
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapamycin-based inhaled therapy for potential treatment of COPD-related inflammation: production and characterization of aerosolizable nano into micro (NiM) particles†
Emanuela Fabiola
Craparo
 *a,
Marta
Cabibbo
a,
Cinzia
Scialabba
a,
Luca
Casula
b,
Francesco
Lai
b and
Gennara
Cavallaro
ac
*a,
Marta
Cabibbo
a,
Cinzia
Scialabba
a,
Luca
Casula
b,
Francesco
Lai
b and
Gennara
Cavallaro
ac
aLaboratory of Biocompatible Polymers, Department of Biological, Chemical and Pharmaceutical Sciences and Technologies (STEBICEF), University of Palermo, Via Archirafi 32, Palermo, 90123, Italy. E-mail: emanuela.craparo@unipa.it
bDepartment of Life and Environmental Sciences, University of Cagliari, Via Ospedale, 72, 09124 Cagliari, Italy
cAdvanced Technology and Network Center (ATeN Center), University of Palermo, Palermo 90133, Italy
First published on 24th November 2023
Abstract
Our paper describes the production and characterization of inhalable microparticles loaded with nanoparticles for the lung administration of rapamycin (Rapa). In detail, core–shell lipid/polymer hybrid nanoparticles loaded with Rapa (Rapa@Man-LPHNPs) were produced with mean size of about 128 nm and slightly negative ζ potential (−13.8 mV). A fluorescent graft polyaspartamide-poly(lactic-co-glycolic acid) copolymer (PHEA-g-RhB-g-PLGA) for use as the polymeric core was obtained by nanoprecipitation, while an appropriate mixture of DPPC and mannosylated phospholipid (DSPE-PEG2000-Man) was used to provide the macrophage-targeting lipid shell. The successful formation of Rapa@Man-LPHNPs was confirmed by TEM and DSC analyses. The loaded drug (4.3 wt% of the total weight) was slowly released from the polymeric core and protected from hydrolysis, with the amount of intact drug after 24 h of incubation in the medium being equal to 74 wt% (compared to 40% when the drug is freely incubated at the same concentration). To obtain a formulation administrable by inhalation, Rapa@Man-LPHNPs were entrapped inside PVA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :LEU microparticles by using the nano into micro (NiM) strategy, specifically by spray drying (SD) in the presence of a pore-forming agent. In this way, NiM particles with geometric and theoretical aerodynamic diameters equal to 4.52 μm and 3.26 μm, respectively, were obtained. Furthermore, these particles showed optimal nebulization performance, having an FPF and an MMAD equal to 27.5% and 4.3 μm, respectively.
:LEU microparticles by using the nano into micro (NiM) strategy, specifically by spray drying (SD) in the presence of a pore-forming agent. In this way, NiM particles with geometric and theoretical aerodynamic diameters equal to 4.52 μm and 3.26 μm, respectively, were obtained. Furthermore, these particles showed optimal nebulization performance, having an FPF and an MMAD equal to 27.5% and 4.3 μm, respectively.
1. Introduction
Pulmonary drug delivery for the treatment of lung diseases offers highly localized drug concentrations, rapid onset of therapeutic action, low enzymatic activity, and better therapeutic control with respect to other administration routes.1 On the other hand, nanomedicines are very promising in localized lung disease treatments via the design of appropriate carriers to allow mucus penetration, macrophagic escape/uptake and pulmonary surfactant corona inhibition, such as through size control and charge tuning, surface modification, co-delivery of mucolytic agents, and decoration with surface ligands.2In several pulmonary pathologies, macrophages show a defective phagocytosis capacity although they are increased in number;3 therefore, they represent the optimal therapeutic target for the treatment of inflammatory respiratory diseases such as chronic obstructive pulmonary disease (COPD).4,5 Because mannose receptors are highly expressed on alveolar macrophages, it has been widely demonstrated that mannose-surface decoration of nanocarriers is an active drug-targeting strategy to treat tumorigenesis, inflammation, and infections in lung diseases.6–10
In almost all chronic lung diseases, activation of the mammalian target of the rapamycin (mTOR) complex was recently correlated to pathogenetic processes such as accelerated inflammation, aging and cell senescence,11,12 and to airway remodeling due to activation of lung fibroblasts.13,14 On the other hand, the mTOR inhibition gives positive effects against the cytokine storm in COVID-19 patients.15 Similarly, an improvement in lung functions was achieved in animal models of pulmonary hypertension treated with mTOR inhibitors.16 Therefore, the mTOR modulation appears to be a promising therapeutic intervention to achieve a favourable prognosis in COPD patients.17,18
In this context, the use of nanomedicine for the inhalation of repurposed drugs, such as rapamycin (Rapa), could allow a more localized and specific immunomodulatory approach to treatment of inflammation in lung diseases, as supported by widespread scientific evidence.19 In fact, systemic toxicity is the crucial problem that precludes the administration of Rapa due to broad immunosuppression with unwanted side effects.20,21 Some attempts at pulmonary Rapa administration have already been described in the literature, showing selective effects on lung macrophages.22,23
Recently, it was demonstrated that Rapa was able to reduce smoke-induced inflammatory-oxidative stress and cell apoptosis, which are the main causes of the progression of COPD,24 and also to alleviate the cigarette smoke extract (CSE)-induced inflammatory response by autophagy activation.12,18,25 Approved as an immunosuppressant, Rapa has been used to treat many conditions such as autoimmune diseases, transplant rejection, and malignant tumors.26 It is used also to promote health and longevity in several age-related diseases.11,27 Rapa is currently one of the most studied and promising drugs for idiopathic pulmonary fibrosis (IPF).28 Furthermore, it is included among the repurposed non-oncology drugs (RNODs) for inhaled therapy against lung cancer due to cell proliferation suppression by inhibition of the mTORC1 pathway.7
In this paper, the production, and the characterization of inhalable microparticles made from biocompatible excipients and containing lipid/polymer hybrid Rapa-loaded nanoparticles, is described. As the excipient to form the micrometer matrix, a mixture of polyvinyl alcohol (PVA) and leucin (LEU) was chosen. As the polymeric component to form the hybrid nanoparticles, an amphiphilic graft copolymer of α,β-poly(N-2-hydroxyethyl)-DL-aspartamide (PHEA) was used,29–31 while as lipid components, a mixture of the endogenous 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-(polyethylene glycol)2000-mannose (DSPE-PEG2000-mannose) was chosen. The latter lipid component was chosen with the aim to obtain mannosylated carriers (Man-LPHNPs) to actively target alveolar macrophages, while the DPPC was chosen due to the already documented interaction of inhalable hybrid DPPC-based nanoparticles with the pulmonary surfactant.1,32
2. Experimental
Materials
Rhodamine B (RhB), polylactic-co-glycolic acid (PLGA, acid free), carbonyldiimidazole (CDI), ammonium bicarbonate (AB), diethylamine (DEA), poly(vinylalcohol) (![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) w = 31000–50000) (PVA), L-leucine (LEU), 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), Dulbecco's phosphate-buffered saline (DPBS), iron(III) chloride hexahydrate, ammonium thiocyanate, amino acid solution, type II mucine from porcine stomach, and egg yolk emulsion, were purchased from Sigma-Aldrich (Milan, Italy). Hydroxyethyl cellulose (HEC) and potassium chloride (KCl) were purchased from Carlo Erba. 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-(polyethylene glycol)2000-mannose (DSPE-PEG2000-mannose) was purchased from Ruixibiotech (Xi'an, China). Rapamycin (Rapa) was purchased from Accel Pharmatech (NJ, USA). Spectra/Por 4 membrane standard RC dialysis tubing was purchased from Spectrum Laboratories Inc. (USA). All solvents were of analytical grade and obtained from Sigma-Aldrich (Italy). Macrophage (Raw 264.7), and human bronchial epithelium (16 HBE) cell lines were purchased from Istituto Zooprofilattico Sperimentale della Lombardia e dell'Emilia Romagna, Italy.
w = 31000–50000) (PVA), L-leucine (LEU), 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), Dulbecco's phosphate-buffered saline (DPBS), iron(III) chloride hexahydrate, ammonium thiocyanate, amino acid solution, type II mucine from porcine stomach, and egg yolk emulsion, were purchased from Sigma-Aldrich (Milan, Italy). Hydroxyethyl cellulose (HEC) and potassium chloride (KCl) were purchased from Carlo Erba. 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-(polyethylene glycol)2000-mannose (DSPE-PEG2000-mannose) was purchased from Ruixibiotech (Xi'an, China). Rapamycin (Rapa) was purchased from Accel Pharmatech (NJ, USA). Spectra/Por 4 membrane standard RC dialysis tubing was purchased from Spectrum Laboratories Inc. (USA). All solvents were of analytical grade and obtained from Sigma-Aldrich (Italy). Macrophage (Raw 264.7), and human bronchial epithelium (16 HBE) cell lines were purchased from Istituto Zooprofilattico Sperimentale della Lombardia e dell'Emilia Romagna, Italy.
α,β-Poly(N-2-hydroxyethyl)-DL-aspartamide (PHEA) and PHEA-g-RhB were obtained as previously reported.33
Synthesis and characterization of PHEA-g-RhB-g-PLGA graft copolymer
An appropriate amount of CDI was added to a dispersion of PLGA in anhydrous DMF (95 mg ml−1), according to R1 = (mol CDI per mol PLGA) = 2, and the resulting dispersion left under stirring at 40 ± 0.1 °C for 4 h. Then, a dispersion of PHEA-g-RhB in anhydrous DMF was prepared (33.3 mg ml−1), in accordance with R2 = (mol PLGA per mol of repeating units of PHEA) = 0.05, in which DEA was added as a catalyst in accordance with R3 = (mol DEA per mol PLGA) = 5. The latter dispersion was added dropwise to the activated PLGA dispersion. The resulting reaction mixture was left under stirring at 40 ± 0.1 °C for 48 hours. After this time, the copolymer was precipitated in a suitable quantity of diethyl ether, separated by centrifugation at 9800 rpm, at 4 °C for 10 min, washed several times with a diethyl ether/dichloromethane mixture (ratio 45:55 v/v), dried under vacuum and kept in a refrigerator (4 °C) under argon. The resulting yield was equal to 45% with respect to the starting PHEA-g-RhB.
PHEA-g-RhB-g-PLGA 1H-NMR (300 MHz, DMF-d7, 25 °C, TMS): δ 1.3 ppm (12-HRhB, CH3CH2–); δ 1.5–1.9 ppm (m, 3-HPLGA –[OCOCH(CH3)]92–); δ 3.0 ppm (m, 2HPHEA –COCHCH2CONH–); δ 3.5 ppm (t, 2HPHEA –NHCH2CH2O–); δ 3.6 ppm (t, 2HPHEA –NHCH2CH2O–); δ 5.0 ppm (m, 1HPHEA –NHCH(CO)CH2–); δ 4.2–4.5 and 5.4–5.8 ppm (1HPLGA, –[OCOCH(CH3)]92–); δ 5.3 ppm (2HPLGA, –[OCOCH2]92–); δ 7.0–8.0 ppm (10HRhB, H-Ar). Derivatization degree in PLGA (DDPLGA) is equal to 4.8 ± 0.2 mol%.
The average molecular weight (w) of PHEA-g-RhB-g-PLGA, determined by SEC analysis, was found to be 156122 Da (w/n = 1.03).
Lipid/polymer hybrid nanoparticles (Man-LPHNPs) production
:10 v/v) and left stirring overnight. When the drug-loaded nanoparticles are produced, the mixture was centrifuged at 20 °C for 15 min at 8000 rpm, and the supernatant recovered.
:25 weight ratio) in a round-bottomed flask and the solvent was then evaporated under vacuum at 40 °C. The obtained lipid film was rehydrated using 25 ml of bidistilled water (to obtain a final lipid concentration of 0.5 wt%) under mechanical stirring at 75 °C for 30 min.
:1 v/v), and then subjected to high-pressure homogenization (HPH) at a pressure of 10000–15000 psi by using an EmulsiFlex-C5 homogenizer (Avestin, Canada). Finally, the obtained dispersion was submitted to ultracentrifugation (Optima XPN Ultracentrifuge, Type 70 Ti Rotor) at 10 °C for 1 hour at 40000 rpm; the resulting pellets (empty Man-LPHNPs or Rapa@Man-LPHNPs) were freeze-dried and stored for further characterization.33 As control samples, un-targeted drug-loaded hybrid nanoparticles were produced by using DPPC/DSPE-PEG2000 vesicles (named, respectively, LPHNPs and Rapa@LPHNPs samples).
Man-LPHNPs characterization
ζ-potential values (mV) were calculated from electrophoretic mobility using the Smoluchowski relationship. Analyses were performed in triplicate.
:9 v/v) and filtered (0.45 μm pore size filters) for HPLC analysis. A Waters Breeze System Liquid Chromatograph system, equipped with a Luna® C18 column (250 × 4.6 mm, 5 μm, from Phenomenex), an autosampler (40 μl as injected volume), and a UV-vis detector (detection wavelength set to 277 nm), was used. The mobile phases were a methanol:water 80:20 v/v mixture at a flow rate of 1 ml min−1 (25 °C). The calibration curve was created by plotting peak areas (at retention time = 14 min) versus Rapa standard concentration values in methanol (range of 0.02–0.001 mg ml−1) and resulted in values y = 136.5x (R2 = 0.9994).
Biological characterization
For quantitative uptake of samples of Man-LPHNPs and LPHNPs, fluorescence analysis was carried out. After incubation with 0.3 mg ml−1 of each sample for 2, 6 and 24 h, cells were lysed in 100 μL of lysis buffer (1% triton X-100, 2% SDS in DPBS) at 37 °C. 75 μL of lysates was transferred to disposable 96-well plates for fluorescence intensity measurements (λEX: 550 nm; λEX: 580 nm) on an Eppendorf spectrofluorophotometer plate reader. The remaining 25 μL of lysates was used to determine the protein content using the bicinchoninic acid kit for protein determination (Sigma-Aldrich), according to the protocol of the manufacturer. For determination of the mean fluorescence intensity, fluorescence signals were corrected for protein in the samples. The results were expressed as the ratio of fluorescence intensity (I.F.) per microgram of protein. The experiment was carried out in triplicate.
Microparticle production
Microparticles were produced by using a Micro-Spray Dryer B-290 (Buchi, Milan). A 150 μm nozzle was selected and constant parameters such as inlet temperature (110 °C), feed rate (15%), and air aspirator (100%) were chosen. The liquid feed for each formulation was prepared by dissolving in water different components as reported in Table 3. Each final dispersion was filtered, spray dried and the obtained powder was recovered with a yield of up to 50 wt% with respect to the theoretical value.To achieve microparticle entrapping of empty Man-LPHNPs (NiM) or Rapa@Man-LPHNPs (NiM@Rapa), the nanoparticles were added to the liquid feed prior to the SD at a concentration of 1 wt% (see Table 1 for the composition of the liquid feed).
| Sample | Z average (nm) | PDI | ζ-potential (mV ± S.D.) |
|---|---|---|---|
| Freshly prepared | |||
| Empty nanoparticles | 94.2 | 0.24 | −14.0 ± 7.3 |
| Rapa@nanoparticles | 120.8 | 0.20 | −18.0 ± 6.4 |
| Empty Man-LPHNPs | 117.1 | 0.30 | −12.0 ± 9.3 |
| Rapa@Man-LPHNPs | 128.5 | 0.28 | −13.8 ± 5.6 |
| Post freeze drying | |||
| Empty Man-LPHNPs | 159.8 | 0.28 | −15.7 ± 7.5 |
| Rapa@Man-LPHNPs | 148.0 | 0.33 | −12.4 ± 6.4 |
All the samples used for NGI measurement were obtained by adding rhodamine (0.005% wt/vol) to the liquid feed.
Microparticle characterization
:50 (v/v) water:ethanol mixture was used to collect the deposited powder and the quantification was carried out by determining RhB content at λ = 562 nm, using a synergy 4 multiplate UV-vis reader (BioTek, Winooski, USA). The nebulization test was repeated at least three times (3 capsules for each actuation) for each sample.
The aerodynamic parameters were then calculated using the generated data to give the emitted dose (ED; drug collected in the mouthpiece, IP, PS and impaction cups) and the ED% (ratio of the ED to the initial mass of sample tested). Determination of the fine particle fraction (FPF%), defined as the mass percentage of the ED with an aerodynamic diameter less than 5.0 μm, was obtained from interpolation of the cumulative aerodynamic diameter distribution curve between stage 1 and stage 2. The cumulative mass amount from each stage was plotted as a percentage of the recovered sample versus the cut-off diameter, not including the mass deposited in the induction port due to the unavailability of a precise upper-size limit for particles deposited in this section. Following the Eur. Ph. 11th ed. Indications, the mass median aerodynamic diameter (MMAD) of the particles, defined as the aerodynamic diameter at which 50% of the particles are larger and 50% are smaller, was then extrapolated from the graph and the geometric standard deviation (GSD) value was determined. Moreover, to determine the dispersibility index (DI), the ratio of the MMAD to the theoretical daer was calculated, where a large DI value indicates low mid-air dispersion of the aerosols exiting the inhaler.42,43
Complex viscosity (η*) on artificial mucus alone or in the presence of PVA:LEU_AB10 or NiM@Rapa was determined at 20 °C by using a rotational rheometer (TA Instruments, New Castle, DE) coupled to 8 mm parallel plate geometry and a controlled Peltier plate, maintaining a gap of 300 μm. Firstly, the linear viscoelastic region of artificial mucus was determined by a strain sweep test ranging from 0.01 to 20%, which was found to be in the range of 5–10%. Then, a frequency sweep (0.01–2 Hz) was performed for all samples at 5% constant strain to determine η*. All rheological tests were conducted in triplicate, and Trios Software v3.3 (TA Instruments) was used for data acquisition and analysis.
3. Results and discussion
Here, inhalable microparticles were produced as potential carriers for rapamycin (Rapa) in the treatment of lung inflammation-based diseases, such as chronic obstructive pulmonary disease (COPD).44,45 Rapa is an immunosuppressant, being a potent inhibitor of the mammalian target of the rapamycin (mTOR) protein complex,20 whose inhalation could minimize its systemic side effects due to oral administration, but which is currently not allowed because its therapeutic use is not approved.46To obtain these particles in the micrometer range, the nano into micro (NiM) strategy was followed by entrapping Rapa-loaded nanostructures into a microparticulate matrix by spray drying (SD). The NiM particles were produced by using excipients able to dissolve into the lungs once inhaled, and releasing Rapa-loaded colloidal carriers, i.e., polymer–lipid hybrid nanoparticles (Man-LPHNPs). The latter comprise a polymeric core, where the drug was entrapped, and a phospholipid shell, capable of improving some characteristics of the resulting hybrid system, such as stability, rapid dispersibility of physiological fluid, and biocompatibility.
Lipid–polymer hybrid nanoparticles (LPHNPs) production
In detail, the polymeric core of the hybrid nanostructures was made from a graft copolymer of α,β-poly(N-2-hydroxyethyl)-DL-aspartamide (PHEA), which is a biocompatible polymer, highly versatile from a chemical point of view, and widely used to produce biocompatible materials for drug delivery.30,47 The fluorescent probe rhodamine B (RhB) and the polyester poly(lactic-co-glycolic acid) (PLGA) were covalently grafted to the PHEA backbone, with the aim of making the resulting polymer fluorescent for technological and biological purposes, and insoluble in water to obtain nanoparticles. The chemical structure of the PHEA-g-RhB-g-PLGA and its synthetic pathway are shown in Fig. 1A. The characterization of PHEA-g-RhB-g-PLGA by FT-IR (Fig. 1B), 1H-NMR (Fig. 1C) and size-exclusion chromatography (SEC) analysis (Fig. 1D) confirmed its functionalization with 4.8 mol% of PLGA. The obtained average molecular weight (w) value of about 156.0 kDa agrees with the expected increase in w by functionalizing the PHEA-g-RhB with 4.8 mol% of PLGA. By using 1H-NMR analysis we have also quantified the functionalization of PHEA with 0.55 mol% of RhB (Fig. S1A†), and confirmed the presence of RhB still linked to the copolymer after the next step of PLGA grafting (Fig. S1B†).
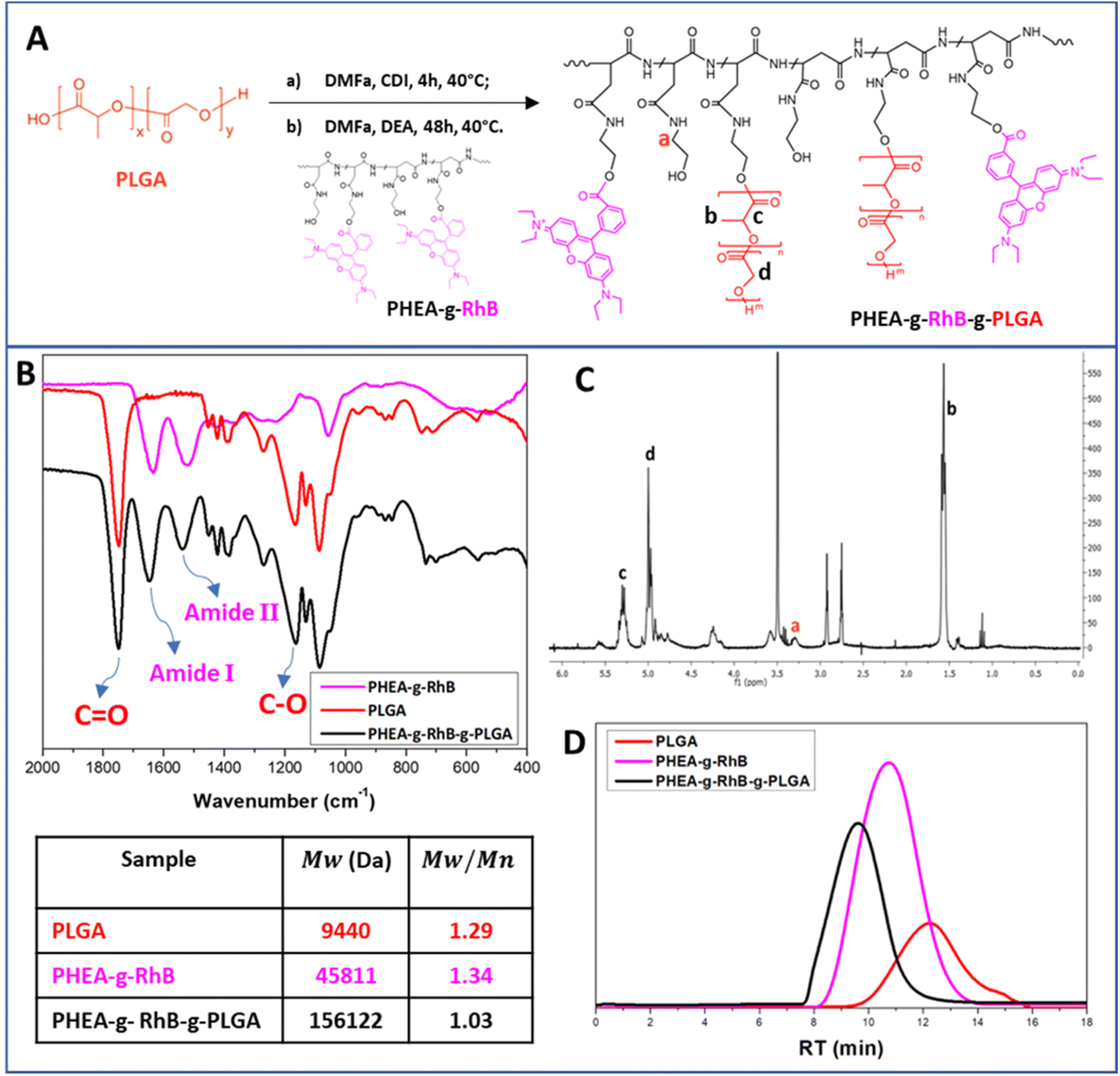 | ||
| Fig. 1 (Panel A) Synthetic pathway to obtain the PHEA-g-RhB-g-PLGA graft copolymer; (Panel B) the FT-IR spectra of PHEA-g-RhB (fuchsia line), PLGA (red line) and PHEA-g-RhB-g-PLGA graft copolymer (black line); (Panel C) 1H-NMR spectrum of PHEA-g-RhB-g-PLGA graft copolymer; (Panel D) SEC chromatograms (left) of PHEA-g-RhB (fuchsia line), PLGA (red line) and PHEA-g-RhB-g-PLGA graft copolymer (black line) and obtained w and polydispersity values (w/n) (right). | ||
Starting from the PHEA-g-RhB-g-PLGA, negatively charged Rapa-containing nanoparticles of about 120 nm were prepared by the nanoprecipitation technique (named Rapa@nanoparticles). These particles showed significantly greater sizes compared to the empty ones (Table 1), indicating that the drug entrapment affects particle size. Compared with previously described Rapa-loaded nanoparticles, obtained from an amphiphilic graft polycaprolactone/PHEA derivative, they are significantly larger in size (120.8 nm vs. 51.1 nm), probably due to a reduction in hydrophobic behavior of the graft PLGA-based copolymer.35
To obtain the lipid/polymer hybrid nanoparticles, liposomal vesicles were produced from the 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-(polyethylene glycol)-mannose sodium salt (DSPE-PEG2000-mannose) by a thin layer evaporation (TLE) technique.33 DPPC was chosen due to its lung biocompatibility/tolerability, being the main endogenous component of the pulmonary surfactant, as well as for its ability to interact with it.32,33,48 Additionally, DSPE-PEG2000-mannose was selected either for its ability to further stabilize the colloidal particles, and above all to favor the macrophage targeting thanks to mannose exposure on the outer shell of the Man-LPHNPs.6,8,49 In fact, macrophages are increased in the lungs of COPD patients, and overexpress mannose receptors such as CD-206, CD-163 and CD-204 on their surface.50
Man-LPHNPs and Rapa@Man-LPHNPs were prepared by mixing the two dispersions containing the polymeric nanoparticles (respectively empty or drug-loaded) and liposomal vesicles. This mixture was then subjected firstly to high-pressure homogenization (HPH), and ultra-centrifugation of the homogenized dispersion to separate the hybrid systems from the nanoparticles or liposomal vesicles alone.
Thanks to the lipid shell, the drying does not cause significant changes in particle size, nor in polydispersity of the empty and drug-loaded Man-LPHNPs after re-dispersion in distilled water (Table 1). The negative ζ-potential values are not significantly influenced by the incorporation of the drug, nor by the freeze-drying or the re-dispersion process in bidistilled water (Table 1).
The quantity of phospholipids, determined by a colorimetric assay and expressed as wt% of the Man-LPHNPs, resulted in 63 ± 4 and 60 ± 3 wt% for the empty and Rapa-loaded samples, respectively, slightly lower than the theoretical values.
DSC thermograms of polymeric nanoparticles (A), liposomal vesicles (B) and Rapa@Man-LPHNPs (C) confirmed the successful formation of the hybrid systems (Fig. 2).
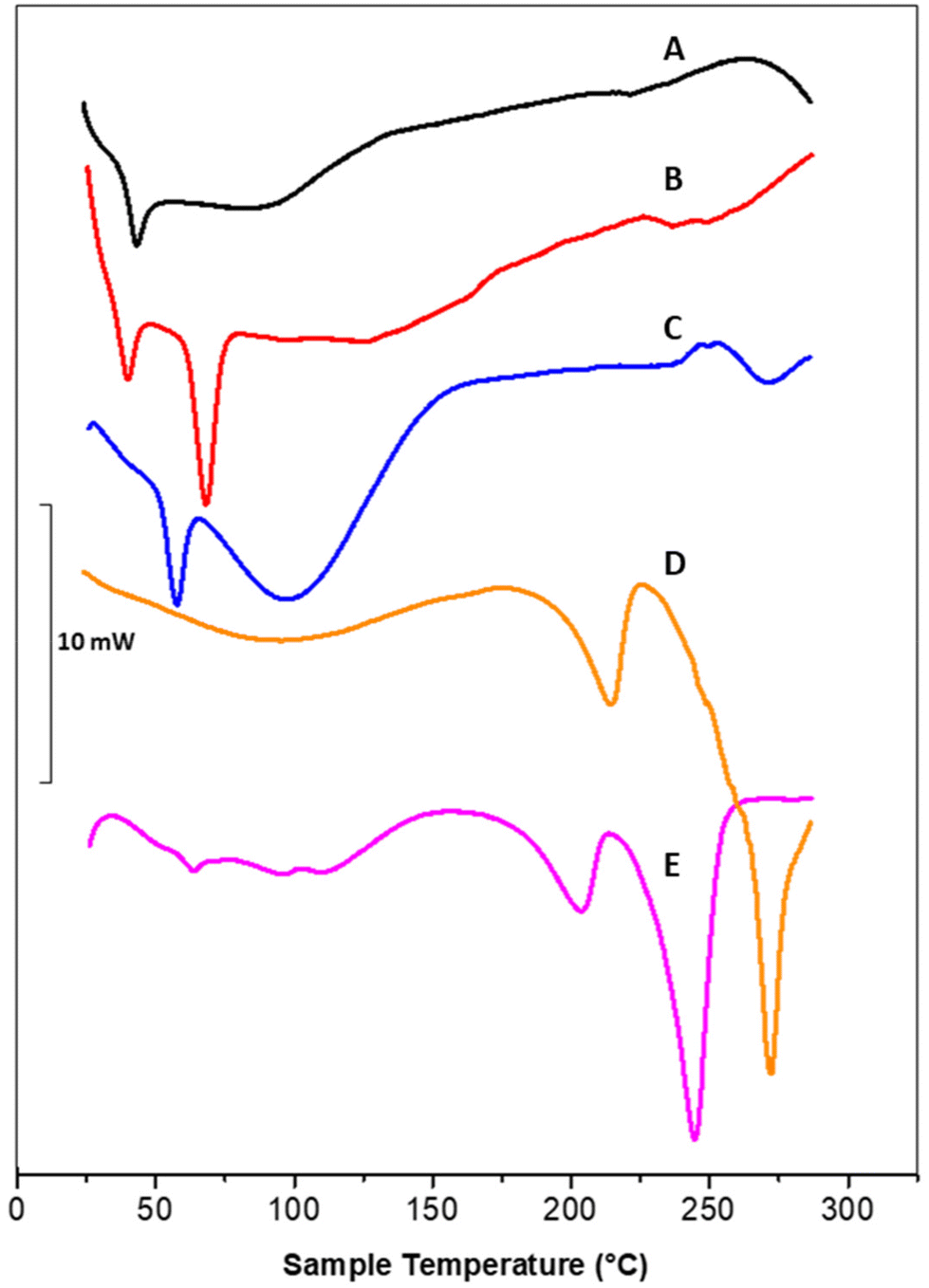 | ||
| Fig. 2 Representative DSC thermograms of: (A) polymeric nanoparticles (black line), (B) liposomes (red line), (C) Rapa@Man-LPHNPs (blue line), (D) PVA:LEU_AB10 microparticle sample (at 75:25 wt/wt) (orange line), and (E) NiM@Rapa sample (fuchsia line). | ||
In particular, the thermogram of the polymeric nanoparticles (black line, A) shows one endothermic peak at 43.8 °C, attributed to the glass transition temperature (tg) of PLGA and a broad peak at 88.6 °C, attributed to the PHEA-g-RhB-g-PLGA melting/degradation; the thermogram of the liposomal vesicles (red line, B) shows two peaks, at 40.4 and 68.8 °C, attributed to the sol–gel phase transitions of DSPE-PEG2000-mannose and DPPC, respectively. The profile of Rapa@LPHFNPs (blue line, C) shows two endothermic peaks that do not overlap with those recorded on nanoparticle and liposomes separately. This result suggests that the formation of hybrid systems successfully occurred by interactions between the lipids and the polymer, affecting the sol–gel transition temperature of phospholipids. On the other hand, the copolymer constituting the core could no longer be clearly visible due to the lower percentage by weight in the total sample, compared to the amount of phospholipids.
Moreover, the Rapa@Man-LPHNPs thermograms do not show any endothermic peaks, which could be attributed to the drug (at about 178.4 °C, data not shown), indirectly indicating the presence of amorphous drug in the Man-LPHNPs.51 The transition temperature values of obtained samples are reported in Table S1.†
To confirm the formation of nanosized particles with the hypothesized core–shell structure, a TEM analysis was performed on the Rapa@Man-LPHNPs aqueous dispersion (Fig. 3).
 | ||
| Fig. 3 TEM photogram of Man-LPHNPs particles. | ||
The image shows the presence of nanosized particles with an internal zone, the polymeric core, and a denser superficial zone, attributed to the phospholipid layer.
The drug loading %, i.e., the mass percentage ratio of incorporated drug to drug-loaded hybrid system, was found to be 4.3 wt%, while the entrapment efficiency (EE%), that is the weight ratio percentage of the entrapped drug and the theoretical value, was 95.6 wt%.
To evaluate the ability of the entrapped drug, corresponding to the 4.3 wt% of the weight of drug-loaded hybrid nanoparticles, to be released after administration, a cumulative release study in simulated lung fluid (modified SLF4, pH 7.4) was performed by using the dialysis method, under sink conditions.36 The cumulative amounts of released drug, expressed as the weight percentage of the loaded drug (that is the 4.3 wt% of the drug-loaded hybrid nanoparticle's weight), are reported in Fig. 4A as a function of the incubation time (red dots).
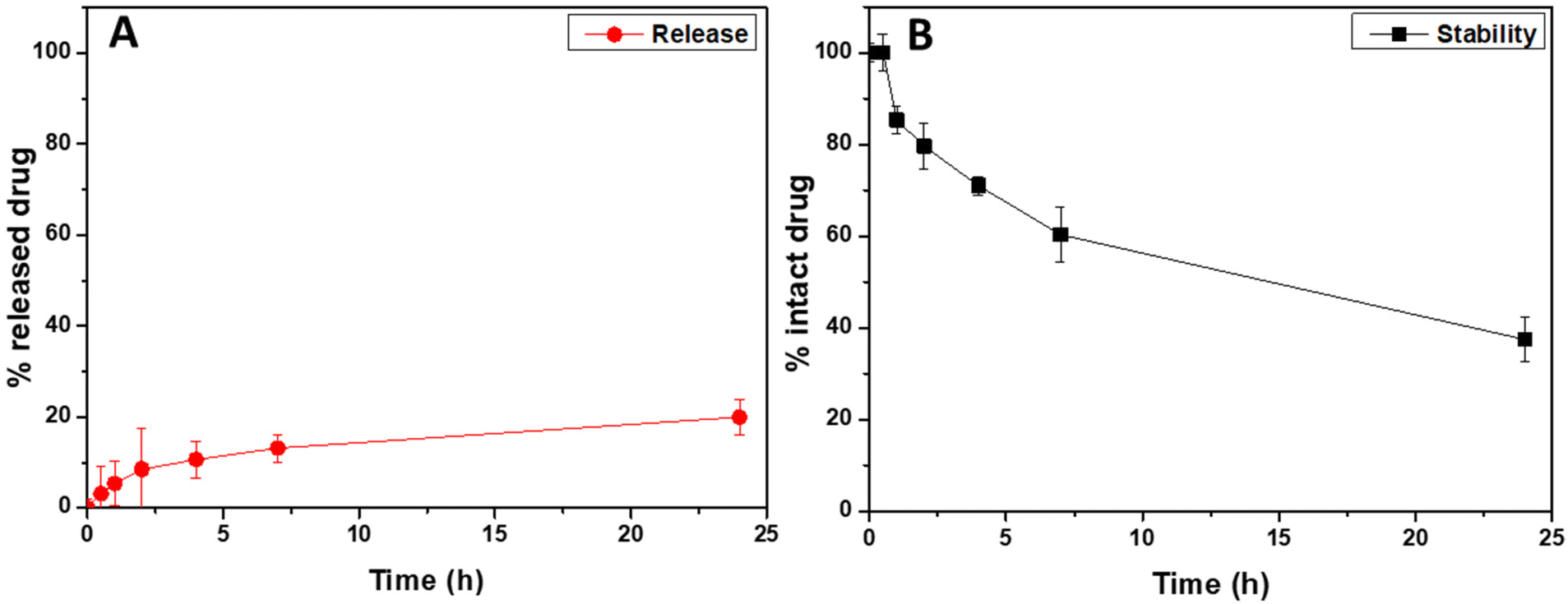 | ||
| Fig. 4 Rapa cumulative release profile from the Rapa@Man-LPHFNPs sample, expressed as wt% of the drug-loaded nanoparticle sample (A), and stability profile of the free drug (B), performed in simulated lung fluid (SLF4, pH 7.4). | ||
As can be clearly seen, the Rapa incorporated into Man-LPHNPs is slowly released in the incubation medium, without any burst effect, with the amount of released drug being about 20 wt% after 24 h of incubation. Compared to hybrid particles based on the PCL/PHEA graft copolymer, a higher drug amount is released due to the less hydrophobic core.35
Furthermore, in addition to the amount of released drug, we quantified the content of the intact drug still entrapped within the drug-loaded hybrid particles after 24 h of incubation, which amounted to approximately 74% by weight of the initially placed amount. Considering that Rapa is known to undergo hydrolysis in physiological fluids, we conducted a drug stability study by incubating free Rapa in SLF4 at the same concentration used for the release study. The stability profile of free Rapa, as illustrated in Fig. 4B (represented by black squares), indicates that approximately 60% by weight of the free Rapa degrades after 24 h of incubation (only the 40% of intact drug was recovered compared to the 74% found in Rapa-loaded hybrid nanoparticles). These findings suggest that loading the drug into the hybrid system provides protection against hydrolytic degradation, resulting in approximately 74% by weight of intact drug content after 24 h of incubation in SLF4 (considering both the released and entrapped drug). This enhanced stability potentially increases drug bioavailability when entrapped within such carriers.35,51 In conclusion, the Man-LPHNPs we have developed prove to be excellent carriers for Rapa, as they not only increase drug stability but also facilitate controlled release of the drug in simulated pulmonary fluid.
As the biocompatibility of the administered formulation is essential for the in vivo fate of the drug-loaded carriers, an in vitro test was carried out. In particular, the viability of bronchial epithelial cells (16-HBE) and macrophages (Raw 264.7) incubated for up to 48 h in the presence of Man-LPHNPs systems, empty or loaded with Rapa, at different drug concentrations, was evaluated. Furthermore, as a control, pegylated hybrid systems (LPHNPs) obtained from a DPPC/DSPE-PEG2000 mixture, empty or loaded with Rapa (named LPHNPs and Rapa@LPHNPs, respectively), were tested. Results, reported in Fig. S2A† for 16-HBE and in Fig. S2B† for Raw 264.7, showed high biocompatibility of all the tested samples, both after 24 and 48 h of incubation.
It is widely known that lung cells mainly involved in the progression of COPD are macrophages. Therefore, the ability of the hybrid mannosylated particles to be more specifically internalized by macrophages, given that mannose is a ligand for the lectin receptor highly expressed on the surface of macrophage cells, was evaluated by exploiting the emission property of RhB in the red region of the light spectrum.
For this purpose, the mannosylated and unmannosylated systems were incubated in the presence of the macrophage and bronchial epithelial cells, and after 2, 6 and 24 h the intracellular fluorescence was quantified (Fig. 5A) and highlighted by fluorescence microscopy (Fig. 5B and C).
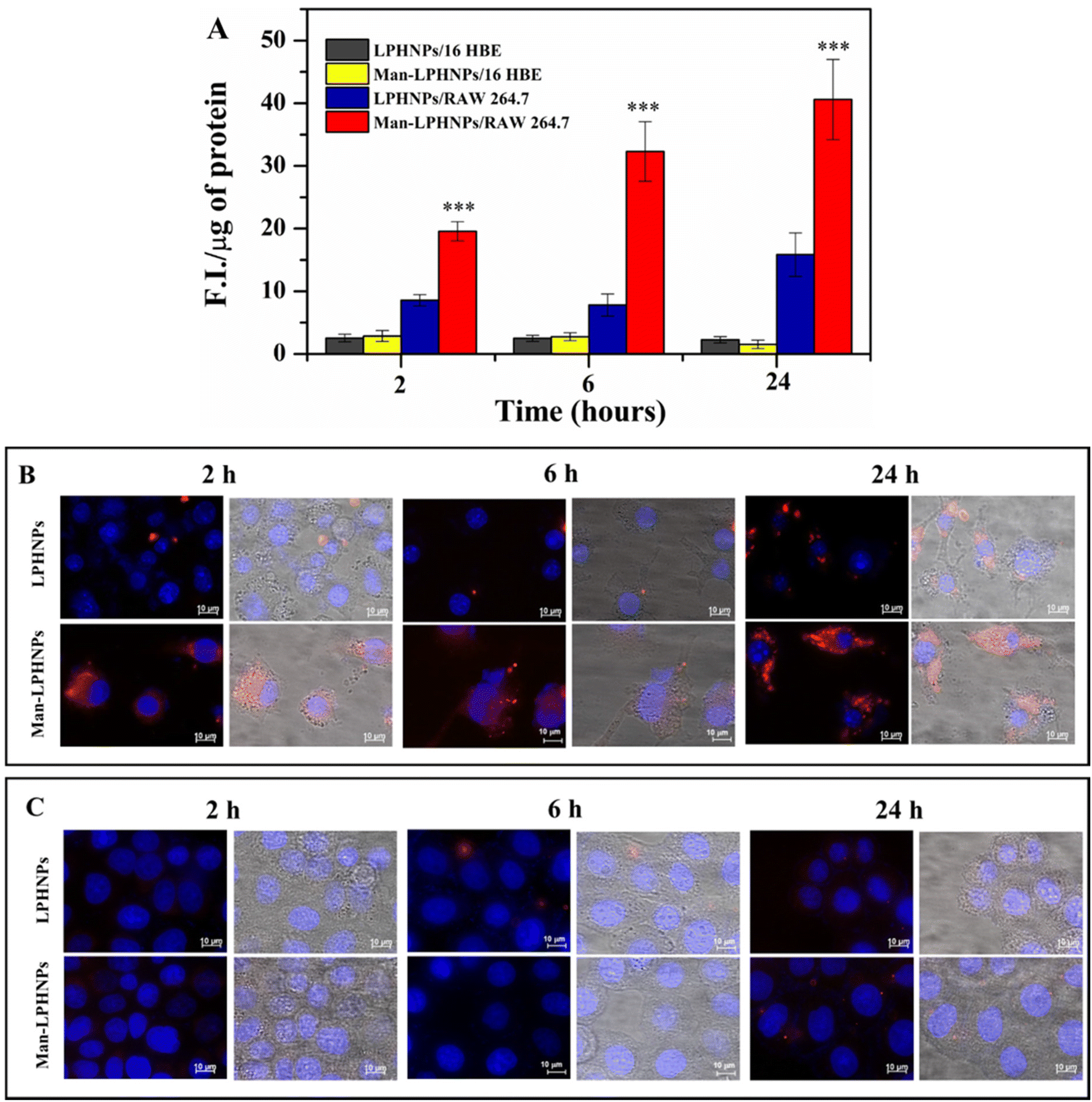 | ||
| Fig. 5 Cell uptake: quantitative fluorescence nanoparticle uptake (Panel A), fluorescence and bright microscopy images of Raw264.7 cells (Panel B) and 16-HBE cells (Panel C) treated with Man-LPHNPs and LPHNPs after 2, 6 and 24 h. The scale bar represents 10 μm. | ||
The results, reported in Fig. 5A, illustrate the normalized fluorescence intensity (F.I.) per μg of proteins present in the same cell lysate. In particular, Man-LPHNPs exhibited a significant advantage in terms of internalization into macrophage cells over the untargeted particles (p < 0.001), after just 2 hours of incubation, and this process is time-dependent, as the fluorescence intensity increases over time reaching a maximum uptake within 24 h. These differences in the uptake within the epithelial cells were not significant among Man-LPHNPs and LPHNPs, where a lower uptake is also observed. Moreover, multi-channel fluorescence micrographs reinforced our findings arising from the quantitative uptake study, highlighting that Man-LPHNPs more efficiently enter RAW 264.7 cell lines since a marked red fluorescence was registered at all the incubation times (Fig. 5B). In contrast, concerning the 16-HBE uptake, the obtained data, in accordance with the quantitative assay, show that a low fluorescence is observed for all systems at the same incubation time (Fig. 5C), demonstrating the key role of mannose in the internalization process in the macrophagic cells. These findings not only validate the effectiveness of our targeting strategy but also confirm the potentiality of the designed carriers for Rapa delivery directly to the target cells, once inhaled, which is a crucial aspect for applications in lung inflammation research.
Inhalable particle production
Given that these freeze-dried systems are re-dispersed in an aqueous fluid very easily, without the formation of aggregates (Table 1), a phenomenon which normally occurs with polymeric nanoparticle systems and that requires high quantities of cryoprotectants, they could potentially be dispersed in a physiological medium and administered by nebulization.However, to administer Rapa@Man-LPHNPs as inhalable powders (through the pulmonary route) microparticles of water-dispersible excipients containing them have been produced using the nano into micro (NiM) strategy. Once inhaled, the NiM might dissolve in contact with the pulmonary fluids, releasing the nanosized structures, whose in vivo fate will depend on the surface characteristics and/or the presence of targeting moieties. In the case of the Rapa@Man-LPHNPs, the hydrophilic shell could facilitate their diffusion through the mucus to reach the pulmonary epithelium and the surface decoration with mannose, and therefore their interaction with airway macrophages.33
A mixture of polyvinyl alcohol (PVA) and L-leucine (LEU) was chosen as biocompatible excipients to form the microparticles matrix, because of their easy processability via spray drying (SD), and the good ability to improve the aerosolization efficiency of microparticles and to reduce the aggregation of entrapped particles.52–54
To optimize the microparticle production process and find the most suitable operating conditions for the realization of NiMs, preliminary experiments were carried out. In particular, microparticles (PVA:LEU_NT) were obtained by using a mixture H2O:EtOH 90:10 (v/v) as feed liquid, a 5% dispersion of PVA:LEU 75:25 wt:wt, a 150 μm nozzle, 110 °C as inlet temperature and 15% flow rate.
Furthermore, to modulate the chemical–physical properties of the particles, ammonium bicarbonate (AB) was added to the liquid feed immediately before being processed, at AB concentrations of 10 wt% (PVA:LEU_AB10) and 20 wt% (PVA:LEU_AB20) with respect to the total PVA:LEU mixture. AB is a pore-forming agent widely used in the SD process to obtain porous particles55,56 as it decomposes above 50 °C giving rise to the gas responsible for the formation of pores/bubbles, which modulate the chemical–physical characteristics as well as the aerodynamic properties of obtained particles.37,57
In Table 2, the composition of the liquid feeds to obtain each microparticle sample is reported.
SEM images of the PVA:LEU_NT sample, and those of PVA:LEU_AB10 and PVA:LEU_AB20 samples, obtained in the presence of 10 and 20 wt% AB, respectively, show powders without aggregates (Fig. 6A).
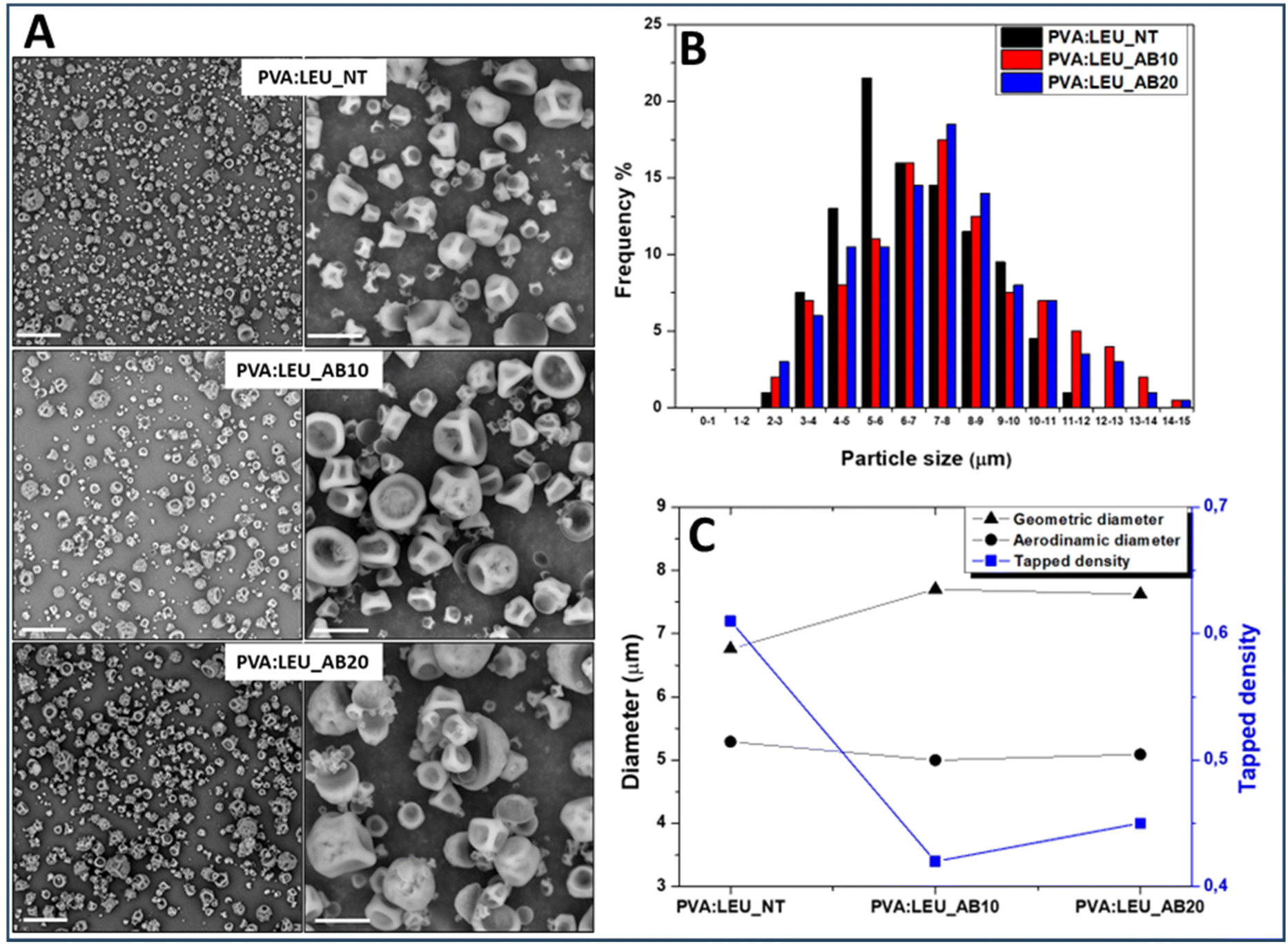 | ||
| Fig. 6 Characterization of microparticle samples as a function of AB amount (0 wt%, PVA:LEU_NT; 10 wt%, PVA:LEU_AB10; and 20 wt%, PVA:LEU_AB20). (Panel A) SEM images (magnification: on the left 1000× and on the right 5000×), the scale bar: 40 μm (left), 10 μm (right); (Panel B) size distribution; (Panel C) geometric diameter (dgeom), tapped density (ρtapped) and theoretical aerodynamic diameter (daer). | ||
The dimensional analysis of the obtained powders, carried out on the SEM images (on a particle number > 500) by ImageJ software, is reported as size distribution curves for each sample (Fig. 6B). As can be seen, as the quantity of AB increases (from 0% to 20%) there is a shift towards the right of the Gaussian, which means larger particle size values, passing from 6.76 ± 2.13 μm for PVA:LEU_NT to 7.62 ± 3.01 μm for PVA:LEU_AB20 (p < 0.001).
The complete decomposition of AB under the operating conditions of the SD process was confirmed by FT-IR (Fig. S3†), since the spectra of the samples are superimposable and do not show peaks attributable to AB.
Using the syringe method,58 the value of the tapped density of each sample (ρtapped) was determined and the theoretical aerodynamic diameter (daer) was calculated (Table S2†). The comparison between the values of dgeom, ρtapped and daer is shown in the graph reported in Fig. 6C.
As can be seen, in the presence of increasing amounts of AB, the obtained particles show significant increase (p < 0.05) of the dg value; at the same time, a significant reduction of the ρtapped value is determined (p < 0.05). As a result of such variations, a significant effect on the particle daer values is obtained, which stabilizes at a value suitable for aerosolization, i.e. at about 5 μm. However, since the PVA:LEU_AB10 and PVA:LEU_AB20 samples do not exhibit significant differences in terms of ρtapped, dgeom and daer, the sample obtained with AB at 10 wt% was selected for the production of NiM.
Two NiM samples were produced by incorporating in the PVA:LEU_AB10 matrix the empty LPHNPs, and the Rapa@Man-LPHNPs (both added at 1% w/v in the feed fluid), which were indicated, respectively, as NiM and NiM@Rapa samples. The SEM images of the obtained samples are shown in Fig. 7A, while the dimensional distributions are shown in Fig. 7B. The yield values, ρtapped, dgeom and daer are also reported in Table S2.†
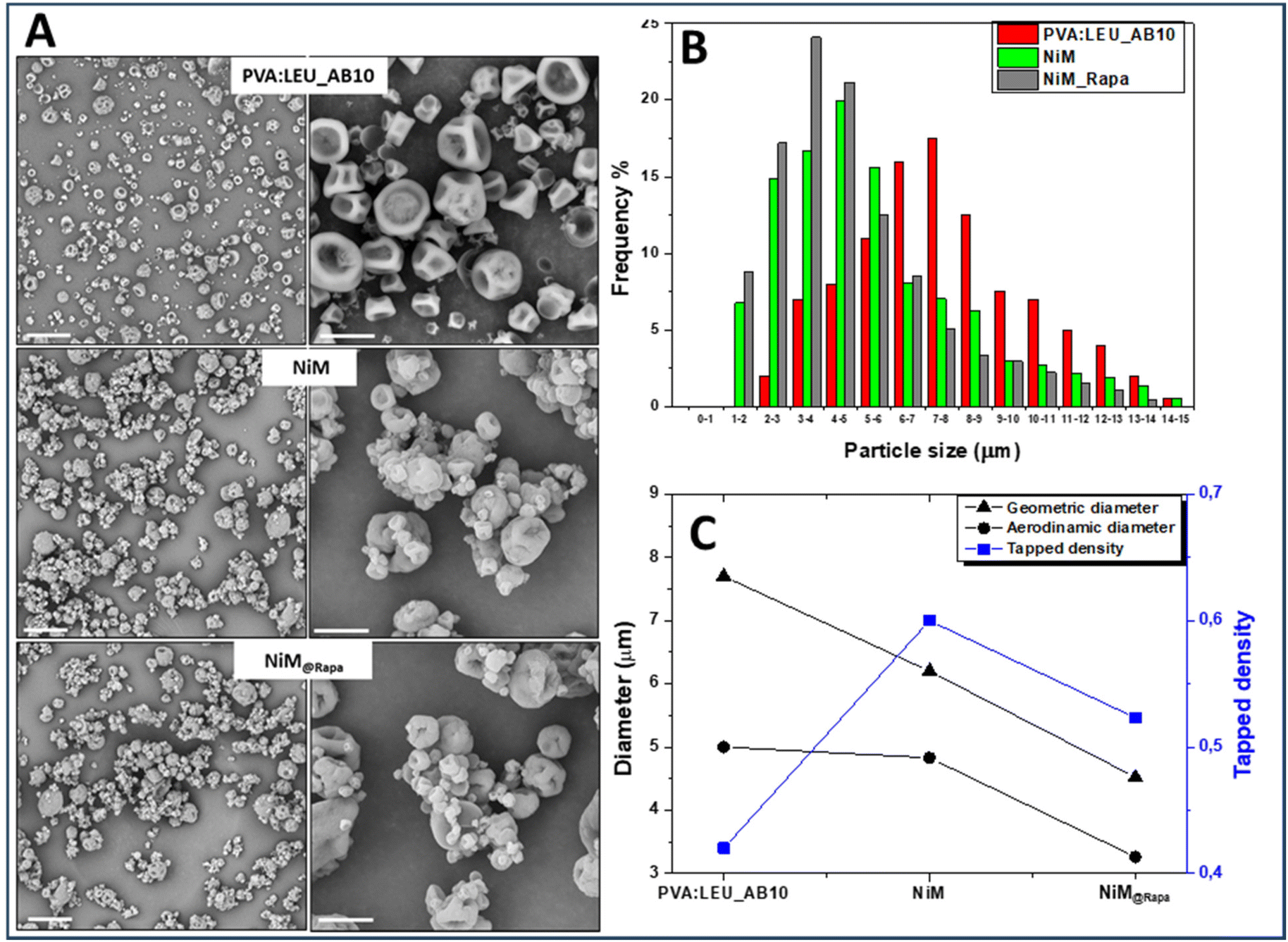 | ||
| Fig. 7 Characterization of microparticle samples, obtained in the absence of Man-LPHNPs (PVA:LEU_AB10), in the presence of empty Man-LPHNPs (NiM) or in the presence of Rapa@Man-LPHNPs (NiM@Rapa). (Panel A) SEM images (magnification: on the left 1000× and on the right 5000×), the scale bar: 40 μm (left), 10 μm (right); (Panel B) size distribution; (Panel C) geometric diameter (dgeom), tapped density (rtapped) and theoretical aerodynamic diameter (daer). | ||
From the SEM images it is evident that the presence of Man-LPHNPs (empty or containing Rapa) modifies the morphology of the microparticle samples, which appear to have a more homogeneous surface than that of the PVA:LEU_AB10 sample (Fig. 7A).
On the other hand, the size distributions reported in Fig. 7B clearly showed that with the addition of the empty and the drug-loaded Man-LPHNPs, there is a shift towards the left of the Gaussian, which means lower particle size values, passing from 7.70 ± 3.60 μm for PVA:LEU_AB10 to 4.52 ± 3.47 μm for NiM@Rapa (p < 0.001).
The DSC analysis of PVA:LEU_AB10 and NiM@Rapa samples, as reported in Fig. 2, confirms that the presence of Man-LPHNPs in the matrix modifies the properties of the matrix excipients, given that the melting temperature values of the PVA:LEU_AB10 microparticle sample (respectively at 215.0 °C and 272.3 °C) shifted to lower values (respectively at 203.8 and 245.0 °C) in the NiM@Rapa sample. On the other hand, these endothermic peaks obtained from the PVA:LEU_AB matrix are perfectly comparable to those obtained from the thermogram of the PVA:LEU physical mixture at 75:25 wt:wt, and corresponding, respectively, to the melting point of PVA and LEU alone.
Furthermore, from the analysis of dgeom, ρtapped and daer values reported in Fig. 7C, by comparing PVA:LEU_AB10 and NiM@Rapa samples, the values of dgeom decrease (7.70 vs. 4.52 μm) while ρtapped values increase (0.420 vs. 0.523 g ml−1). These variations lead to an overall reduction in the daer, from 5.00 μm for the PVA:LEU_AB10 sample to 3.26 μm for the NiM@Rapa sample, indicating that the presence of Man-LPHNPs determines a decreasing effect on the aerodynamic characteristics of the obtained particles. This value falls perfectly within the ideal size range for effective aerosolization and inhalation of these particles into the lungs.
To further investigate the aerosolization behavior, the microparticles were then tested using the DPI Breezhaler® connected to the NGI, and the drug deposition (using RhB as tracking agent) and aerodynamic parameters were determined. As can be seen in Fig. 8, PVA:LEU_NT and PVA:LEU_AB10 samples show a similar behavior in terms of deposition on the NGI, accumulating predominantly in the first stages, whereas, the increase in the porogen concentration (PVA:LEU_AB20) leads to a lower deposition in the these stages (Fig. 8), indicating a deterioration of their aerosolization properties. Interestingly, although they show significant differences in morphology, dgeom, and daer values, the NiM particles showed an analogous deposition to the corresponding microparticles made only of excipients (PVA:LEU_AB10).
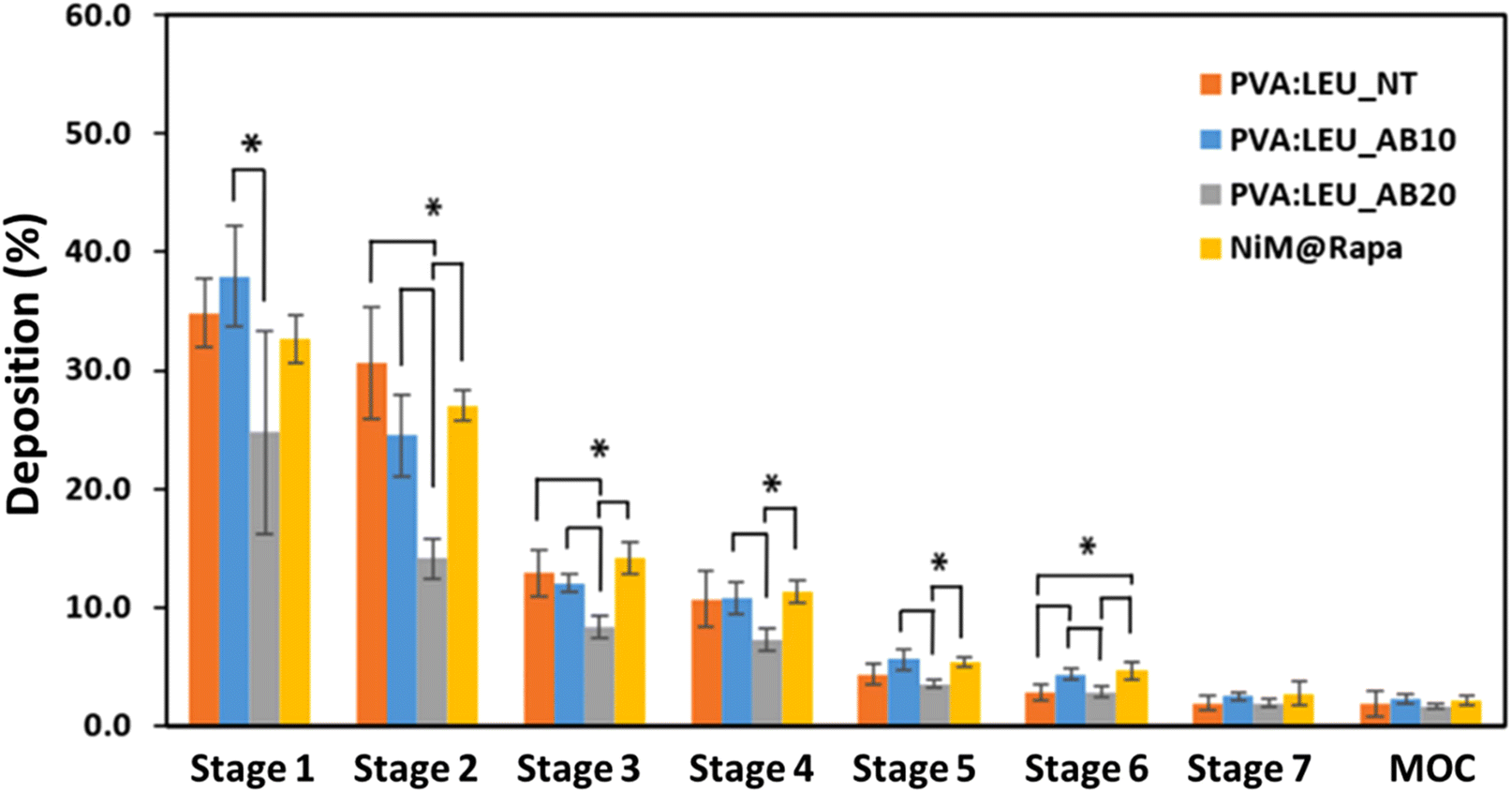 | ||
| Fig. 8 Deposition of MP samples on the stages of the NGI after testing with Breezhaler® at a flow rate of 90 l min−1 (*p < 0.05) (MOC = micro-orifice collector). | ||
The resulting aerodynamic parameters (Table 3) revealed that all the tested samples reached a high ED – and thus high aerosolization efficiency – with slightly lower and superimposable values for PVA:LEU_AB10 and NiM@Rapa samples. With respect to the FPF%, values ranging from 26 to ∼28 were observed, with no significant difference among the samples. Lastly, the MMAD resulted in being lower than 5 μm in all cases with resulting DI values lower than unitary, proving the feasibility of the produced microparticles to be administered to the lungs. These results, in terms of FPF and MMAD, show a better aerosolization behavior for these particles than those described elsewhere as carriers for Rapa, obtained using both mannitol-based NiM particles35 and polymeric microparticles.37
| PVA:LEU_NT |
PVA:LEU_AB10 |
PVA:LEU_AB20 |
NiM@Rapa | |
|---|---|---|---|---|
| ED% | 82.4 ± 3.8*,§ | 71.2 ± 5.4* | 77.4 ± 7.2§ | 70.3 ± 8 |
| FPD (mg) | 6.2 ± 1.2 | 6.2 ± 1.2 | 5.9 ± 0.5 | 5.6 ± 0.5 |
| FPF (%) | 26.0 ± 4.8 | 28.2 ± 6.1 | 26.2 ± 0.7 | 27.5 ± 1.9 |
| MMAD (μm) | 4.7 ± 0.2 | 4.5 ± 0.2 | 4.1 ± 0.2 | 4.3 ± 0.2 |
| GSD | 2.6 ± 0.2 | 2.9 ± 0 | 2.9 ± 0.1 | 2.7 ± 0 |
| DI | 0.9 | 0.9 | 0.8 | 0.9 |
Given the optimal results in terms of nebulization performances, the NiM@Rapa particles were then further studied to evaluate their ability, once deposited on the lungs, to release the Rapa@Man-LPHNPs without aggregates and to modify the characteristics of the pathological mucus.
The re-dispersion test, made by dispersing a known amount of NiM@Rapa in milliQ H2O, showed mean size values lower than 200 nm and ζ-potential value superimposable to that obtained by analyzing the freshly prepared Man-LPHNPs. Therefore, the SD process does not modify the Rapa@Man-LPHNPs aggregation state, giving a homogeneous dispersion that is free of aggregates.
Finally, to evaluate the effect of the NiM@Rapa on the rheological properties of the artificial mucus, a frequency sweep analysis was carried out, and the complex viscosity (η) was determined as a function of the applied shear stress on the mucus alone, and in the presence of NiM@Rapa or PVA:LEU_AB10 microparticles. The artificial mucus was prepared according to a previously published paper.33 Results are reported in Fig. 9.
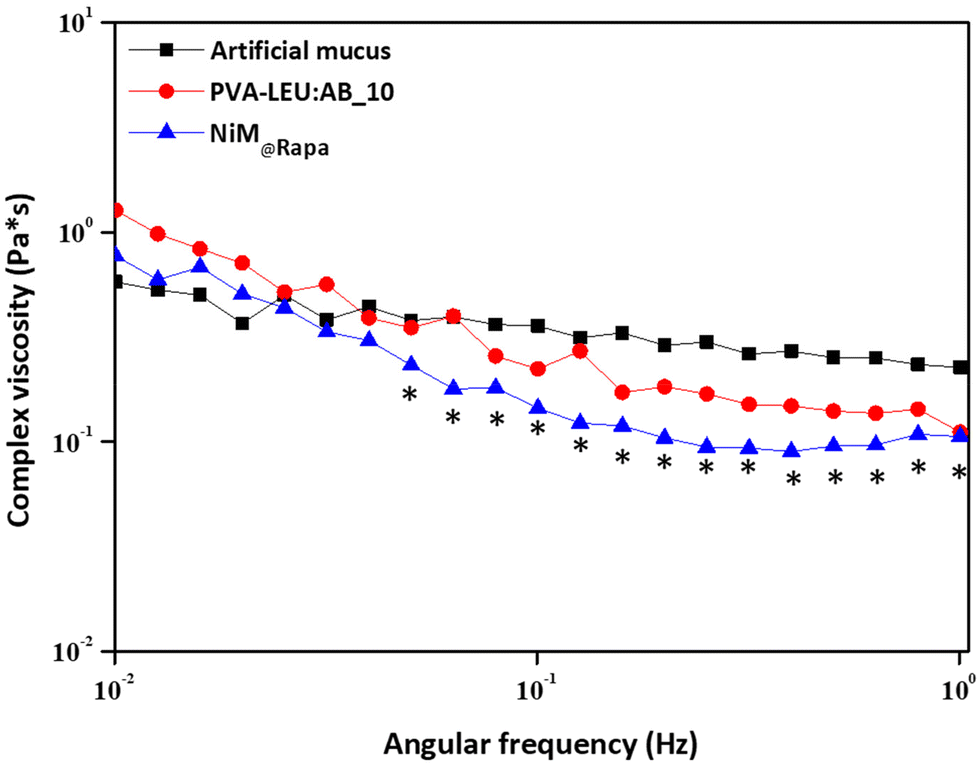 | ||
| Fig. 9 Complex viscosity (η) as a function of the applied shear stress of the mucus alone, or in the presence of the PVA:LEU_AB10 or the NIM@Rapa samples (*p < 0.05). | ||
As can be observed, the η value of the artificial mucus has an almost constant trend, with a slight reduction as a function of the applied shear stress. In the presence of the NiM@Rapa sample, the η decreases significantly at higher values of applied shear stress (*p < 0.05). Although a change in the complex viscosity trend is also recorded in the presence of particles that do not contain Man-LPHNPs (PVA:LEU_AB10 microparticles), this is not significant with respect to the mucus alone. Therefore, the presence of the Man-LPHNPs decreases the viscosity of the mucus, i.e., reducing the interactions between the mucus components and potentially favoring their diffusion through its meshes.
4. Conclusions
In this work, inhalable nano into micro (NiM) particles containing rapamycin (Rapa) have been prepared to obtain a new targeted delivery system potentially useful for the treatment of COPD-related inflammation.Firstly, Rapa-loaded lipid–polymer hybrid nanoparticles (Man-LPHNPs) were prepared, consisting of a polymeric core, obtained by starting from the copolymer PHEA-g-RhB-g-PLGA, and a lipid shell containing DPPC and DSPE-PEG2000-mannose as phospholipid components.
The core–shell structure was confirmed by TEM investigation. These systems showed colloidal size and slightly negative zeta potential. Furthermore, the obtained Man-LPHNPs showed a significant capability to load rapamycin, effectively shielding the payload from hydrolytic degradation, and ensuring controlled drug release in simulated pulmonary fluid. Additionally, the surface functionalization with mannose allows higher internalization by macrophage cell lines.
To produce inhalable microparticles, the Man-LPHNPs were subsequently incorporated by spray drying (SD) into a micrometer matrix consisting of PVA–LEU. Ammonium carbonate (AB) at 10 wt% has been successfully used to ameliorate the chemical–physical and technological properties of the obtained particles. The SEM investigation confirms the production of homogeneous microparticles without aggregates.
A nebulization test with NGI showed high values of FPF% and MMAD below 5 μm of the obtained particles, confirming their potential applicability for the administration of Rapa as inhalable dry powder. Furthermore, rheological analyses indicated that the system does not appear to be mucoadhesive, given the reduced ability of NiM systems to interact with the mucus.
In conclusion, the overall results proved that NiM systems are to be considered as a promising and innovative approach for inhalation therapy as they show adequate properties in terms of characteristics for aerosolization, low interaction with mucus and drug release profile from the Man-LPHNPs incorporated into NiM@Rapa.
Author contributions
The project was conceptualized and supervised by E. F. C. and G. C. All formal analyses were conducted by M. C., C. S. and L. C. Data curation was done by E. F. C. and F.L. The manuscript was written by E. F. C., L. C. and C. S., and was reviewed and edited by all contributing authors.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank the ATeNCenter of University of Palermo – Laboratory of Preparation and Analysis of Biomaterials for analysis instruments, and Mr Francesco Paolo Bonomo for technical support.The research leading to these results has received funding from the European Union – NextGenerationEU through the Italian Ministry of University and Research under PNRR - M4C2-I1.3 Project PE_00000019: “Health Extended ALliance for Innovative Therapies, Advanced Lab-research, and Integrated Approaches of Precision Medicine – HEAL ITALIA” to Gennara Cavallaro. CUP: B73C22001250006.
The views and opinions expressed are those of the authors only and do not necessarily reflect those of the European Union or the European Commission. Neither the European Union nor the European Commission can be held responsible for them.
References
- A. I. Gomez, M. F. Acosta, P. Muralidharan, J. X. J. Yuan, S. M. Black, D. Hayes and H. M. Mansour, Pulm. Pharmacol. Ther., 2020, 64, 101975 CrossRef CAS PubMed.
- W. Wang, Z. Huang, Y. Huang, X. Zhang, J. Huang, Y. Cui, X. Yue, C. Ma, F. Fu, W. Wang, C. Wu and X. Pan, Adv. Drug Delivery Rev., 2022, 185, 114309 CrossRef CAS PubMed.
- Y. He, Y. Liang, R. Han, W. L. Lu, J. C. W. Mak and Y. Zheng, J. Controlled Release, 2019, 314, 48–61 CrossRef CAS PubMed.
- M. Mehta, Deeksha, N. Sharma, M. Vyas, N. Khurana, P. K. Maurya, H. Singh, T. P. Andreoli de Jesus, H. Dureja, D. K. Chellappan, G. Gupta, R. Wadhwa, T. Collet, P. M. Hansbro, K. Dua and S. Satija, Chem.-Biol. Interact., 2019, 304, 10–19 CrossRef CAS PubMed.
- N. Ahamad, A. Kar, S. Mehta, M. Dewani, V. Ravichandran, P. Bhardwaj, S. Sharma and R. Banerjee, Biomaterials, 2021, 274, 120875 CrossRef CAS PubMed.
- A. Costa, B. Sarmento and V. Seabra, Eur. J. Pharm. Sci., 2018, 114, 103–113 CrossRef CAS PubMed.
- P. Kumbhar, A. Manjappa, R. Shah, N. K. Jha, S. K. Singh, K. Dua, J. Disouza and V. Patravale, J. Controlled Release, 2022, 341, 1–15 CrossRef CAS PubMed.
- M. Mukhtar, N. Csaba, S. Robla, R. Varela-Calviño, A. Nagy, K. Burian, D. Kókai and R. Ambrus, Pharmaceutics, 2022, 14, 1543 CrossRef CAS PubMed.
- H. Chung, J. Y. Park, K. Kim, R. J. Yoo, M. Suh, G. J. Gu, J. S. Kim, T. H. Choi, J. W. Byun, Y. W. Ju, W. Han, H. S. Ryu, G. Chung, D. W. Hwang, Y. Kim, H. R. Kang, Y. R. Na, H. Choi, H. J. Im, Y. S. Lee and S. H. Seok, ACS Nano, 2022, 16, 12262–12275 CrossRef CAS PubMed.
- M. Mukhtar, H. Ali, N. Ahmed, R. Munir, S. Talib, A. S. Khan and R. Ambrus, Expert Opin. Drug Delivery, 2020, 17, 1239–1257 CrossRef CAS PubMed.
- S. C. Johnson, P. S. Rabinovitch and M. Kaeberlein, Nature, 2013, 493, 338–345 CrossRef CAS PubMed.
- P. J. Barnes, J. Baker and L. E. Donnelly, Clin. Sci., 2022, 136, 733–746 CrossRef CAS PubMed.
- Z. Zhou, S. Liang, Z. Zhou, J. Liu, J. Zhang, X. Meng, F. Zou, H. Zhao, C. Yu and S. Cai, Respir. Res., 2023, 24, 8 CrossRef CAS PubMed.
- L. Liu, Y. Qin, Z. Cai, Y. Tian, X. Liu, J. Li and P. Zhao, Phytomedicine, 2021, 92, 153759 CrossRef CAS PubMed.
- Y. Y. Yong, L. Zhang, Y. J. Hu, J. M. Wu, L. Yan, Y. R. Pan, Y. Tang, L. Yu, B. Y. K. Law, C. L. Yu, J. Zhou, M. Li, D. L. Qin, X. G. Zhou and A. G. Wu, Clin. Immunol., 2022, 244, 109093 CrossRef CAS PubMed.
- A. C. Racanelli, S. A. Kikkers, A. M. K. Choi and S. M. Cloonan, Autophagy, 2018, 14, 221–232 CrossRef PubMed.
- M. Bodas and N. Vij, Respir. Res., 2017, 18, 83 CrossRef PubMed.
- Y. Wang, J. Liu, J.-S. Zhou, H.-Q. Huang, Z.-Y. Li, X.-C. Xu, T.-W. Lai, Y. Hu, H.-B. Zhou, H.-P. Chen, S.-M. Ying, W. Li, H.-H. Shen and Z.-H. Chen, J. Immunol., 2018, 200, 2571–2580 CrossRef CAS PubMed.
- S. N. Tammam, S. El Safy, S. Ramadan, S. Arjune, E. Krakor and S. Mathur, J. Controlled Release, 2021, 337, 258–284 CrossRef CAS PubMed.
- H. Hua, Q. Kong, H. Zhang, J. Wang, T. Luo and Y. Jiang, J. Hematol. Oncol., 2019, 12, 1–19 CrossRef PubMed.
- E. R. Bentley and S. R. Little, Adv. Drug Delivery Rev., 2021, 178, 113971 CrossRef CAS PubMed.
- A. Gupta, G. Pant, K. Mitra, J. Madan, M. K. Chourasia and A. Misra, Mol. Pharm., 2014, 11, 1201–1207 CrossRef CAS PubMed.
- S. R. Carvalho, A. B. Watts, J. I. Peters, S. Liu, S. Hengsawas, M. S. Escotet-Espinoza and R. O. Williams, Eur. J. Pharm. Biopharm., 2014, 88, 136–147 CrossRef CAS PubMed.
- D.-d. Zong, X.-m. Liu, J.-h. Li, R.-y. Ouyang, Y.-j. Long, P. Chen and Y. Chen, Respir. Res., 2021, 22, 1–12 CrossRef PubMed.
- S.-w. Xu, Y.-j. Zhang, W.-m. Liu, X.-f. Zhang, Y. Wang, S.-y. Xiang, J.-c. Su and Z.-b. Liu, Exp. Lung Res., 2023, 1–10 Search PubMed.
- H. Zhang, X. Zhou, X. Chen, Y. Lin, S. Qiu, Y. Zhao, Q. Tang, Y. Liang and X. Zhong, Inflammation Res., 2019, 68, 957–968 CrossRef CAS PubMed.
- Y. Xiaofei, L. Tingting, W. Xuan and H. Zhiyi, Front. Pharmacol., 2022, 13, 1043474 CrossRef PubMed.
- M. A. Islam, M. K. Kibria, M. B. Hossen, M. S. Reza, S. A. Tasmia, K. F. Tuly, M. P. Mosharof, S. R. Kabir, M. H. Kabir and M. N. H. Mollah, Sci. Rep., 2023, 13, 4685 CrossRef CAS PubMed.
- G. Giammona, G. Pitarresi, E. F. Craparo, G. Cavallaro and S. Buscemi, Colloid Polym. Sci., 2001, 279, 771–783 CrossRef CAS.
- G. Pitarresi, F. S. Palumbo, R. Calabrese, E. F. Craparo and G. Giammona, J. Biomed. Mater. Res., Part A, 2008, 84, 413–424 CrossRef CAS PubMed.
- E. F. Craparo, G. Pitarresi, M. L. Bondi, M. P. Casaletto, M. Licciardi and G. Giammona, Macromol. Biosci., 2008, 8, 247–259 CrossRef CAS PubMed.
- Y. Xu, E. Parra-Ortiz, F. Wan, O. Cañadas, B. Garcia-Alvarez, A. Thakur, H. Franzyk, J. Pérez-Gil, M. Malmsten and C. Foged, J. Colloid Interface Sci., 2023, 633, 511–525 CrossRef CAS PubMed.
- E. F. Craparo, M. Cabibbo, C. Scialabba, G. Giammona and G. Cavallaro, Biomacromolecules, 2022, 23, 3439–3451 CrossRef CAS PubMed.
- J. C. M. Stewart, Anal. Biochem., 1980, 104, 10–14 CrossRef CAS PubMed.
- E. F. Craparo, S. E. Drago, F. Quaglia, F. Ungaro and G. Cavallaro, Drug Delivery Transl. Res., 2022, 12, 1859–1872 CrossRef CAS PubMed.
- M. R. C. Marques, R. Loebenberg and M. Almukainzi, Dissolution Technol., 2011, 15–28 CrossRef CAS.
- E. F. Craparo, M. Cabibbo, S. E. Drago, L. Casula, F. Lai and G. Cavallaro, Int. J. Pharm., 2022, 628, 122325 CrossRef CAS PubMed.
- F. Buttini, G. Colombo, P. C. L. Kwok and W. T. Wui, Inhalation Drug Delivery: Techniques and Products, 2013, pp. 91–119 Search PubMed.
- S. Khalili, S. Ghanbarzadeh, A. Nokhodchi and H. Hamishehkar, Res. Pharm. Sci., 2018, 13, 283–287 CrossRef PubMed.
- K. Ahookhosh, S. Yaqoubi, M. Mohammadpourfard, H. Hamishehkar and H. Aminfar, Int. J. Pharm., 2019, 566, 157–172 CrossRef CAS PubMed.
- Q. Liao, I. C. H. Lam, H. H. S. Lin, L. T. L. Wan, J. C. K. Lo, W. Tai, P. C. L. Kwok and J. K. W. Lam, Int. J. Pharm., 2020, 584, 119444 CrossRef CAS PubMed.
- H. Yu, J. Teo, J. W. Chew and K. Hadinoto, Int. J. Pharm., 2016, 499, 38–46 CrossRef CAS PubMed.
- L. Casula, F. Lai, E. Pini, D. Valenti, C. Sinico, M. C. Cardia, S. Marceddu, G. Ailuno and A. M. Fadda, Pharmaceutics, 2021, 13, 1300 CrossRef CAS PubMed.
- B. K. Kennedy and J. K. Pennypacker, Ann. Am. Thorac. Soc., 2016, 13, S398–S401 CrossRef PubMed.
- Z. Yan, Z. Xiaoyu, S. Zhixin, Q. Di, D. Xinyu, X. Jing, H. Jing, D. Wang, Z. Xi, Z. Chunrong and W. Daoxin, Sci. Rep., 2016, 6, 1–11 CrossRef PubMed.
- A. Haeri, M. Osouli, F. Bayat, S. Alavi and S. Dadashzadeh, Artif. Cells, Nanomed., Biotechnol., 2018, 46, 1–14 CrossRef CAS PubMed.
- G. Cavallaro, E. F. Craparo, C. Sardo, G. Lamberti, A. A. Barba and A. Dalmoro, Int. J. Pharm., 2015, 495, 719–727 CrossRef CAS PubMed.
- Q. Liu, J. Xue, X. Zhang, J. Chai, L. Qin, J. Guan, X. Zhang and S. Mao, Acta Biomater., 2022, 147, 391–402 CrossRef CAS PubMed.
- L. Pinheiro do Nascimento, N. Tsapis, F. Reynaud, D. Desmaële, L. Moine, J. Vergnaud, S. Abreu, P. Chaminade and E. Fattal, Eur. J. Pharm. Biopharm., 2022, 170, 112–120 CrossRef CAS PubMed.
- Y. Kaku, H. Imaoka, Y. Morimatsu, Y. Komohara, K. Ohnishi, H. Oda, S. Takenaka, M. Matsuoka, T. Kawayama, M. Takeya and T. Hoshino, PLoS One, 2014, 9, 1–8 CrossRef PubMed.
- E. F. Craparo, M. Cabibbo, A. Conigliaro, M. M. Barreca, T. Musumeci, G. Giammona and G. Cavallaro, Pharmaceutics, 2021, 13, 503 CrossRef CAS PubMed.
- G. Pilcer and K. Amighi, Int. J. Pharm., 2010, 392, 1–19 CrossRef CAS PubMed.
- A. Nieto-Orellana, H. Li, R. Rosiere, N. Wauthoz, H. Williams, C. J. Monteiro, C. Bosquillon, N. Childerhouse, G. Keegan, D. Coghlan, G. Mantovani and S. Stolnik, J. Controlled Release, 2019, 316, 250–262 CrossRef CAS PubMed.
- L. Chen, T. Okuda, X. Y. Lu and H. K. Chan, Adv. Drug Delivery Rev., 2016, 100, 102–115 CrossRef CAS PubMed.
- A. Chvatal, R. Ambrus, P. Party, G. Katona, O. Jójárt-Laczkovich, P. Szabó-Révész, E. Fattal and N. Tsapis, Int. J. Pharm., 2019, 559, 68–75 CrossRef CAS PubMed.
- S. Hamedani, S. Yaqoubi, R. Safdari, H. Hamishehkar and A. Nokhodchi, J. Drug Deliv. Sci. Technol., 2022, 78, 103958 CrossRef CAS.
- Y. Hu, M. Li, M. Zhang and Y. Jin, Int. J. Pharm., 2018, 551, 212–222 CrossRef CAS PubMed.
- A. M. Healy, B. F. McDonald, L. Tajber and O. I. Corrigan, Eur. J. Pharm. Biopharm., 2008, 69, 1182–1186 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3bm01210g |
| This journal is © The Royal Society of Chemistry 2024 |