
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContinuous flow microfluidic system with magnetic nanoparticles for the spectrophotometric quantification of urea in urine and plasma samples†
Kenia
Chávez-Ramos
 * and
María del Pilar
Cañizares-Macías
* and
María del Pilar
Cañizares-Macías
Laboratorio de Métodos de Flujo Continuo, Departamento de Química Analítica, Facultad de Química, Universidad Nacional Autónoma de México, Ciudad de México 04510, Mexico. E-mail: keniacr@ciencias.unam.mx
First published on 10th October 2024
Abstract
Urea, synthesized exclusively in the liver, is primarily transported through the bloodstream to the kidneys, where it is excreted in urine, accounting for 80–90% of nitrogen excretion in humans. Elevated blood urea levels, indicative of kidney dysfunction, make it a crucial biomarker for assessing renal function. Previous studies on urea detection using microdevices have largely focused on conductometric methods. In this study, we demonstrated the application of a continuous flow miniaturized system for rapid spectrophotometric urea quantification using polydimethylsiloxane (PDMS) microdevices. The microdevice featured two distinct zones: an enzymatic reaction zone, where urease-conjugated magnetic nanoparticles were immobilized, and a detection zone, where reagents were incorporated to produce a colored reaction product via a modified Berthelot reaction. Integrating magnetic nanoparticles as a solid support for the enzyme enabled the reuse of PDMS microdevices without compromising the analytical signal. Spectrophotometric detection was performed in an additional microdevice acting as a microflow cell coupled with optical fibers. A calibration curve was constructed using urea standards diluted in phosphate buffer solution (PBS), yielding a linear range of 0.12–3.00 mg dL−1. The method demonstrated detection and quantification limits of 0.04 mg dL−1 and 0.12 mg dL−1, respectively. Precision and accuracy assessments yielded a repeatability of 0.90% and intermediate precision of 4.52%, with recovery rates near 100%. The method was applied to plasma and urine samples, showing urea concentrations within normal physiological ranges and an analysis throughput of 36 measurements per hour.
Introduction
Kidney failure occurs when the kidneys lose their ability to effectively filter waste products, excess fluids, and minerals from the bloodstream. As kidney function declines, toxic waste accumulates in the body, leading to complications such as elevated blood pressure, fluid retention, and anemia due to decreased red blood cell production. Unfortunately, kidney failure is irreversible, and once this stage is reached, patients require dialysis or a kidney transplant for survival.1 Without timely intervention, the condition can be fatal. Kidney failure contributes significantly to morbidity and mortality rates, representing one of the leading causes of hospitalization and emergency care.2 Chronic kidney disease, a precursor to kidney failure, affects an estimated 5–15% of the global population.3Urea determination in plasma, serum, and urine samples is a routine clinical laboratory test to assess kidney function.4 Urea, the primary end-product of nitrogen metabolism in mammals, accounts for 80–90% of human nitrogen excretion. Synthesized exclusively in the liver, urea is transported via the bloodstream to the kidneys and excreted in urine.5 Normal plasma or serum urea concentrations range from 15 to 45 mg dL−1, while urine urea concentrations can be up to 50 times higher.6 Elevated urea levels in these biological fluids indicate kidney dysfunction or urinary tract obstruction, making urea detection essential for timely diagnosis and effective disease management.7,8
Urea detection and quantification are mostly performed using enzymatic methods. These methods involve the hydrolysis of urea catalyzed by the enzyme urease (URS), resulting in the production of ammonium ions (NH4+) and carbon dioxide (CO2). The generated products are typically detected using electroanalytical techniques. Alternatively, spectrophotometric detection can be achieved through the Berthelot method, which relies on the formation of a colored indophenol complex. In this method, the ammonium ions (NH4+) are converted to ammonia (NH3) under alkaline conditions. Ammonia then reacts with hypochlorite (OCl−) to form monochloramine (NH2Cl). The monochloramine subsequently reacts with two phenol molecules to yield an indophenol dye, which can be detected at wavelengths between 630 and 720 nm.9–11 Consequently, urea quantification is performed indirectly by measuring the concentration of NH4+ ions produced during enzymatic hydrolysis.
Most reported methodologies for urea detection have focused on biosensors and electroanalytical techniques, including potentiometry, amperometry, and conductimetry.12–14 Advances in microfluidic technology have further enhanced urea detection and quantification, significantly reducing reagent consumption and analysis time while maintaining accuracy and sensitivity.15 However, the critical challenge in electroanalytical techniques is the fabrication of electrodes within the microchannel device, which is crucial for achieving high sensitivity and reliable detection.4,7,16 In contrast, spectrophotometric detection of urea via the Berthelot reaction in microfluidic systems has received relatively limited attention.
Few studies have reported on microchip-based urea detection systems. For example, Remiszewska et al.17 developed a microanalytical system for urea determination in cell cultures, utilizing urease immobilized on the surface of a microreactor fabricated through low-temperature co-fired ceramic (LTCC) technology. This system detects NH4+ ions using a modified Berthelot method. However, several limitations are associated with this approach. The microfabrication process is intricate and time-consuming, involving the assembly of 14 independently designed layers to form the final device. Additionally, the chemical modification of the ceramic surface for microreactor fabrication requires multiple steps, including hydration, silanization, reaction with glutaraldehyde, and the immobilization of urease. These steps must be meticulously executed each time a new microdevice is fabricated, as any variation in the immobilization process can lead to reduced reproducibility and repeatability. Furthermore, the microdevices are disposable, with the immobilized enzyme remaining viable for up to 30 days, after which sensitivity decreases by 14% compared to the initial measurement. Despite requiring only 1 μL min−1 of reagent and sample for detection, the system has a relatively low throughput, with a measurement rate of just six samples per hour.
In this work, we present the design, microfabrication, and evaluation of reusable polydimethylsiloxane (PDMS) microdevices for urea quantification in urine and plasma samples using a modified Berthelot method. This approach addresses several limitations reported in previous studies. Rapid and cost-effective fabrication of the microdevices was achieved through photolithography and soft lithography techniques. We introduced magnetic nanoparticles (MNPs) conjugated with urease as solid support, facilitating easier manipulation within the microchannels and significantly increasing the surface area available for the enzymatic reaction. As a result, analysis time was notably reduced. Urea quantification in various urine and plasma samples was also compared with a batch method, validating the microdevice's performance and functionality.
Experimental
Reagents
For the microfluidic devices microfabrication and characterization, the following reagents were used: SU-8 3035 permanent epoxy negative photoresist (Microchem Inc.), 2-propanol (Sigma-Aldrich), propylene glycol monomethyl ether acetate (Sigma-Aldrich), and polydimethylsiloxane kit Sylgard 184 (Dow Corning Corporation).For microdevice urea detection, the following reagents were used: sodium acid phosphate (Na2HPO4) and potassium dihydrogen phosphate (KH2PO4) for the preparation of the phosphate buffered saline (PBS) 0.05 mol L−1 pH 7.0, salicylic acid (C7H6O3), sodium nitroprusside (Na2[C5N6OFe]), urea (CO(NH2)2), urease from Canavalia ensiformis (Jack Bean Type III, 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 150 U g−1), and Tween 80, all of them were purchased from Sigma-Aldrich. Commercial chlorine sample (Cloralex®) with a 5% sodium hypochlorite (NaClO), sodium hydroxide (NaOH) (Fermont), and LinkAmine MAGNETIC 200 nm kit (Nanoimmunotech).
150 U g−1), and Tween 80, all of them were purchased from Sigma-Aldrich. Commercial chlorine sample (Cloralex®) with a 5% sodium hypochlorite (NaClO), sodium hydroxide (NaOH) (Fermont), and LinkAmine MAGNETIC 200 nm kit (Nanoimmunotech).
Microdevice fabrication and characterization
The design of the microchannels was created using Adobe Illustrator CS6 software within a 70 mm circumference and subsequently printed onto an acetate negative, serving as the optical mask. Microdevices were fabricated through photolithography and soft lithography techniques using polydimethylsiloxane (PDMS) and sealed onto a thin PDMS layer via corona treatment (BD-20AC, Electro-Technic Products), following a procedure previously described by our group.18 The microdevices were sectioned into smaller pieces, and cross-sectional images were captured using a DM750 microscope (Leica Microsystems) with a 4× objective. Microchannel dimensions, including width and height, were characterized using Leica LAS EZ software in conjunction with a 5 mm microscope reticle (0.05 × 100 mm, The Microscope Depot).Sample collection
Blood and urine samples were taken with prior informed consent approved by the Research Ethics Committee of the Universidad Nacional Autónoma de México, according to the Ley General de Salud articles 41 Bis, 98 and 103 and Ley General de Salud en materia de investigación para la salud articles 13 and 14 in force in Mexico Legislation. Data collected from human participants are not available for confidentiality reasons.Plasma samples: blood samples were collected from patients aged 21 to 50 years using BD Vacutainer® tubes containing EDTA K2 as an anticoagulant. Plasma was immediately separated by centrifugation at 4500 rpm for 10 minutes and stored at −20 °C.
Urine samples: the same patients were asked to collect their first-morning urine sample in sterilized containers. Three milliliters of each urine sample were filtered through a 0.45 μm pore syringe filter (Sigma-Aldrich) and stored at 4 °C.
Urease (URS) conjugation to magnetic nanoparticles (MNPs)
The urease enzyme (URS) was covalently immobilized onto carboxyl-functionalized magnetic nanoparticles (MNPs) using the LinkAmine kit.19 To activate 5 mg of the carboxylated MNPs, 500 μL of a solution containing 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and N-hydroxysuccinimide (NHS) was added, followed by incubation at 37 °C for 30 minutes with gentle agitation (Eppendorf Thermomixer HCM100-Pro, DLAB Scientific). Following activation, the nanoparticles were washed three times to remove excess reagents. Subsequently, 6 mg mL−1 of urease solution (500 μL) was conjugated to the activated MNPs by incubation for 2 hours at 37 °C with gentle agitation. The resulting MNPs–URS were isolated via centrifugation (CR-68x, CAPP Rondo) and washed thoroughly to remove unbound enzymes. The supernatant, corresponding to the remanent 6 mg mL−1 urease solution, was stored at 4 °C for later quantification of the unbound URS via the Lowry protein assay (Tables S1–S2 and Fig. S1 in ESI material†). To block any remaining active carboxyl groups on the MNPs, a bovine serum albumin (BSA) solution was added, followed by incubation for an additional 2 hours at 37 °C with gentle stirring. After blocking, the conjugated MNPs–URS were washed and resuspended in 500 μL of storage buffer, aliquoted in small volumes (50 μL) to maintain enzyme stability, and stored at 4 °C for further use.Transmission electron microscopy (TEM) analysis
Transmission electron microscopy (TEM) was employed to assess the successful conjugation of urease to magnetic nanoparticles by comparing the MNPs before and after URS attachment. For sample preparation, 0.6 μL of MNPs were diluted in 2 mL of isopropanol and dispersed for 5 minutes in an ultrasonic bath (M1800, Branson). Subsequently, 5 μL of the nanoparticle dispersion was mixed with 2 mL of isopropanol containing 0.3 μL of Tween-80 to enhance particle distribution. TEM imaging was conducted using a transmission electron microscope (JEOL JEM-2010), operated at an acceleration voltage of 200 kV. Images were captured with a high-speed digital camera (Gatan Orius SC200). The acquired micrographs were analyzed using Gatan Microscopy Suite software to evaluate the morphology and confirm enzyme attachment to the MNPs.Experimental setup
The microdevice was placed on a stack of five neodymium disc magnets, each with a diameter of 1 cm. The second inlet of the microfluidic device was sealed, and an appropriate volume of MNPs–URS solution (concentration 10 mg mL; 142.49 μg URS per mg MNPs corresponding to 5.72 U per mg MNPs) was injected through the inlet 1 using a micropipette (Finnpipette F1, Thermo Scientific). Sonication was performed for 30 seconds before injection to ensure proper dispersion of the nanoparticles. Following this, the microdevice was positioned on a temperature controller (Mero TCU-125, Dolomite), and the stack of five neodymium disc magnets was placed on top, aligning with the enzymatic reaction zone as illustrated in Fig. 1(a). Once the magnetic field immobilized the MNPs–URS, reagents were introduced sequentially through the microdevice inlets using syringe pumps (NE-300, New Era Pump Systems) at a continuous flow rate of 2 μL min−1, resulting in a total flow rate of 8 μL min−1: (1) distilled water, (2) urea solution diluted in PBS (concentration range: 0.12–3 mg dL−1), (3) sodium hypochlorite in a basic medium, and (4) sodium salicylate with sodium nitroprusside (catalyst) in a basic solution. Approximately 5 μL of the reaction product (or blank) were collected at the microdevice outlet using a micropipette tip. The collection time for each sample was 40 seconds, after which spectrophotometric analysis was conducted. For multiple sample analyses, a waiting time of approximately 4 minutes was required under continuous flow to allow for sufficient flushing of the previous sample before initiating the next measurement. | ||
| Fig. 1 Schematic representation of the methodology. (a) Experimental setup to urea quantification using MNPs–URS in continuous flow: inlets (1) distilled water (carrier), (2) urea solution in PBS, (3) basic sodium hypochlorite solution, and (4) basic salicylate solution with nitroprusside as a catalyst. The flow rate for each solution was 2 μL min−1. The reaction product was collected in micropipette tips at the outlet for subsequent spectrophotometric detection. (b) PDMS microdevice detection system with a flow Z cell design microchannel (length of optical path = 1 cm, width = 362 ± 7 μm, and height = 96 ± 6 μm). | ||
Spectrophotometric detection system
Absorbance measurements of the collected reaction product were performed using a PDMS microdevice configured as a Z-shaped flow cell. Fig. 1(b) presents a schematic of the detection system. The flow cell featured a microchannel with an optical path length of 1 cm, a width of 362 ± 7 μm, and a height of 96 ± 6 μm. The microdevice was designed with integrated guides to accommodate optical fibers (FIBER 200-UV, Ocean Optics) coupled to a multi-LED light source (BluLoop, Ocean Optics) and a UV-vis spectrometer (USB4000, Ocean Optics) positioned on opposite sides of the flow cell. The colored reaction product was introduced into the microchannel via pipetting until the optical path was filled. Absorbance was measured at a wavelength of 690 nm using the SpectraSuite software. Each absorbance measurement required approximately one minute to be completed.Establishment of reaction conditions
Two parameters—the enzyme lifetime retained in the MNPs and the enzymatic reaction temperature—were evaluated to select the reaction conditions for the developed methodology. Lastly, a full factorial experimental design was conducted for three factors, as detailed below.Study of enzyme lifetime
Using a urea standard solution in PBS (2.5 mg dL−1), a reaction was conducted in the microdevice with the following conditions: 8 μL of MNPs–URS (0.46 U of urease), 0.5% sodium hypochlorite in a basic medium (0.1 mol L−1 NaOH), and 0.25 mol L−1 sodium salicylate with 0.025 mol L−1 sodium nitroprusside in a basic medium (0.1 mol L−1 NaOH). The reaction was maintained at a constant temperature of 37 °C. Absorbance measurements (λ = 690 nm) were recorded at time intervals ranging from 5 to 240 minutes in triplicate. Additionally, a reaction blank was measured in triplicate under the same conditions. This experiment was conducted for three days, and the absorbance values were plotted as a function of time.Influence of temperature on enzymatic and detection reactions
Absorbance measurements were performed in triplicate for both a reaction blank and a urea standard solution in PBS (2.5 mg dL−1) at two different temperatures, 37 °C and 60 °C. The reaction conditions included 8 μL of MNPs–URS (equivalent to 0.46 U of enzyme activity), 0.5% sodium hypochlorite in a basic medium (0.1 mol L−1 NaOH), and a solution of 0.25 mol L−1 sodium salicylate with 0.025 mol L−1 sodium nitroprusside in a basic medium (0.1 mol L−1 NaOH).Design of experiments for the selection of reagents concentrations and immobilized enzyme amount
A full factorial experimental design (23) with three central points was employed to optimize the analytical signal using a standard urea solution in PBS (3 mg dL−1). The salicylate concentration was constant at 0.25 mol L−1 in a basic medium (0.1 mol L−1 NaOH), setting the temperature at 37 °C. Three parameters expected to influence both the enzymatic and detection reactions were selected and assigned high (+1), low (−1), and central (0) values, as outlined in Table S3.†The design matrix was generated using STATGRAPHICS software, covering all possible combinations of the selected factors, resulting in 11 experiments. The absorbance of the reaction product and the blank were measured for each experiment. The analytical signal was determined by subtracting the blank absorbance from the reaction product absorbance, and this difference was used for subsequent analysis.
Construction of the calibration curve
A calibration curve was constructed by performing serial dilutions from a standard of 10 mg dL−1 urea in PBS, yielding concentrations ranging from 0.075 to 3 mg dL−1. Each standard solution was introduced into the microdevice in continuous flow mode, starting from the lowest concentration to the highest, under the selected reaction conditions: 8 μL of MNPs–URS (0.46 U), 0.1% sodium hypochlorite in a basic medium (0.1 mol L−1 NaOH), and a salicylate/nitroprusside solution (0.25 mol L−1 and 0.05 mol L−1, respectively) in a basic medium (0.1 mol L−1 NaOH). The flow rate for each reagent was set to 2 μL min−1, resulting in a total flow rate of 8 μL min−1. The temperature was maintained at 37 °C.A volume of 5 μL of the reaction product was collected at the microdevice outlet using a micropipette tip. Absorbance was immediately measured at λ = 690 nm. This procedure was performed in triplicate for each urea concentration. To account for background interference, the mean absorbance of the reaction blank (n = 10) was subtracted from the absorbance values of each urea concentration. The corrected absorbance values were then plotted against urea concentration to generate the calibration curve, which was used to determine the linear detection range. Additionally, the limit of detection (LOD) and limit of quantification (LOQ) were calculated using eqn (1) and (2), based on the standard deviation of the y-intercept (S0) and the slope (b1) of the calibration curve.
 | (1) |
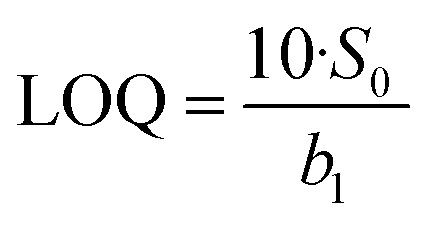 | (2) |
Assessment of methodology precision
A urea solution in PBS with a 1.5 mg dL−1 concentration was prepared in duplicate and analyzed over seven days under the previously established reaction conditions. Absorbance values were measured for each preparation. A one-way analysis of variance (ANOVA) was performed to evaluate the repeatability and intermediate precision of the methodology used in the microdevice.Recovery percentages
Recovery percentages were determined using a pair of plasma and urine samples. Dilutions were prepared to ensure the non-fortified samples had a urea concentration close to 0.5 mg dL−1. Subsequently, 1 mg dL−1 of urea standard was added to achieve a fortified sample concentration close to 1.5 mg dL−1 (midpoint of the calibration curve).The methodology was performed for the non-fortified and fortified samples in sextuplicate within the established reaction conditions. Absorbance values were recorded, and the average absorbance value of the reaction blank was subtracted. Using the previously constructed calibration curve, the actual concentrations of both solutions were determined, and the recovery percentage was calculated based on eqn (3).
 | (3) |
C F is the urea concentration in the fortified sample, CN is the urea concentration in the non-fortified sample, and FC is the added urea standard for fortification; in this case, FC = 1 mg dL−1.
Comparison with batch methodology
A batch quantification method for urea, employing larger volumes of the same reagent solutions of microdevice assay without the additions of MNPs, was used to construct a urea calibration curve, encompassing concentrations between 0.013 and 0.35 mg dL−1, as specified in the “Construction of the batch calibration curve” section of the ESI material.† Precision studies for the batch methodology were conducted by preparing a 0.175 mg dL−1 urea standard in PBS in duplicate over seven days. Absorbance values were recorded, and repeatability and intermediate reproducibility were assessed using one-way ANOVA. Finally, recovery percentages were determined using plasma and urine samples for fortification in sextuplicate, following the same procedure as employed for the microdevice methodology.Sample quantification
Urea quantification was performed on eight pairs of urine and plasma samples from the same patient. The samples were diluted with PBS and then introduced into a microdevice. The established reaction conditions were used for urea quantification in triplicate. The average of the blank was subtracted from the absorbance measurement to obtain corrected absorbance. This value was used to quantify the urea concentration using the calibration curve and considering the dilution factor. Finally, on the same day, the same samples were also analyzed using the batch method to facilitate a comparative assessment of the results obtained from the microdevice methodology.Cleaning of microdevices
Upon reaching the 100 minutes operational lifetime of the immobilized enzyme on the magnetic nanoparticles, the microdevices were cleaned as follows: the magnet tower positioned atop the microdevice was removed. The microchannels were flushed with distilled water through each inlet at a flow rate of 15 μL min−1 (total flow rate of 60 μL min−1) for a minimum of 10 minutes to ensure complete removal of all MNPs–URS. The microdevice was then immersed in distilled water and subjected to ultrasonic cleaning for 15 minutes to remove any residual magnetic nanoparticles from the microchannels. Finally, the microchannels were dried using compressed air, and the microdevice was stored for subsequent use.Results and discussion
Characteristics of the microdevice
The constructed PDMS microdevice is depicted in Fig. 2(a). The microchannel design consists of two main components: (1) the enzymatic reaction zone, where MNPs–URS were introduced and retained within the microchannels using magnets, and a urea standard diluted in PBS (0.05 mol L−1, pH 7.0) was added using distilled water as the carrier. This zone extends up to just before the entry of the sodium hypochlorite solution, and (2) the detection reaction zone, where sodium hypochlorite and salicylate/nitroprusside solutions in an alkaline medium were introduced for the modified Berthelot reaction (Fig. 2(b)). The resulting reaction product was collected for subsequent spectrophotometric detection. The microchannels measured were 176 ± 6 μm in width and 92 ± 6 μm in height, as shown in the cross-sectional image in Fig. 2(c).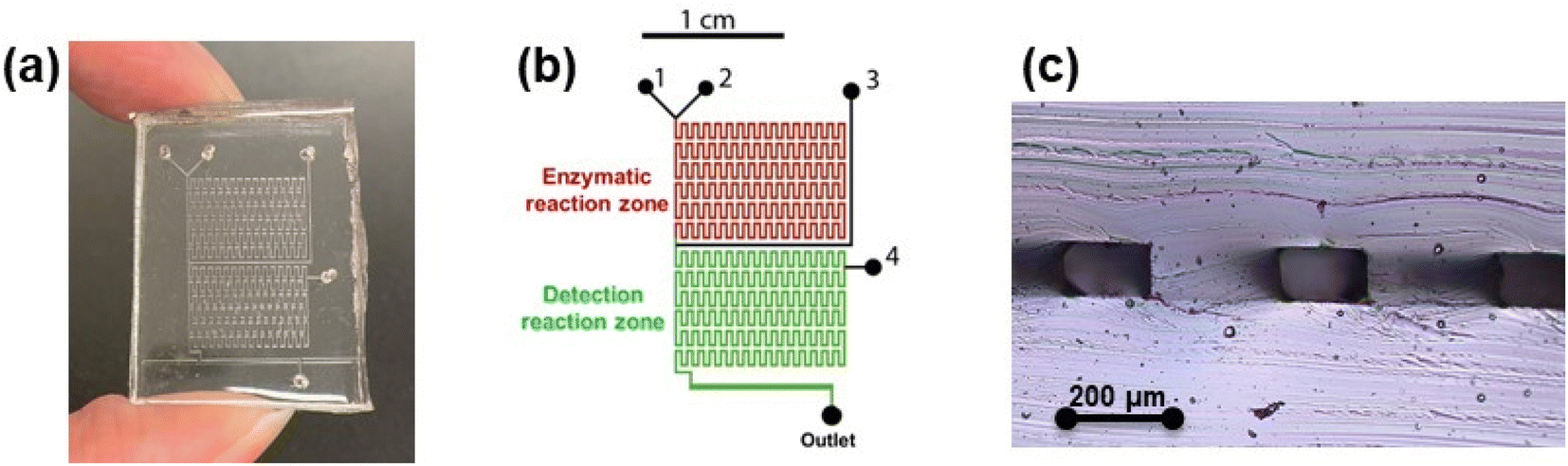 | ||
| Fig. 2 Microdevice characteristics. (a) Fabricated PDMS microdevice. (b) Microchannels design with two identified main zones: the enzymatic and the detection reaction zones. (c) Cross-sectional view of microchannels: width 176 ± 6 μm and height 92 ± 6 μm. | ||
Transmission electron microscopy (TEM) analysis
TEM analysis revealed MNPs without urease conjugation (Fig. 3(a)) had a smaller size (10.72 ± 1.86 nm) compared to those conjugated with urease, and they appeared well-defined, making them easily distinguishable. The MNPs–URS exhibited encapsulation of the nanoparticles, forming vesicle-like structures with varying diameters where the enzyme attached to the surface, increasing the average size to 230.62 ± 97.46 nm (Fig. 3(b)). This confirms the successful conjugation of urease to the MNPs. | ||
| Fig. 3 TEM images of the magnetic nanoparticles. (a) MNPs without urease conjugation. (b) Urease-conjugated MNPs (URS–MNPs). | ||
Establishment of reaction conditions
 | ||
| Fig. 4 Establishment of the reaction conditions. (a) Absorbance measurements for a 2.5 mg dL−1 urea standard in PBS for 5 to 240 min on three different days; the dotted line corresponds to the average of the reaction blank (n = 3). (b) Comparison of absorbance measurements for a 2.5 mg dL−1 urea standard in PBS and reaction blank at 37 °C and 60 °C (n = 3). (c) Pareto chart of standardized effects and (d) cube plot resulting from the 23 full factorial design of experiments with three center points; [salicylate] = 0.25 mol L−1 at 37 °C. | ||
150 U g−1). However, due to the limitation in the temperature controller, which did not allow for independent temperature control across different reaction zones in the microdevice, a single temperature had to be chosen for the entire process. The results showed a slight increase in the analytical signal when the reaction temperature was raised to 60 °C, both for the reaction blank (0.503 ± 0.012) and the urea standard in PBS at a concentration of 2.5 mg dL−1 (1.029 ± 0.016), compared to the absorbance measurements for the blank (0.391 ± 0.013) and urea standard (0.912 ± 0.006) at 37 °C, as shown in Fig. 4(b). Nevertheless, the blank/standard ratio remained very similar, 2.33 at 37 °C and 2.05 at 60 °C, indicating no significant increase in the method's sensitivity. Therefore, a temperature of 37 °C was selected for subsequent analyses.
 | ||
| Fig. 5 Calibration curve for urea quantification. (a) Example of the analytical signals (absorption spectra) for the reaction blank and the reaction product employing 2 mg dL−1 of urea in PBS. (b) Calibration curve, linear range 0.12 to 3.00 mg dL−1, LOD = 0.04 mg dL−1, LOQ = 0.12 mg dL−1 (n = 3). | ||
| Parameter | Microdevice | Batch |
|---|---|---|
| Linear range | 0.12–3.00 mg dL−1 | 0.013–0.35 mg dL−1 |
| Linear equation | y = 0.3467x + 0.0167 | y = 2.763x + 0.0119 |
| R 2 = 0.9979 | R 2 = 0.9974 | |
| LOD | 0.040 mg dL−1 | 0.006 mg dL−1 |
| LOQ | 0.12 mg dL−1 | 0.02 mg dL−1 |
| Repeatability | 0.90% | 0.88% |
| Intermediate precision | 4.52% | 2.83% |
| % recovery | Plasma sample: 101.81 ± 1.19% | Plasma sample: 102.23 ± 2.37% |
| Urine sample: 101.48 ± 1.69% | Urine sample: 101.73 ± 1.94% | |
| Analysis time | 1 min 40 s | 16 min |
| Reagents volume | 1.3 μL | 100–500 μL |
| Waste volume | 5 μL | 2000 μL |
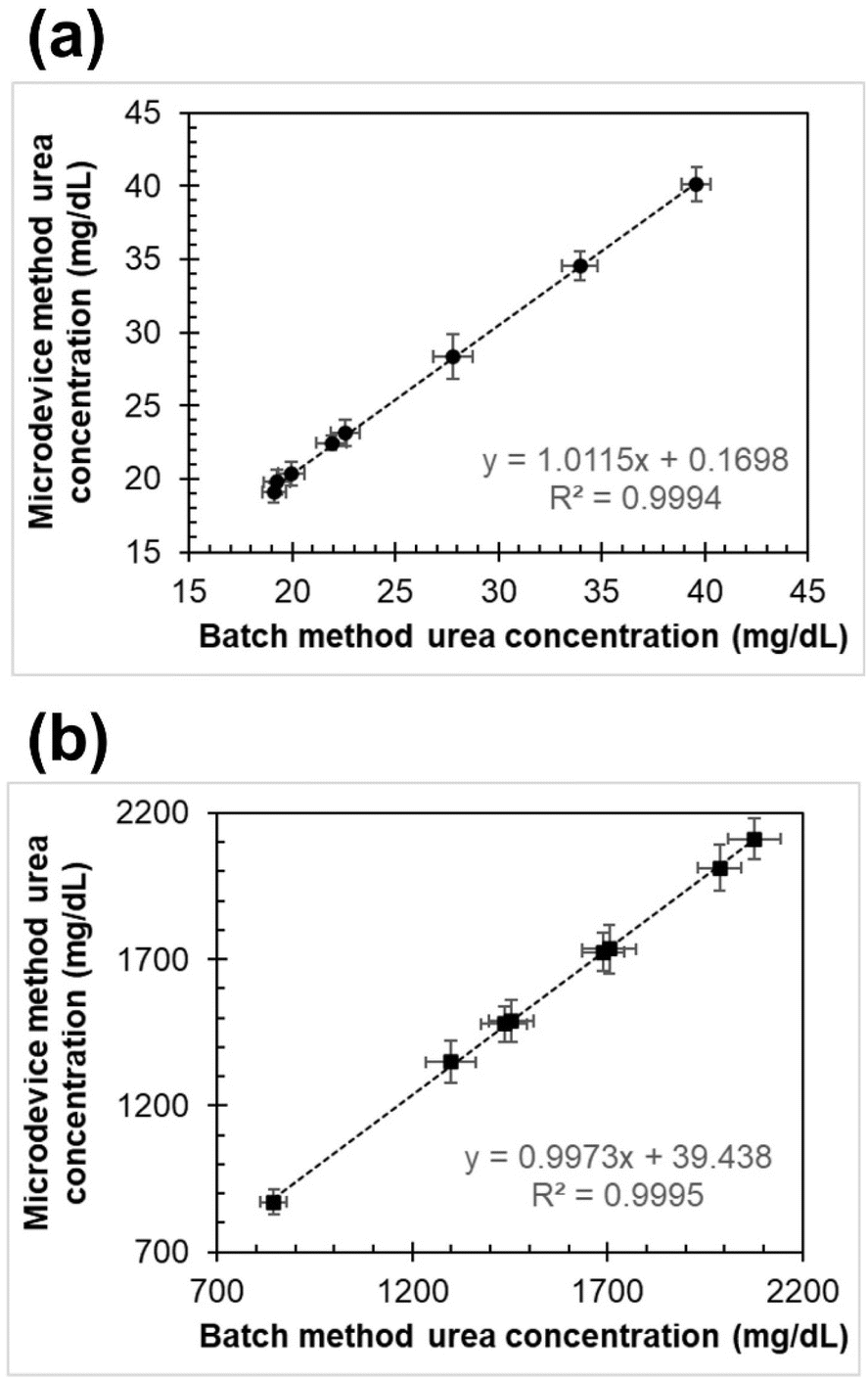 | ||
| Fig. 6 Comparison of urea quantification in eight pairs of (a) plasma and (b) urine samples using the microdevice and batch quantification methods (n = 3). | ||
Given that the typical average concentration of urea in plasma ranges from 15 to 45 mg dL−1 and can be up to 50 times more concentrated in urine compared to plasma (750–2250 mg dL−1),6 the values obtained from the samples fall within the normal range for this clinical analysis, confirming the proper functioning of the developed methodology in the microdevice.
| Method | Sample volume (μL) | Linear range (mmol L−1) | Sensitivity (S) in linear range | LOD (mmol L−1) | Analysis rate | Bioreactor/biosensor lifetime | Ref. |
|---|---|---|---|---|---|---|---|
| a Peak areas were measured; NR = not reported. | |||||||
| Integrated 3D printed heaters for microfluidic applications: FIA ammonium analysis | 100 (ammonium) | 0.03–0.3 | 74.46 mmol−1 L | 0.009 | 37 samples per h | — | 21 |
| PDMS-thymol/nitroprusside composite based sensor for ammonium monitoring in water samples | 500 (ammonium) | 0–0.6 | 3.82 mmol−1 L | 0.02 | 12 samples per h | Composites were stable during 12 moths | 22 |
| Integrated microfluidic system based on the berthelot reaction | 1 (ammonium) | 0.03–1.2 | 0.73 mmol−1 L | NR | 6 samples per h | — | 23 |
| Integrated micro ammonia analysis system (IMAAS) with microreactor and in-plane type optical detector | 100–150 (ammonium) | 0.02–0.2 | NR | NR | 24 samples per h | — | 24 |
| NH4+ ATIR optical biosensor with immobilized urease | 50 (urea) | 0.1–1 | 0.05 mmol−1 L | 0.02 | 30 samples per h | NR | 25 |
| Bioreactor based on LTCC system for urea determination in cell cultures | 1 (urea) | 0–1 | 1.4 mmol−1 L | 0.002 | 6 samples per h | After 30 days sensitivity drops to 86% | 17 |
| Berthelot reaction and microfluidic device to quantify the ammonia and urea dually in aqueous systems | 10 (urea) | 1.7–8.3 | 0.40 mmol−1 L | 0.35 | NR | — | 26 |
| Microfluidic platform with urease-conjugated magnetic nanoparticles (MNPs–URS) for urea quantification | 1.3 (urea) | 0.02–0.5 | 2.1 mmol−1 L | 0.006 | 7 samples in triplicate per h 36 measurements per h | After 100 min absorbance value drops to 83.2% | This work |
Additionally, the sample volume used in this work (1.3 μL) is among the lowest reported, and it achieves the second-highest analysis rate (36 measurements per h). The LOD value is also the second-lowest reported. The developed microfluidic platform further benefits from being reusable. Although the immobilized urease enzyme on magnetic nanoparticles maintains repeatable absorbance values for only 100 minutes before losing about 16% sensitivity, it allows up to 60 consecutive measurements within this time frame.
Conclusions
A reusable PDMS microfluidic platform was designed, characterized, and evaluated for urea quantification using spectrophotometric detection with urease-conjugated magnetic nanoparticles. The reaction was performed in a continuous flow, with an average analysis time of 1 minute 40 seconds per sample using only 8 μL of MNPs–URS (0.46 U of enzyme) with a conjugated enzyme lifetime of 100 minutes. The developed methodology demonstrated precision and accuracy, with a suitable linear range and LOD and LOQ values for urea quantification in plasma and urine. Compared to a batch methodology, the microdevice yielded similar results for both urea and plasma samples quantification, offering notable advantages, including the requirement of only 1.3 μL of sample and reagents per analysis, with a reaction time of 40 seconds and a detection time of 60 seconds, resulting in 5 μL of waste. Furthermore, compared to other reported microdevice methodologies for urea quantification, this approach improved the analysis rate, obtaining 36 measurements per hour for the same sample and the added benefit of microdevice reusability after removing and cleaning MNPs–URS. The results indicate that this microfluidics methodology holds promising potential for future applications. However, a critical improvement remains necessary to achieve full automation, which includes integrating on-line spectrophotometric detection without introducing significant signal variation.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and in ESI materials.†Author contributions
Kenia Chávez-Ramos: conceptualization, methodology, validation, formal analysis, investigation, visualization, writing original draft, writing review & editing. María del Pilar Cañizares-Macías: conceptualization, methodology, resources, supervision, funding acquisition, writing review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank to DGAPA-UNAM for supporting this work through the Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica (PAPIIT Grant IT200718). K. C. R. acknowledges CONAHCYT Mexico for a PhD fellowship (CVU 662463).References
- T. Dhondup and Q. Qian, Electrolyte and Acid-Base Disorders in Chronic Kidney Disease and End-Stage Kidney Failure, Blood Purif., 2017, 43(1–3), 179–188 CrossRef PubMed.
- A. Méndez-Durán, J. F. Méndez-Bueno, T. Tapia-Yáñez, A. Muñoz-Montes and L. Aguilar-Sánchez, Epidemiology of chronic kidney failure in Mexico, Diálisis Traspl., 2010, 31(1), 7–11 CrossRef.
- L. De Nicola and C. Zoccali, Chronic kidney disease prevalence in the general population: Heterogeneity and concerns, Nephrol. Dial. Transplant., 2016, 31(3), 331–335 CrossRef PubMed.
- S. Srivastava, P. R. Solanki, A. Kaushik, M. A. Ali, A. Srivastava and B. D. Malhotra, A self assembled monolayer based microfluidic sensor for urea detection, Nanoscale, 2011, 3(7), 2971–2977 RSC.
- P. S. Francis, S. W. Lewis and K. F. Lim, Analytical methodology for the determination of urea: current practice and future trends, Trends Anal. Chem., 2002, 21(5), 389–400 CrossRef.
- A. J. Taylor and P. Vadgama, Analytical reviews in clinical biocbemistry: the estimation of urea, Ann. Clin. Biochem., 1992, 29, 245–264 CrossRef PubMed.
- A. Ali, M. S. Alsalhi, M. Atif, A. A. Ansari, M. Q. Israr, J. R. Sadaf, E. Ahmed, O. Nur and M. Willander, Potentiometric urea biosensor utilizing nanobiocomposite of chitosan-iron oxide magnetic nanoparticles, J. Phys.: Conf. Ser., 2013, 414, 012024 CrossRef.
- V. Kumar and K. D. Gill, Estimation of Urea in Serum and Urine, in Basic Concepts in Clinical Biochemistry: A Practical Guide, Springer, Singapore, 2018 Search PubMed.
- S. A. Gordon, A. Fleck and J. Bell, Optimal conditions for the estimation of ammonium by the Berthelot reaction, Ann. Clin. Biochem., 1978, 15, 270–275 CrossRef PubMed.
- P. L. Searle and I. Meta, The Berthelot or lndophenol Reaction and Its Use in the Analytical Chemistry of Nitrogen, Analyst, 1984, 109, 549–568 RSC.
- A. Sensor, K. W. Kimble, J. P. Walker, D. N. Finegold and S. A. Asher, Progress toward the development of a point-of-care photonic crystal ammonia sensor, Anal. Bioanal. Chem., 2006, 385, 678–685 CrossRef PubMed.
- L. Quadrini, S. Laschi, C. Ciccone, F. Catelani and I. Palchetti, Electrochemical methods for the determination of urea: Current trends and future perspective, Trends Anal. Chem., 2023, 168, 117345 CrossRef.
- S. N. Botewad, D. K. Gaikwad, N. B. Girhe, H. N. Thorat and P. P. Pawar, Urea biosensors: A comprehensive review, Biotechnol. Appl. Biochem., 2023, 70, 485–501 CrossRef PubMed.
- F. Mashhadban-k, L. Gorgani and G. Najafpour-darzi, Enzymatic electrochemical biosensors for urea detection: A review, Sens. Actuators, A, 2024, 374, 115499 CrossRef.
- A. Niculescu, C. Chircov, C. Alexandra and A. M. Grumezescu, Fabrication and Applications of Microfluidic Devices: A Review, Int. J. Mol. Sci., 2021, 22(4), 2011 CrossRef.
- W. Limbut, S. Loyprasert, C. Thammakhet, P. Thavarungkul, A. Tuantranont, P. Asawatreratanakul, C. Limsakul, B. Wongkittisuksa and P. Kanatharana, Microfluidic conductimetric bioreactor, Biosens. Bioelectron., 2007, 22, 3064–3071 CrossRef.
- E. Remiszewska, K. Malecha, J. Kruk, J. Jankowska-Śliwińska, W. Torbicz, A. Samluk, K. D. Pluta and D. G. Pijanowska, Enzymatic method of urea determination in LTCC microfluidic system based on absorption photometry, Sens. Actuators, B, 2019, 285, 375–384 CrossRef.
- K. Chávez Ramos and M. del P. Cañizares Macías, Microdevice based on centrifugal effect and bifurcation law for separation of plasma from on-line diluted whole blood, Anal. Bioanal. Chem., 2021, 413, 5361–5372 CrossRef.
- Nanoimmunotech, LinkAmine kit MAGNETIC 200 nm – 5 mL, internet, cited 2024 Oct 11, pp. 1–14, available from: https://www.dianova.com/pdf/datasheet_03000315_v150709.pdf Search PubMed.
- M. Almeida Bezerra, V. Azevedo Lemos, D. Menezes de Oliveira, C. Galvão Novaes, J. Alves Barreto, J. Pereira Santos Alve, U. M. Ferreira da Mata Cerqueira, Q. Oliveira dos Santos and S. Alves Araújo, Automation of continuous flow analysis systems – a review, Microchem. J., 2020, 155, 104731 CrossRef.
- E. Fornells, E. Murray, S. Waheed, A. Morrin, D. Diamond, B. Paull and M. Breadmore, Integrated 3D printed heaters for microfluidic applications: Ammonium analysis within environmental water, Anal. Chim. Acta, 2020, 1098, 94–101 CrossRef PubMed.
- M. C. Prieto-Blanco, N. Jornet-martínez, Y. Moliner-martínez, C. Molins-legua and R. Herráez-hernández, Development of a polydimethylsiloxane – thymol/nitroprusside composite based sensor involving thymol derivatization for ammonium monitoring in water samples, Sci. Total Environ., 2015, 504, 105–112 CrossRef PubMed.
- A. Daridon, M. Sequeira, G. Pennarun-Thomas, H. Dirac, J. P. Krog, P. Gravesen, J. Lichtenberg, D. Diamond, E. Verpoorte and N. F. de Rooij, Chemical sensing using an integrated microfluidic system based on the Berthelot reaction, Sens. Actuators, B, 2001, 76(1–3), 235–243 CrossRef.
- J.-S. Park, K.-B. Park, K.-S. Shin, H.-D. Park, M.-C. Kim and J.-R. Kim, Design, fabrication and characterization of an integrated micro ammonia analysis system (IMAAS) with microreactor and in-plane type optical detector based on the Berhelot reaction, Sens. Actuators, B, 2006, 117(2), 516–522 CrossRef.
- B. Kovács, G. Nagy, R. Dombi and K. Tóth, Optical biosensor for urea with improved response time, Biosens. Bioelectron., 2003, 18, 111–118 CrossRef.
- K. S. Deepak, A. Balapure, P. R. Priya, P. S. Kumar, K. Dubey, A. Javed, S. Chattopadhyay and S. Goel, Development of a microfluidic device for the dual detection and quantification of ammonia and urea from the blood serum, Sens. Actuators, A, 2024, 369, 115174 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ay01593b |
| This journal is © The Royal Society of Chemistry 2024 |