrophotometer at 530/590 nm (Tecan).2.17. Quantitation of surface expression of TfR1
For the analysis of TfR1 surface expression, ∼1 × 105 of HEK-293 or MDA-MB-468 cells were collected by centrifugation (200g, 5 min, 4 °C) after 24 h of incubation in a 96-well flat-bottom tissue culture plate (Thermo Fisher Scientific). Then, cells were washed twice with 200 μL of ice-cold FACS buffer (PBS with 2% fetal calf serum and 2 mM EDTA) and transferred to a 96-well round-bottom tissue culture plate. Samples were then blocked with Fc block (BD Biosciences, clone: 24G2, Cat# 553142, RRID: AB_394656) for 30 min on ice and stained with fluorochrome-labeled monoclonal antibody recognizing TfR1, anti-CD71-PE (eBioscience, clone: OKT-9, Cat# 12-0719-42, RRID: AB_10717077) or an isotype control diluted in FACS buffer for 30 min at 4 °C in the dark. The labeled cells were washed twice with ice-cold FACS buffer and analyzed by flow cytometry using the BD Accuri C6 Plus (BD Biosciences). Data were analyzed using FlowJo software (FlowJo, RRID: SCR_008520). For each experiment, 100000 events per sample were recorded. The experiment was repeated twice, with samples in triplicate.
3. Results and discussion
3.1. HsaFtH-RK design, isolation, and characterization
In general, the internal cavity surface of ferritins is negatively charged. Therefore, it is perfectly designed for binding of positively charged metal ions.26 In this work, we aimed to reverse the internal charge of the ferritin cavity to enable the loading of negatively charged molecules, mainly fluorescent probes, nucleic acids, and a plethora of bioactive compounds. To achieve this, there are two possible approaches: (i) rational redesign of the whole internal ferritin surface or (ii) selective replacement of the C-terminus formed by the E-helix and nonstructured chain containing three negatively charged amino acids (aspartic and glutamic acid residues oriented inside the ferritin cage).24 In this study, we decided to engineer the protein C-terminus, in particular because this approach is less likely to cause structural damage to the quaternary structure of ferritin.The outer/inner surface charges of the wildtype (wt) HsaFtH and HsaFtH-RK were calculated, and the model confirms a substantial change in the inner surface charge distribution (Fig. 1A and B), while the external surface remains unaltered (Fig. S1†). The extraordinary thermal stability of ferritins can be exploited for their facile isolation. During the heat-mediated isolation, we did not observe any differences in thermal stability of wt HsaFtH and HsaFtH-RK, suggesting that the engineering procedure did not alter the major structural features of HsaFtH. This was further validated by TEM, showing that HsaFtH-RK is expressed as a supramolecular assembly indistinguishable from wt HsaFtH (Fig. 1C and D). DLS analyses revealed a slight increase in the hydrodynamic diameter of HsaFtH-RK (15.7 nm) compared to HsaFtH (13.5 nm) (Fig. S2†), which could be attributed to some portion of loosening of contacts between individual subunits of HsaFtH-RK due to deletion of the former nonstructured amino acid chain. As shown in Fig. 1E and F, the size distribution is homogenous in a wide range of pH, underpinning the exceptional stability of HsaFtH-RK. The desired size reduction, indicating a proper nondeleterious disassembly of both ferritins, was observed at pH 1 and 2.
 | ||
| Fig. 1 Physicochemical characterization of the produced HsaFtH and HsaFtH-RK. Coulomb surface charge calculations of the ferritins’ internal surface show a clear reduction of negative charge (blue) of (A) HsaFtH, which is replaced by positive charges (red) in (B) HsaFtH-RK. TEM micrographs showing indistinguishable supramolecular assembly of (C) HsaFtH and (D) HsaFtH-RK. Scale bar, 100 nm. DLS analysis of the stability of (E) HsaFtH and (E) HsaFtH-RK supramolecular assembly in a wide range of pH. | ||
We further examined whether the replacement of negatively charged amino acids in the E-helix interferes with iron uptake and storage. By Prussian blue staining, we detected non-altered ferroxidase activity and iron accumulation irrespective of HsaFtH engineering (Fig. 2A and B). In addition, the CBB-stained gel indicated good purity of the produced ferritins. Further evaluation of ferroxidase activity kinetics confirmed that E-helix engineering did not alter the functional properties of HsaFtH-RK, and its ferroxidase activity is comparable to wt HsaFtH, while no catalytic activity was found for Fer8, which is an L-chain ferritin lacking the catalytic site (Fig. 2C). Taken together, it was found that the engineering procedure did not perturb the structural and biochemical properties of HsaFtH-RK. Importantly, the provided data show that the engineered terminal part of the E-helix of HsaFtH represents an ideal site for follow-up studies focused on the rational design of ferritins for a broad spectrum of applications in which their structural and biochemical properties must be retained.
 | ||
| Fig. 2 Evidence of effective iron loading in HsaFtH-RK and RNA presence in its internal cavity usable as a sponge for superior loading of intercalation molecules. (A) Ferroxidase activity and the ability to store Fe3+ shown by Prussian blue staining. (B) CBB staining of ferritins on the same gel. (C) Measurement of ferroxidase activity kinetics of various ferritins. As a negative control, we used Fer8, a recombinant horse L-chain ferritin lacking the ferroxidase catalytic site. (D) 1% agarose gel of HsaFtH-RK subjected to NaOH disassembly, liberating the encapsulated RNA, followed by neutralization with HCl. RNaseI treatment resulted in degradation of the liberated RNA. (E) HsaFtH (annotated as H) and HsaFtH-RK (annotated as RK) were loaded with EtBr, PropI, AO, Dox, Elli, DiOC6 and AzoC. HsaFtH-RK exhibits superior loading capacity for all tested molecules. (F) Unaltered structures of HsaFtH and HsaFtH-RK were confirmed by CBB staining. Ferritin oligomerization state is indicated (M – monomer, D – dimer, T – trimer). | ||
3.2. HsaFtH-RK contains RNA loaded during recombinant production in E. coli
The most common approach to actively encapsulate a payload to the ferritin cavity is based on acidic or alkaline reversible disassembly of the 24-meric cage to monomeric subunits.1 Therefore, we further investigated whether the newly introduced RKRK cluster does not interfere with the correct assembly of HsaFtH-RK. For this purpose, we treated the HsaFtH-RK with 1 M NaOH to achieve disassembly, followed by the addition of 1 M HCl to neutralize the environment surrounding HsaFtH-RK to pH 7.5. Importantly, the large size of ferritins allows their facile separation in agarose gels commonly used for the separation of nucleic acids. During this experiment, we also examined possible residual nucleic acid contamination by EtBr staining. To our great surprise, we observed a fast-migrating band in the HsaFtH-RK reassembled by NaOH/HCl treatment (Fig. 2D). It was found that the substance is DNase resistant, but the band disappears after treatment with RNase, suggesting the presence of RNA in the HsaFtH-RK internal cavity. It has been previously demonstrated that mutations in the loop region between D and E-helix,27 as well as attachment of peptide sequences to the C-terminus of ferritins, can induce a FLOP orientation of E-helices that becomes directed outward.20,28,29 In such case, RNA might be bound at the external surface of HsaFtH-RK, thus being accessible to RNase. In contrast, if E-helices retain their native FLIP orientation, RNA might be located within the internal cavity of HsaFtH-RK and be protected against RNase activity (schematized in Fig. S3A†). Hence, we further compared the RNase-treated and non-treated HsaFtH-RK, staining the gels with EtBr, AO and ellipticine (Elli). In all cases, the fluorescence intensities remained equal irrespective of RNase exposure (Fig. S3B†), validating the presence of the RNA payload inside the HsaFtH-RK cavity. This finding led us to speculate whether the inherent RNA load could act as a sponge with the ability to render HsaFtH-RK a superior capacity to bind and deliver RNA/DNA-binding molecules. As shown with a spectrum of nucleic acid-interacting molecules, HsaFtH-RK indeed exhibited a markedly higher fluorescence compared to HsaFtH (Fig. 2E and F), confirming the interaction between loaded compounds and the intracavital RNA sponge of HsaFtH-RK. It is worth noting that recombinant HsaFtH and HsaFtH-RK are more prone to the formation of dimers and trimers known to be formed by horse spleen apoferritin,30–32 as can be seen in native PAGE (Fig. 2B).3.3. Surface modification of HsaFtH-RK with BOC
It has been previously shown that proteins’ surface modification with hydrophobic compounds, such as adamantyl moieties or multi-BOC (tri-BOC arginine), promote protein degradation through the proteasome pathway.33,34 Thus, we hypothesized that the introduction of hydrophobic patches to the HsaFtH-RK surface might activate the protein degradation machinery towards enhanced release of the encapsulated payload. In addition, we expected that BOC modification would markedly improve the binding of hydrophobic compounds to the HsaFtH-RK surface, enhancing its loading capacity. This could be of crucial importance for the delivery of hydrophobic substances, which is notoriously problematic due to their precipitation in aqueous media.The modification of HsaFtH-RK with BOC proceeded easily. However, we found that even minor traces of the ammonium sulfate used for protein salting-out inhibit the reaction. Importantly, the degree of surface modification can be easily monitored due to the effect of BOC on the electrophoretic mobility of HsaFtH-RK in agarose and native PAGE (Fig. 3A). It is worth noting that hydrophobization of the surface of HsaFtH-RK did not induce any precipitation or aggregation. Next, we analyzed whether BOC modification interferes with the loading of RNA-binding fluorescent compounds (EtBr and Elli). Compared to HsaFtH, HsaFtH-RK exhibited markedly higher loading capacity for both tested compounds. In addition, BOC surface modification did not affect the loading of HsaFtH-RK with the tested substances (Fig. 3B).
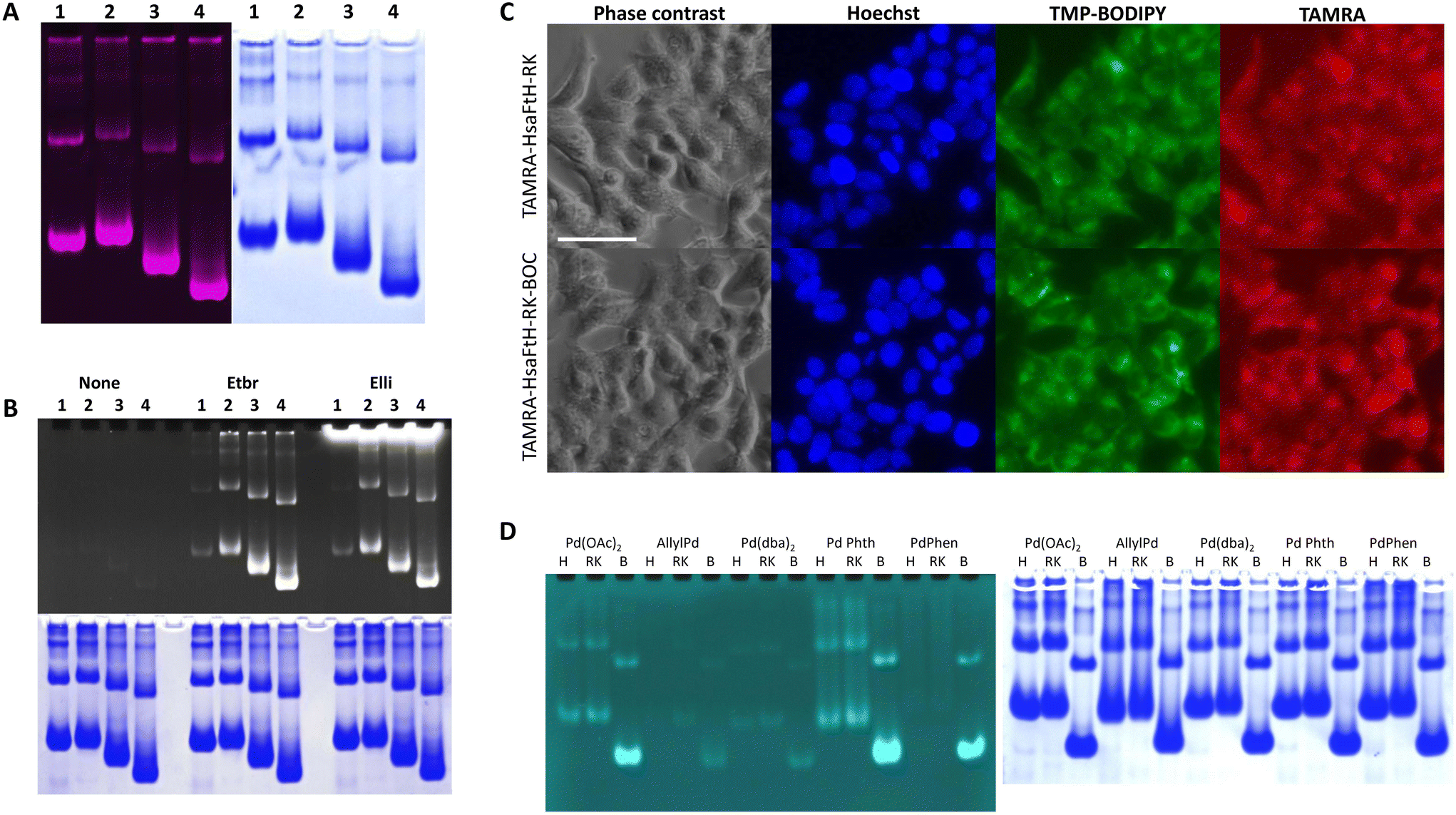 | ||
| Fig. 3 HsaFtH-RK surface modification with BOC. (A) TAMRA-HsaFtH (lane 1), TAMRA-HsaFtH-RK (lane 2), TAMRA-HsaFtH-RK treated with 1 mM (lane 3) or 5 mM (lane 4) BOC separated on native PAGE. After TAMRA visualization (left part), the same gel was stained with CBB to visualize proteins (right part). (B) HsaFtH and HsaFtH-RK were incubated with EtBr or Elli. Their loading was visualized through their intrinsic fluorescence (upper part), while proteins were stained with CBB (lower part). 1, HsaFtH; 2, HsaFtH-RK; 3, HsaFtH-RK-BOC (1 mM); and 4, HsaFtH-RK-BOC (5 mM). (C) Uptake of BOC-modified (5 mM) and nonmodified TAMRA-HsaFtH-RK in HEK-293 cells. Nuclei and membranous compartments were counterstained with Hoechst 33342 and trimethylphenyl-BODIPY (TMP-BODIPY), respectively. Scale bar, 50 μm. (D) HsaFtH (annotated as H), HsaFtH-RK (annotated as RK) and BOC-modified (5 mM) HsaFtH-RK (annotated as B) were loaded with: palladium acetate (Pd(OAc)2); allyl palladium (Allyl Pd); palladium(0) bis(dibenzylideneacetone) (Pd(dba)2); palladium phthalocyanine (Pd Phth); and palladium phenanthroline (PdPhen). Palladium complexes were visualized with propargyl ether of umbelliferone (left part). Proteins were stained with CBB (right part). | ||
We further aimed to examine whether BOC modification impairs the cellular uptake of modified ferritins. As shown with the unmodified TAMRA-HsaFtH-RK and TAMRA-HsaFtH-RK-BOC, we were not able to distinguish any apparent differences in intracellular distribution after 6 h incubation (Fig. 3C). In addition, during investigation of the kinetics of cellular uptake, it was found that after 6 h, the TAMRA signal approaches its saturation, with only negligible increases in intensity up to 21 h incubation (Fig. S4†). We anticipate that the lack of evidence of improved degradation of BOC-modified HsaFtH-RK is associated with the fact that HsaFtH-RK remains accumulated in the endolysosomal compartment, while proteasome machinery is predominantly located within the cytoplasm.35 It is expected that further engineering of HsaFtH-RK to introduce domains enabling efficient endosomal escape could therefore significantly improve the intracellular fate of HsaFtH-RK towards the controlled release of a payload outside the endolysosomal compartments. We are eager to further work on this aspect.
Ferritins are considered ideal vehicles for the delivery of organometallic compounds with inherent cytotoxic or bio-orthogonal catalytic activity.36 Since these compounds are frequently highly hydrophobic, we further examined if the BOC-modified HsaFtH-RK could serve as a vehicle for their loading. For this purpose, we employed our recently developed technique for the detection of protein-organometallic compound interactions in native PAGE, utilizing the palladium-catalyzed Tsuji–Trost reaction to convert nonfluorescent umbelliferone propargyl ether to UV-excitable umbelliferone.37 The technique is based on separation of the protein/organometallic complexes in native PAGE. The gel is then soaked in an aqueous solution of propargyloxy umbelliferone, washed, treated with a solution of sodium borohydride, and observed under a transilluminator. The reducing agent facilitates cleavage of the propargyloxy group by palladium and liberation of the deprotected umbelliferone, which is brightly fluorescent under UV. The proteins can be later detected by staining with CBB. As shown in Fig. 3D, highly hydrophobic complexes of palladium with phenanthroline and phthalocyanine (Fig. S5†) exhibited significantly higher affinity to BOC-modified HsaFtH-RK compared to HsaFtH and nonmodified HsaFtH-RK. Similar phenomenon was found for palladium acetate, validating our hypothesis that BOC modification of ferritins could serve as an approach to considerably improve their loading capacity for hydrophobic compounds. Alternatively, this platform can be utilized for intracellular delivery of catalytically active palladium complexes for bio-orthogonal chemistry.
Due to the poor solubility of organic compounds in water, we also tested the stability of HsaFtH-RK in organic solvents commonly used to facilitate solubilization of drugs.38 In this assay, EtBr was utilized as an indicator of HsaFtH-RK dissociation that results in leakage of loaded RNA and quenching of the fluorescence signal. Interestingly, we found that HsaFtH-RK remains fully soluble with no effect on the HsaFtH-RK quaternary structure, and no RNA shedding due to the solvents (EtOH, DMSO, DMF and ACN) up to their 30% concentration (v/v) (Fig. S6†). This finding confirms that HsaFtH-RK possesses exceptional stability that can be exploited for its loading with poorly soluble bioactive compounds.
3.4. Encapsulation of ectopic nucleic acids into HsaFtH-RK for RNA interference
We further evaluated the possibility of replacing the intrinsically bound E. coli RNA with ectopic siRNA. It was found that after loading of various types of ferritins [commercial apoferritin (Apo) isolated from horse spleen, HsaFtH, and HsaFtH-RK] with Cy3-siRNA through reversible disassembly using NaOH/HCl procedure, HsaFtH-RK was the only ferritin capable of encapsulating siRNA. In addition, encapsulation efficiently protected the loaded siRNA payload against RNase degradation (Fig. 4A). Furthermore, we found that besides siRNA, HsaFtH-RK can be simply loaded with DNA oligonucleotides or LNA, which was not achieved with wt HsaFtH (Fig. S7†). It is worth noting that the obtained results contradict previously published work showing the encapsulation of siRNA into wt HsaFtH,19 and although we employed a variety of procedures, we have never achieved any fluorescence signal using wt HsaFtH for the loading of nucleic acids. In the next step, we evaluated the ability of HsaFtH-RK to deliver Cy3-siRNA into d2EGFP+ HEK-293 cells and compared its efficiency with that of commercially available transfection reagent Metafectene SI+ (Fig. 4B). The obtained results revealed that using both approaches, Cy3-siRNA can be efficiently delivered into the intracellular region of cells. However, there is a pronounced differential subcellular localization pattern of the Cy3-siRNA signal. In the case of HsaFtH-RK, the siRNA is distributed in a pattern associated with HsaFtH-RK corresponding to accumulation within endolysosomal compartments. In contrast, Metafectene SI+-delivered Cy3-siRNA localizes in a bright dot-like pattern within the cellular interior. | ||
| Fig. 4 Examination of siRNA loading and knock-down efficiency. (A) Cy3-siRNA was loaded to Apo, HsaFtH, and HsaFtH-RK through NaOH/HCl-mediated reversible disassembly, then treated with RNase A. (B) Intracellular delivery of Cy3-siRNA via HsaFtH-RK or Metafectene SI+ to d2EGFP+ HEK-239 cells. Scale bar, 20 μm. (C) Fluorescence readouts of EGFP RNA interference 48 h after delivery of anti-EGFP siRNA using HsaFtH-RK or Metafectene SI+ in d2EGFP+ HEK-293 cells. Scale bar, 20 μm. | ||
Therefore, to evaluate whether HsaFtH-RK-delivered siRNA is capable of activating the RNA interference pathway, we loaded HsaFtH-RK with anti-EGFP siRNA. As demonstrated in Fig. 4C, no downregulation of d2EGFP in d2EGFP+ HEK-293 cells was observed, with the exception of Metafectene SI+-transfected siRNA. We rationalized that there can be several reasons for the failure of HsaFtH-RK to induce RNA interference, with the HsaFtH-RK subcellular localization being the most obvious. In all experiments, we observed TAMRA-HsaFtH-RK localized mostly in the endolysosomal compartment, which is consistent with ferritin's natural function to carry iron to cells that is released upon endosome acidification, followed by release of empty ferritin into circulation39 or degradation. Hence, to facilitate HsaFtH-RK endolysosomal escape, we treated the cells with the lysosomotropic agent CHQ.40 However, even with CHQ administration, we did not observe any improvement in RNA interference efficiency (Fig. 4C). We also speculated that the siRNA release from HsaFtH-RK might be markedly slower compared to Metafectene SI+. Thus, we extended the experiment to 72 h administration. In addition, as fluorescence microscopy might not be sufficiently sensitive to detect minor changes in d2EGFP expression, we employed FACS. However, FACS confirmed that while Metafectene SI+ inhibits the expression of d2EGFP, no such observable effect is caused by the siRNA delivery by HsaFtH-RK (even with applied CHQ) (Fig. S8†).
Importantly, the de-acidification of lysosomes by CHQ can be detected as an increase in red fluorescence of TAMRA, which is pH sensitive and decreases its intensity in acidic pH, which corresponds to the pH of the lysosomal environment (Fig. S9†). As shown in Fig. S10,† cells treated with TAMRA-HsaFtH exhibited discrete subcellular localization surrounding the nucleus, with colocalization with QC-stained endolysosomes (Fig. S11†). After administration of CHQ, which leads to an increase in lysosomal pH,41 significant increase in TAMRA-HsaFtH-RK fluorescence intensity was observed (Fig. S12†). This phenomenon confirms the lysosomotropic activity of CHQ, which is however insufficient to trigger siRNA release from HsaFtH-RK and the endolysosomal compartments. Another issue that might affect the delivery of payload to cells is the retention of the HsaFtH inside the cells. Since ferritins are designed to reside in the cells only temporarily, we speculated that the retention time might be insufficient for the efficient release of siRNA from HsaFtH-RK. Hence, we also monitored the retention of Cy5-siRNA encapsulated in HsaFtH-RK. As demonstrated in Fig. S13,† we observed a progressive decrease in fluorescence intensity of Cy5-siRNA loaded in HsaFtH-RK, while cells transfected with Metafectene SI+ retained bright fluorescence up to 72 h. Therefore, the obtained results indicate that inefficient retention of the siRNA loaded in HsaFtH-RK could contribute to the lack of detectable RNA interference. The third issue that corroborates our conclusions is the extraordinary stability of HsaFtH-RK, which might allow diffusion of small molecules, such as drugs, from the internal cavity into the intracellular space through subunit junctions (3–5 Å), but might effectively prevent the escape of larger macromolecules. The most stable Pyrococcus furiosus ferritin withstands temperatures up to 120 °C.42 We have found that HsaFtH and HsaFtH-RK are stable up to 80 °C. However, although being extremely thermostable, we anticipated that the quaternary structure of ferritins could be potentially disrupted by proteolytic enzymes that could therefore contribute to release of a payload. Thus, we analyzed the susceptibility of HsaFtH to proteolytic cleavage and found that despite BSA being efficiently cleaved by trypsin and proteinase K, HsaFtH-RK remains considerably stable with only minor shift in its electrophoretic mobility (Fig. S14†).
Taken together, the obtained data might explain the failure to perform siRNA-mediated RNA interference with HsaFtH-RK. Although HsaFtH-RK is unique vehicle with a plethora of advantageous properties, including high stability, production of highly homogenous nanoparticles, and efficient loading of siRNA, we clearly show that further molecular engineering or chemical modifications to promote fast endosomal escape of siRNA are needed. For this purpose, the properties of some viruses that need to escape the endolysosomal compartment after internalization to deliver their nucleic acids to the cytoplasm can be adapted.43 One viable approach might benefit from the use of protein transduction domains of cell-penetrating peptides, such as dimeric TAT (dfTAT).44 Another approach could benefit from co-administration of HsaFtH-RK with small molecules able to disrupt the endosomal membranes (such as UNC7938 published by Yang et al.45). As previously published, the combination of dfTAT and UNC7938 caused substantially improved delivery of macromolecules to the cytoplasm.46 It can be speculated that additional engineering of HsaFtH-RK to introduce dfTAT, and its cotreatment with UNC7938, could promote delivery to the cytoplasm, where BOC moieties should target HsaFtH-RK to be degraded by proteasomes with subsequent siRNA release. Alternatively, HsaFtH might be conjugated with adamantane moieties,33 PEST sequences,47 or with a set of hydrophobic amino acids fused to the N-terminus to form N-degrons as determinants driving proteasomal degradation.48 An alternative might be the introduction of cathepsin (lysosome-specific protease) cleavage sites into a loop connecting the B–C helix that would induce lysosome-specific degradation of the protein and effective siRNA release.
3.5. Investigation of Dox loading through the sponge-like effect of loaded RNA
The sponge-like ability of HsaFtH-RK to absorb RNA-binding compounds prompted us to further evaluate its applicability for loading and delivery of the chemotherapeutic compound Dox. Dox encapsulation was performed by passive diffusion of Dox into the internal cavity of HsaFtH and HsaFtH-RK at increased temperature (60 °C). As shown in gels presented in Fig. 5A, both HsaFtH and HsaFtH-RK were capable of encapsulating Dox without any perturbation to their quaternary structure. It is worth noting that the whole Dox loading process can be simply monitored by the naked eye or use of UV transilluminator (Fig. 5B). Successful loading of Dox into the ferritin cavity results in a dramatic change of color from the bright red of free Dox to crimson, indicating loading. Furthermore, the loading of Dox also results in quenching of intrinsic Dox fluorescence, as demonstrated by UV transilluminator photographs. Upon SEC purification and diafiltration, both encapsulates were utilized to examine cellular uptake and cytotoxic properties. | ||
| Fig. 5 Evaluation of passive loading of Dox. (A) Visualization of Dox encapsulation into HsaFtH and HsaFtH-RK. Dox loading was monitored colorimetrically (Dox absorption, upper part) and through intrinsic fluorescence of Dox under UV exposure (middle part), while proteins were stained with CBB (lower part). Lanes 1 and 7 show the reaction mixture before incubation. Lanes 2 and 8 show the reaction mixture after 1 h incubation at 60 °C, and lanes 3 and 5 show the reaction mixture after SEC purification. Lanes 4 and 6 show HsaFtH and HsaFtH-RK without Dox. Ferritin oligomerization state is indicated (M – monomer, D – dimer, T – trimer). (B) Monitoring of the Dox loading into HsaFtH and HsaFtH-RK under UV transilluminator (upper part) and ambient light (lower part). | ||
As shown in Fig. 6A, HsaFtH-RK-delivered Dox exhibited markedly higher fluorescence intensity compared to the administration of cells with wt HsaFtH, which is in line with its higher loading efficiency. This finding also confirms the efficient uptake of HsaFtH-RK. In addition, the increased fluorescence intensity of Dox is suggestive of its enhanced retention within the HsaFtH-RK cavity that is most likely due to Dox binding to the intracavital RNA sponge. Further QC staining revealed that some amount of Dox is gradually released from HsaFtH-RK into the cytoplasm. Overall, this could be highly advantageous for the development of drug nanoformulations with long-term sustained-release kinetics.49 To examine the cytotoxicity of Dox loaded in HsaFtH-RK and HsaFtH, we first attempted to normalize the assay to equal concentrations of free vs. loaded Dox. However, it was found that the fluorescence intensity of Dox is suppressed due to aggregation-induced fluorescence quenching.50 Hence, we decided to analyze the calibration curves of Dox dissolved in DMSO, which was consequently used as solvent for the extraction of Dox encapsulated in HsaFtH-RK. Utilization of the organic solvent promoted the extraction of Dox from the RNA sponge in HsaFtH-RK and prevented the formation of aggregates, allowing direct comparison of free and HsaFtH-RK-loaded Dox. After extraction, the fluorescence of Dox was meassured at 485/595 nm. The absorption and fluorescence spectra of free Dox dissolved in DMSO are shown inFig. S15†. Finally, as a proof of concept, we performed a comparative cytotoxic screening of free Dox, HsaFtH-Dox and HsaFtH-RK-Dox in HEK-293 and MDA-MB-468 cells, which represent healthy (HEK-293) and cancer (MDA-MB-468) cells. First, we analyzed the surface expression of ferritin receptor TfR1 in both lines to confirm that both cell lines can effectively internalize the H-chain ferritins (Fig. S16†), which is a crucial prerequisite for a test of biological properties of any H-chain ferritin. As demonstrated in Fig. 6B, free Dox administration exhibited slightly higher cytotoxic activity, in particular, in lower applied concentrations. In contrast, HsaFtH-RK-Dox exhibited a more gradual decrease in viability of both cell lines, with markedly lower cytotoxic effects in the lower applied concentrations of Dox, indicating the above-discussed sustained release kinetics. Interestingly, compared to HsaFtH-Dox, HsaFtH-RK-Dox exhibited higher cytotoxic activity, highlighting its advantageous properties for the delivery of RNA/DNA-binding molecules. However, it should be noted that the obtained data cannot be generalized to every cell type, and the cytotoxic properties would be further affected by a target receptor expression (TfR1 in this case) and the intrinsic susceptibility of cells to a payload.
 | ||
| Fig. 6 HsaFtH-RK efficiently delivers Dox and changes its cytotoxicity profile. (A) Cellular delivery and intracellular accumulation of free Dox, HsaFtH-Dox, and HsaFtH-RK-Dox in HEK-293 cells. The micrographs were captured after 48 h incubation. Scale bar, 50 μm. Viability of (B) HEK-293 and (C) MDA-MB-468 cells treated with free Dox, HsaFtH-Dox, and HsaFtH-RK-Dox after 72 h exposure. The viability was examined in triplicate and assessed as a decrease in resorufin fluorescence, after resazurin-to-resorufin conversion in viable cells. The error bars indicate the standard deviation. | ||
Taken together, the spongelike effect of RNA intrinsically contained in the HsaFtH-RK cavity could be simply exploited for highly efficient loading of bioactive compounds with RNA/DNA-binding properties. It must be noted that such compounds exhibit distinct nucleic acid-binding constants, and thus, the binding and subsequent release in physiological conditions must be always investigated in detail. We demonstrate, for the first time, that recombinant protein/nucleic acid hybrid can have a plethora of advantageous properties that can be utilized in a various biomedical applications. Among them, HsaFtH-RK could be of utmost interest to construct biosensing devices for the detection of molecules with high affinity to nucleic acids or, potentially, to construct modalities for diagnostic imaging utilizing fluorophore/contrast agent-labelled nucleic acids. Future steps will lead to further improvement of HsaFtH-RK properties (in particular for siRNA delivery) and consequent investigation of the behaviour of HsaFtH in vivo in terms of bioaccumulation, immunogenicity and efficiency of payload delivery. We believe that our novel HsaFtH-RK is a unique platform for further engineering toward achieving these goals.
4. Conclusion
Recombinant genetically engineered HsaFtH-RK containing an RKRK cluster at the C-terminus fully assembles with E-helices, presumably in a FLIP orientation toward the internal cavity of the supramolecular assembly. During the recombinant production, HsaFtH-RK binds short bacterial RNAs that remain entrapped inside its positively charged cavity. This payload can act as a sponge, dramatically increasing the amount of spontaneously loaded molecules with nucleic acid affinities. We also show that the HsaFtH-RK surface can be modified with BOC moieties to make the surface hydrophobic without any sign of protein aggregation, and such modification is useful for the loading of poorly water-soluble drugs and organometallic compounds. Importantly, the bacterial RNA inside HsaFtH-RK can be removed by NaOH/HCl reversible disassembly and be replaced by siRNA or other small nucleic acids with relevant size. However, our experiments indicate that in its current form, HsaFtH-RK is not a suitable transport vehicle for siRNA to induce RNA interference. HsaFtH-RK is simply too stable to release the siRNA in lysosomal pH. Moreover, it remains restricted to an endolysosomal network of vesicles seen as a perinuclear cloud, whilst siRNA has to be translocated to the cytoplasm, where it can be incorporated in an active RISC complex. The issue is common to all DNA/RNA delivery techniques such as transfection, where an active lysosome disruption is required for effective transfection. However, the lysosomal pH is sufficient to protonate Dox and allow its dissociation from the ferritin-carried RNA and diffusion to the cell interior, where the cytotoxic effect is exerted. We believe that our study provides a solid basis for the further molecular and chemical engineering work that is needed to address these issues.Data availability
The raw data required to reproduce these findings are available upon request from the corresponding authors.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the Czech Health Research Council (Project No. NU20-03-00477), the Czech Science Foundation (Project No. 22-14568S), League Against Cancer Prague and Internal Grant Agency of Mendel University in Brno (Project No. AF-IGA2022-IP018). We gratefully acknowledge the Core Facility Cryo-electron Microscopy and Tomography at CEITEC Masaryk University and the Core Facility Genomics at CEITEC Masaryk University supported by MEYS CR (LM2018127) and the European Regional Development Fund Project “UP CIISB” (No. CZ.02.1.01/0.0/0.0/18_046/0015974) for sharing the facilities to obtain the scientific data.References
- Z. Heger, S. Skalickova, O. Zitka, V. Adam and R. Kizek, Nanomedicine, 2014, 9, 2233–2245 CrossRef CAS PubMed.
- M. Liang, K. Fan, M. Zhou, D. Duan, J. Zheng, D. Yang, J. Feng and X. Yan, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14900–14905 CrossRef CAS PubMed.
- N. Pontillo, F. Pane, L. Messori, A. Amoresano and A. Merlino, Chem. Commun., 2016, 52, 4136–4139 RSC.
- A. I. Kuruppu, L. Zhang, H. Collins, L. Turyanska, N. R. Thomas and T. D. Bradshaw, Adv. Healthcare Mater., 2015, 4, 2816–2821 CrossRef CAS PubMed.
- J. M. Dominguez-Vera and E. Colacio, Inorg. Chem., 2003, 42, 6983–6985 CrossRef CAS PubMed.
- M. Okuda, Y. Suzumoto and I. Yamashita, Cryst. Growth Des., 2011, 11, 2540–2545 CrossRef CAS.
- L. L. Chen, G. L. Bai, S. P. Yang, R. Yang, G. H. Zhao, C. S. Xu and W. N. Leung, Food Res. Int., 2014, 62, 1147–1153 CrossRef CAS.
- P. Huang, P. F. Rong, A. Jin, X. F. Yan, M. G. Zhang, J. Lin, H. Hu, Z. Wang, X. Y. Yue, W. W. Li, G. Niu, W. B. Zeng, W. Wang, K. C. Zhou and X. Y. Chen, Adv. Mater., 2014, 26, 6401–6408 CrossRef CAS PubMed.
- A. Makino, H. Harada, T. Okada, H. Kimura, H. Amano, H. Saji, M. Hiraoka and S. Kimura, Nanomedicine, 2011, 7, 638–646 CrossRef CAS PubMed.
- R. L. Yang, Y. Q. Li, X. Y. Wang, J. J. Yan, D. H. Pan, Y. P. Xu, L. Z. Wang and M. Yang, RSC Adv., 2019, 9, 28548–28553 RSC.
- R. L. Fan, S. W. Chew, V. V. Cheong and B. P. Orner, Small, 2010, 6, 1483–1487 CrossRef CAS PubMed.
- K. W. Pulsipher, S. Honig, S. Deng and I. J. Dmochowski, J. Inorg. Biochem., 2017, 174, 169–176 CrossRef CAS PubMed.
- C. Sun, Y. Yuan, Z. Xu, T. Ji, Y. Tian, S. Wu, J. Lei, J. Li, N. Gao and G. Nie, Bioconjugate Chem., 2015, 26, 193–196 CrossRef CAS PubMed.
- I. H. Lee, B. Ahn, J. M. Lee, C. S. Lee and Y. Jung, Analyst, 2015, 140, 3543–3550 RSC.
- S. Kanbak-Aksu, M. Nahid Hasan, W. R. Hagen, F. Hollmann, D. Sordi, R. A. Sheldon and I. W. Arends, Chem. Commun., 2012, 48, 5745–5747 RSC.
- V. Pekarik, M. Peskova, R. Guran, J. Novacek, Z. Heger, K. Tripsianes, J. Kumar and V. Adam, Dalton Trans., 2017, 46, 13690–13694 RSC.
- B. Maity, K. Fukumori, S. Abe and T. Ueno, Chem. Commun., 2016, 52, 5463–5466 RSC.
- K. L. Fan, X. H. Jia, M. Zhou, K. Wang, J. Conde, J. Y. He, J. Tian and X. Y. Yan, ACS Nano, 2018, 12, 4105–4115 CrossRef CAS PubMed.
- L. Li, M. Munoz-Culla, U. Carmona, M. P. Lopez, F. Yang, C. Trigueros, D. Otaegui, L. B. Zhang and M. Knez, Biomaterials, 2016, 98, 143–151 CrossRef CAS PubMed.
- E. J. Lee, S. J. Lee, Y. S. Kang, J. H. Ryu, K. C. Kwon, E. Jo, J. Y. Yhee, I. C. Kwon, K. Kim and J. Lee, Adv. Funct. Mater., 2015, 25, 1279–1286 CrossRef CAS.
- Z. W. Yuan, B. Wang, Y. L. Teng, W. Ho, B. Hu, K. O. Boakye-Yiadom, X. Y. Xu and X. Q. Zhang, Nanoscale, 2022, 14, 6449–6464 RSC.
- B. L. Zhang, X. H. Chen, G. H. Tang, R. F. Zhang, J. Y. Li, G. M. Sun, X. Y. Yan and K. L. Fan, Nano Today, 2022, 46, 1–15 Search PubMed.
- N. Pediconi, F. Ghirga, C. Del Plato, G. Peruzzi, C. M. Athanassopoulos, M. Mori, M. E. Crestoni, D. Corinti, F. Ugozzoli, C. Massera, A. Arcovito, B. Botta, A. Boffi, D. Quaglio and P. Baiocco, Bioconjugate Chem., 2021, 32, 1105–1116 CrossRef CAS PubMed.
- E. C. Theil, Inorg. Chem., 2013, 52, 12223–12233 CrossRef CAS PubMed.
- Y. R. Qu, K. Davey, Y. Sun, A. Middelberg and J. X. Bi, ACS Appl. Bio Mater., 2022, 5, 3167–3179 CrossRef CAS PubMed.
- M. Uchida, S. Kang, C. Reichhardt, K. Harlen and T. Douglas, Biochim. Biophys. Acta, Gen. Subj., 2010, 1800, 834–845 CrossRef CAS PubMed.
- R. Jappelli, A. Luzzago, P. Tataseo, I. Pernice and G. Cesareni, J. Mol. Biol., 1992, 227, 532–543 CrossRef CAS PubMed.
- A. Luzzago and G. Cesareni, EMBO J., 1989, 8, 569–576 CrossRef CAS PubMed.
- S. E. Kim, K. Y. Ahn, J. S. Park, K. R. Kim, K. E. Lee, S. S. Han and J. Lee, Anal. Chem., 2011, 83, 5834–5843 CrossRef CAS PubMed.
- S. A. Saeed and T. R. Boyde, FEBS Lett., 1981, 123, 111–114 CrossRef CAS PubMed.
- D. N. Petsev, B. R. Thomas, S. T. Yau, D. Tsekova, C. Nanev, W. W. Wilson and P. G. Vekilov, J. Cryst. Growth, 2001, 232, 21–29 CrossRef CAS.
- D. N. Petsev, B. R. Thomas, S. Yau and P. G. Vekilov, Biophys. J., 2000, 78, 2060–2069 CrossRef CAS PubMed.
- T. K. Neklesa, H. S. Tae, A. R. Schneekloth, M. J. Stulberg, T. W. Corson, T. B. Sundberg, K. Raina, S. A. Holley and C. M. Crews, Nat. Chem. Biol., 2011, 7, 538–543 CrossRef CAS PubMed.
- Y. Shi, M. J. Long, M. M. Rosenberg, S. Li, A. Kobjack, P. Lessans, R. T. Coffey and L. Hedstrom, ACS Chem. Biol., 2016, 11, 3328–3337 CrossRef CAS PubMed.
- A. von Mikecz, J. Cell Sci., 2006, 119, 1977–1984 CrossRef CAS PubMed.
- B. Tesarova, M. Charousova, S. Dostalova, A. Bienko, P. Kopel, R. Kruszynski, D. Hynek, P. Michalek, T. Eckschlager, M. Stiborova, V. Adam and Z. Heger, Int. J. Biol. Macromol., 2019, 126, 1099–1111 CrossRef CAS PubMed.
- V. Pekarik, M. Peskova, J. Duben, M. Remes and Z. Heger, Sci. Rep., 2020, 10, 1–10 CrossRef PubMed.
- M. H. Abraham, R. E. Smith, R. Luchtefeld, A. J. Boorem, R. S. Luo and W. E. Acree, J. Pharm. Sci., 2010, 99, 1500–1515 CrossRef CAS PubMed.
- B. G. Atkinson, R. L. Dean, J. Tomlinson and T. W. Blaker, Biochem. Cell Biol., 1989, 67, 52–57 CrossRef CAS PubMed.
- L. E. Gallagher, O. A. Radhi, M. O. Abdullah, A. G. McCluskey, M. Boyd and E. Y. W. Chan, Cell Death Dis., 2017, 8, 14 Search PubMed.
- S. Y. Lu, T. Sung, N. W. Lin, R. T. Abraham and B. A. Jessen, PLoS One, 2017, 12, 1–22 Search PubMed.
- J. Tatur, P. L. Hagedoorn, M. L. Overeijnder and W. R. Hagen, Extremophiles, 2006, 10, 139–148 CrossRef CAS PubMed.
- I. Le Blanc, P. P. Luyet, V. Pons, C. Ferguson, N. Emans, A. Petiot, N. Mayran, N. Demaurex, J. Faure, R. Sadoul, R. G. Parton and J. Gruenberg, Nat. Cell Biol., 2005, 7, 653–664 CrossRef CAS PubMed.
- A. Erazo-Oliveras, K. Najjar, L. La Dayani, T. Y. Wang, G. A. Johnson and J. P. Pellois, Nat. Methods, 2014, 11, 861–867 CrossRef CAS PubMed.
- B. Yang, X. Ming, C. Cao, B. Laing, A. Yuan, M. A. Porter, E. A. Hull-Ryde, J. Maddry, M. Suto, W. P. Janzen and R. L. Juliano, Nucleic Acids Res., 2015, 43, 1987–1996 CrossRef CAS PubMed.
- J. Allen, K. Najjar, A. Erazo-Oliyeras, H. M. Kondow-McConaghy, D. J. Brock, K. Graham, E. C. Hager, A. L. J. Marschall, S. Dubl, R. L. Juliano and J. P. Pellois, ACS Chem. Biol., 2019, 14, 2641–2651 CrossRef CAS PubMed.
- M. Rechsteiner and S. W. Rogers, Trends Biochem. Sci., 1996, 21, 267–271 CrossRef CAS PubMed.
- S. M. Sriram, B. Y. Kim and Y. T. Kwon, Nat. Rev. Mol. Cell Biol., 2011, 12, 735–747 CrossRef CAS PubMed.
- J. V. Natarajan, A. Darwitan, V. A. Barathi, M. Ang, H. M. Htoon, F. Boey, K. C. Tam, T. T. Wong and S. S. Venkatraman, ACS Nano, 2014, 8, 419–429 CrossRef CAS PubMed.
- N. S. H. Motlagh, P. Parvin, F. Ghasemi and F. Atyabi, Biomed. Opt. Express, 2016, 7, 2400–2406 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3bm01257c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |