DOI:
10.1039/D4AY00943F
(Paper)
Anal. Methods, 2024,
16, 5280-5287
Development and validation of the RP-HPLC method for quantification of tavaborole
Received
20th May 2024
, Accepted 9th July 2024
First published on 11th July 2024
Abstract
The stability-indicating approach for tavaborole quantification was developed and validated to establish a precise, linear, accurate, and robust HPLC method. The development section includes optimizing the detection wavelength, the mobile phase ratio, and the type of column used to achieve the best possible separation and sensitivity for analysis. The chromatographic conditions were established, considering peak symmetry, resolution, and retention time. The mobile phase composition, comprising a buffer: acetonitrile (75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25, %v/v), with an injection volume of 15 μL, showed suitable elution and recovery at 265 nm. A constant column oven temperature of 35 °C and a 1 mL min−1 flow rate were maintained. The pH of the buffer was changed to 3.0 by using orthophosphoric acid. Linearity was observed from 5 to 1000 ppm (r2 = 1.00000). The capacity (retention) factor (k) of 3.43 was observed, indicating significant interaction and good separation. Forced degradation (FD) or stress tests were performed for chemical and physical photolytic stress conditions, and the results observed were within the specified limits. The stability in the analytical solution was observed for up to 35 hours at 5 °C, confirming the stability of the solution. Validation of the developed HPLC method confirmed the system's suitability, precision, linearity, accuracy, FD, robustness, and results. All validation criteria for the technique were within acceptable limits.
:25, %v/v), with an injection volume of 15 μL, showed suitable elution and recovery at 265 nm. A constant column oven temperature of 35 °C and a 1 mL min−1 flow rate were maintained. The pH of the buffer was changed to 3.0 by using orthophosphoric acid. Linearity was observed from 5 to 1000 ppm (r2 = 1.00000). The capacity (retention) factor (k) of 3.43 was observed, indicating significant interaction and good separation. Forced degradation (FD) or stress tests were performed for chemical and physical photolytic stress conditions, and the results observed were within the specified limits. The stability in the analytical solution was observed for up to 35 hours at 5 °C, confirming the stability of the solution. Validation of the developed HPLC method confirmed the system's suitability, precision, linearity, accuracy, FD, robustness, and results. All validation criteria for the technique were within acceptable limits.
1. Introduction
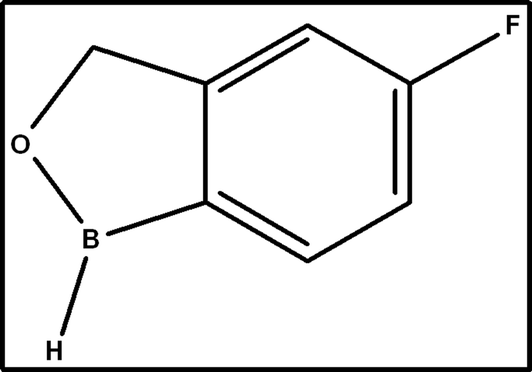
Onychomycosis, affecting millions worldwide, targets nails. Dermatophytes, molds, and even yeast can be the major cause of this common infection. Although onychomycosis generally follows a benign chronic clinical course, it can lead to complications in certain patient groups, particularly those with diabetes and peripheral vascular disease. This condition is identified by nail dystrophy, discoloration, and thickening, with a prevalence of 50% among patients over 70 years of age.1,2 Psoriasis, peripheral vascular disease, Ram's horn nail (onychogryphosis), immunosuppression, hyperhidrosis, diabetes, aging, onycholysis, or nail damage are some factors leading to onychomycosis.3,4 TVB is a novel broad spectrum boron-containing topical antifungal drug substance that belongs to the oxazole class developed for the treatment of toenail onychomycosis. The chemical formula of TVB is 5-fluoro-1,3-dihydro-1-hydroxy-2,1-benzoxaborol with a molecular weight of 151.93 g mol−1 (Fig. 1).5,6 TVB is known for its antibacterial attributes and was specifically selected for its applications in fungal infection treatment.7,8 The antifungal activity of TVB is due to the 5-fluoro group, and its hydrophilicity was improved by substituting a 1-phenyl with 1-hydroxy group.9 The compound TVB demonstrated sustained release behavior even in the presence of keratin by penetrating through the nail compared to other oxaborole (10%) and ciclopirox (8%) nail lacquer,10 confirming the low protein binding activity. TVB 5% topical solution has received USFDA approval for treating onychomycosis triggered by Trichophyton mentagrophytes or Trichophyton rubrum.11 These are the significant dermatophytes that cause onychomycosis. Tavaborole explicitly blocks protein synthesis by inhibiting leucylaminoacyl transfer RNA synthetase.12
 |
| | Fig. 1 Structure of tavaborole. | |
Previously, Tampucci et al. developed the HPLC-UV method to analyze TVB during experimental transungual permeation studies. To quantify TVB, samples were taken from the receiving phase, and the bovine hoof membrane was extracted to quantify the TVB. This approach involved a simple solid–liquid extraction procedure. The method exhibited good linearity, with the coefficient of variation for intraday and interday precision below 2%, indicating good sensitivity and precision.13 Puppala et al. reported oxidative degradation of TVB in RP-UPLC when trifluoroacetic acid buffer and acetonitrile were used as the mobile phase. The TVB remained stable after acidic and alkali treatments, as well as in thermal and photolytic conditions. Exposure to oxidizing conditions, mimicked by H2O2, revealed the drug's instability under stress conditions.14
Sheela et al. reported the UV-based analytical method for TVB determination via techniques, i.e., D0 and D1 can accurately determine TVB levels in various reagents, including methanol, water, and phosphate buffer (pH 2, 4, 7), HCl, NaOH, and borate buffer (pH 9), respectively.15 Existing research suggests a gap for a simple, accurate, robust, and highly sensitive method to quantify TVB.
The novel RP-HPLC approach for TVB quantification described here provides key advantages, including optimized separation efficiency, increased sensitivity, and cost-effectiveness. Parameters such as mobile phase composition, column selection, and detection wavelength were carefully optimized. The method utilizes an Xterra RP18 column, a mobile phase consisting of a potassium dihydrogen phosphate buffer and acetonitrile, and a higher detection wavelength. This configuration improves peak resolution, reduces retention time, and minimizes interference. Additionally, the method exhibits greater robustness and reproducibility, making it suitable for routine analytical applications. The optimized mobile phase is cost-effective and provides better method stability, enhancing efficiency and applicability in various practical scenarios. The methods described here have been validated according to the ICH recommended guidelines.16,17
2. Material and method
2.1. Chemicals and reagents
Acetonitrile, OPA, KH2PO4, and NaOH, were obtained from Merck, Germany. HPLC water, HCl, and H2O2 were purchased from Qualigens, India. TVB was received from Starshine Chemicals, China. All the reagents used during analysis were of Emplura or analytical grade unless indicated otherwise.
2.2. Instrumentation and chromatographic conditions
The analysis was carried out by Shimadzu Prominence-I (Prominence-I LC-2030 plus; LabSolutions Software) with a UV and PDA detector and Infinity 1260 (Infinity 1260, Open lab 2.3 Software) equipped with an autosampler, employing an Xterra RP18 column (5 μm, 4.6 × 150 mm).
2.3. Chromatographic method development
To achieve optimal separation and sensitivity, some parameters were systematically investigated. These included the flow rate of the mobile phase, its composition, the volume of sample injected into the system, and the temperature at which the separation process was carried out. Each parameter has been carefully adjusted and tested to determine the optimal performance conditions considering resolution and detection limits. The chromatographic conditions were optimized, considering factors such as peak symmetry, tailing factor, theoretical plate, and retention time.18,19 TVB has a partition coefficient of 1.28, indicating that it is hydrophobic, and a dissociation constant value of 8.36, indicating ionization in basic solutions. However, the ionization can be controlled using an acidic mobile phase.20 The mobile phase composition comprising buffer solutions of potassium dihydrogen phosphate (KH2PO4, pH – 3.0, 25 mM) which is considered mobile phase A and organic solvents such as acetonitrile (100%) or methanol (100%) were used as mobile phase B and used in different ratios, i.e., 50:50% v/v; 60:40% v/v and 75:25% v/v. The pH was modified to 3.0 with diluted OPA. Column temperature (35 and 40 °C) and injection volume (20–50 μL) were evaluated at 1 mL min−1 flow rate.
2.4. Preparation of standard solution
The TVB stock solution was precisely prepared to achieve a concentration of 1000 ppm. 100 mg of TVB was accurately weighed using an analytical balance and dissolved in 5 mL of methanol. Methanol volume was previously measured using a graduated pipette to ensure volumetric accuracy. The stock solution was subsequently diluted to 100 mL with water as the diluent to achieve the concentrations required for analysis. Working solutions were then prepared by diluting the initial stock solution into various concentrations, which were tailored to the specific concentration required for studies.
3. Method validation
The HPLC methodology developed for quantifying TVB underwent validation per the ICH Q2 (R2).17 The validation was thoroughly evaluated to ensure the reliability and robustness of the method. Parameters including accuracy, precision, linearity, robustness, stress testing, and solution stability were measured to determine the method for its envisioned analytical outcomes.
3.1. Specificity and forced degradation
Specificity is confirmed when the analyte peak is distinctly separated from other peaks. To evaluate the ability of the HPLC method to distinguish the standard, sample, and blank injections were performed.21 FD is a method used to induce the degradation of TVB under settings exhibiting greater severity than the accelerated conditions and undergoes decomposition to produce degradation components that serve as an indicator of the drug molecule stability and the chemical behavior of the drug, which further support the pharmaceutical product development and their packaging.22–24 As per the ICH guideline, the FD study aims to disclose potential degradation products to assist in evaluating the stability and reveal degradation pathways. It also validates the stability-indicating methods used.25 Stress conditions recommended by the ICH for degradation studies include acid, base, photolytic, thermal, and humidity-induced degradation.16,26 The primary goal is to detect any losses of analytes and the subsequent formation of degradation products.
Acid degradation was carried out with 0.5 N HCl. To the sample solution, 10% of the final volume was added and meticulously stirred. Subsequently, 0.5 N NaOH was used to neutralize the mixture, and the volume was adjusted and analyzed immediately. The alkali degradation was carried out using 0.5 N NaOH. 10% of the final volume was added to the sample solution, mixed thoroughly neutralized with 0.5 N HCl, and injected immediately into the HPLC system. Peroxide degradation was carried out with 30% of H2O2. 10% of the final volume was added to the sample solution, stirred well, kept at room temperature for 1 hour, and immediately injected into the HPLC system. Thermal degradation was carried out by keeping the TVB at 105 °C for 3 hours and then analyzed. Humidity degradation was performed by keeping the weighed TVB in the humidity chamber at 95% relative humidity for 24 hours and then analyzed. The API sample underwent photodegradation and was exposed to UV-A (200 W h m−2) and visible light (1.2 million lux hours) at 25 °C. A control API sample (wrapped in aluminum foil) was also kept in the photostability chamber.
3.2. System suitability
The HPLC technique was evaluated to determine its suitability for the intended analytical application and to verify its reproducibility. A solution containing TVB at a known concentration of 500 ppm was analyzed in six replicates for this evaluation. The suitability of the method was determined by assessing the tailing factor and column efficiency in terms of theoretical plates, i.e.,  1500, and % RSD should not exceed 2.0%.
1500, and % RSD should not exceed 2.0%.
3.3. Precision
System precision was estimated from the 06 replicates of the standard solution on the same day and under the same chromatographic conditions. For method precision, the six replicates of the sample solution (500 ppm) were assessed within a day, and the RSD and % assay were determined. Six replicates of 500 ppm sample solution were analyzed on different days, varied HPLC, and different columns for intermediate precision. The RSD (≤2%) of the peak areas and % assay for TVB were measured.
3.4. Linearity, range, LOD, and LOQ
The linear response was measured by preparing various linearity concentrations ranging from 1% (5 ppm) to 200% (1000 ppm). By plotting the peak area against analyte concentrations, a calibration plot was constructed, and an equation was generated for the quantification of TVB based on the peak areas obtained during analysis. The correlation coefficients (r2), slope and intercept were determined from their respective calibration plots. The r2 should  0.999. LOD is the minimum detectable sample concentration but not quantifiable under specified conditions, while LOQ is the lowest determinable concentration. A statistical approach was used to establish the limits of detection and quantification. This involved the use of a calibration curve containing analyte concentrations in close proximity to the respective limits. The limits were computed using the equations for detection and quantitation limit by applying 3.3σ/s and 10σ/s, respectively.27
0.999. LOD is the minimum detectable sample concentration but not quantifiable under specified conditions, while LOQ is the lowest determinable concentration. A statistical approach was used to establish the limits of detection and quantification. This involved the use of a calibration curve containing analyte concentrations in close proximity to the respective limits. The limits were computed using the equations for detection and quantitation limit by applying 3.3σ/s and 10σ/s, respectively.27
3.5. Accuracy
The sample solution was prepared and analyzed in triplicate at each level by spiking the TVB standard in diluent at about 1% (5 ppm) to about 200% (1000 ppm) of the sample concentration. The percentage recovered was calculated to demonstrate the accuracy of the analyte. Individual and average percent recovery had to be between 98.0% and 102.0%, and the % RSD had to be below 2.0% at each level.28
3.6. Robustness
The robustness evaluation of the method for determining TVB involved minor alterations in chromatographic conditions in the factors described in Table 4. This evaluation was conducted to confirm whether the method can maintain the reliability and integrity of the analytical process.29 System suitability parameters including tailing factor ( 2) and theoretical plates (
2) and theoretical plates ( 1500) must meet the requirements under various robustness conditions. The assay value should also not deviate >±2% from the control condition.
1500) must meet the requirements under various robustness conditions. The assay value should also not deviate >±2% from the control condition.
3.7. Solution stability
Stability in analytical solution (SIAS) was evaluated by keeping the samples in an HPLC sample tray at 5 °C. The stability was analyzed initially and at varied time spans for up to 35 hours, ensuring that the % cumulative RSD did not exceed 2.0%. In addition to monitoring the % RSD, parameters such as peak area, retention time, and peak shape were monitored to ensure SIAS. Performing SIAS ensures accurate results and regulatory compliance by confirming that the analyte remains stable during analysis.
4. Result and discussion
4.1. Method development and optimization
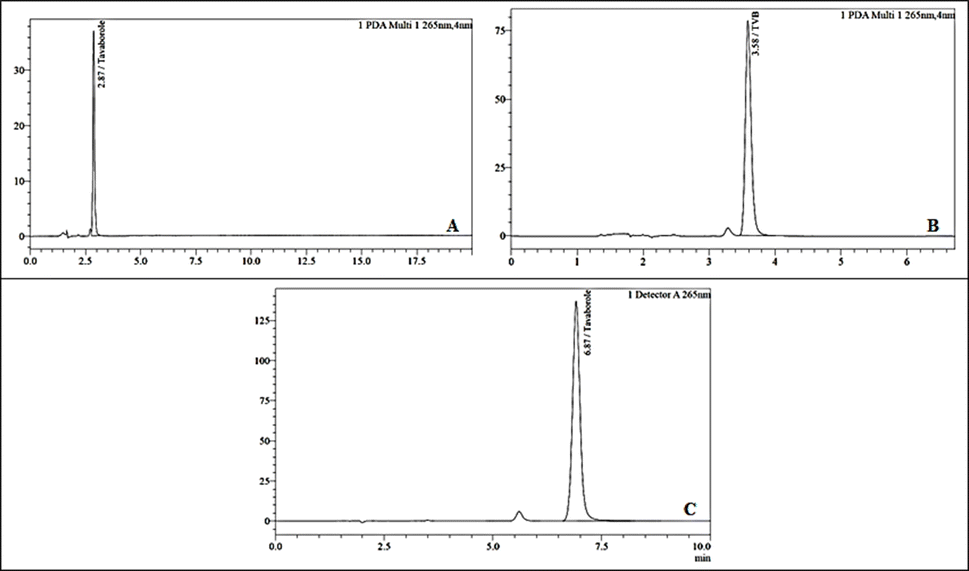
A novel RP-HPLC approach for the quantification of TVB was developed and validated to establish a precise, linear, accurate, and robust method. This analysis achieved exceptional separation and sensitivity through meticulous optimization of the mobile phase, column selection, and wavelength. Several parameters have been carefully considered to ensure optimal separation and sensitivity. These parameters included peak interference, peak purity, peak shape (peak symmetry) and retention time. Peak interference was minimized to ensure clear and distinct separation of analytes. Peak purity was assessed to confirm that each peak represented a single component without co-elution. The peak shape was optimized to achieve sharp and symmetrical peaks, enhancing the method's sensitivity and accuracy.30 The careful selection of an appropriate stationary phase plays a crucial role in developing a method using HPLC. The consistent nature of the column ensures not only its effectiveness and reproducibility but also uniformity throughout.31 The column used during development was Xterra RP18 5 μ, 4.6 × 150 mm. The mobile phase composition, comprising buffer solutions potassium dihydrogen phosphate (25 mm, pH – 3.0) considered as mobile phase A, and organic solvents such as acetonitrile (100%) considered as mobile phase B was used in different ratios i.e., 50:50% v/v; 60:40% v/v and 75:25% v/v, significantly influenced peak separation with the tR of 2.87 min, 3.58 min, and 6.87 min respectively. Initially, the TVB retention was observed at 2.87 min when the acetonitrile concentration was 50%, and when the acetonitrile concentration was 40%, the retention was observed at 3.59 minutes; in both cases, there was co-elution and poor separation, which can lead to loss of sensitivity and interference. The retention factor “k” was observed to be 3.43, indicating significant interaction and good separation.
Furthermore, peaks may not be well resolved, making it difficult to distinguish individual components in the sample. With a decreasing proportion of acetonitrile to 25%, the tR increased to 6.12 min and gave better results (Fig. 2, Table 1). Furthermore, the COT was established at 35 °C and 40 °C, and the flow rate varied among 50 μL, 25 μL, 20 μL, and 15 μL. The OPA was used to adjust the pH of the buffer to 3.0. The mobile phase composition of 75:25 (%v/v) potassium dihydrogen phosphate: acetonitrile, with the optimized flow rate of 15 μL injection volume, showed good elution and recovery at 1 mL min−1 of flow rate and a COT of 35 °C. During the method development phase, the primary goal was to identify a pharmaceutical compound employing simple and robust analytical techniques. Furthermore, the method needed to be cost-efficient, reproducible, and adaptable for routine analytical use.
 |
| | Fig. 2 TVB chromatogram by varying mobile phase composition: (A) 50:50; (B) 60:40; and (C) (75:25) pump ratio of mobile phase (A:B). | |
Table 1 Optimization of mobile phase
|
|
Acceptable value |
Results |
| Mobile phase (A:B) |
— |
50:50 |
60:40 |
75:25 |
|
t
R (min) |
— |
2.87 |
3.58 |
6.12 |
| Tailing factor |
0.9 and 1.4 |
1.23 |
1.32 |
1.11 |
| Theoretical plates |
>2000 |
5235 |
6548 |
7863 |
The previous method by Tampucci et al. utilized a Luna PFP column (150 × 4.6 mm, 5 μm) with a mobile phase of 70% phosphoric acid solution (10 mM, pH 2.0) and 30% acetonitrile, a detection wavelength of 220 nm, and an observed retention time of 8.3 minutes.13 The new method employs an Xterra RP18 column (150 × 4.6 mm, 5 μm), with a mobile phase of potassium dihydrogen phosphate buffer (25 mM, pH 3.0) and acetonitrile (75:25), and a detection wavelength of 265 nm. The use of the Xterra RP18 column offers improved separation efficiency and peak resolution and is comparatively cost-effective and easily available, while the higher detection wavelength of 265 nm provides better sensitivity and selectivity, reducing interference from other components. Additionally, the mobile phase is cost-effective, as it uses less acetonitrile, and the pH 3.0 buffer provides better method stability. Furthermore, a retention time of approximately 6.8 minutes was observed, enhancing throughput and efficiency by reducing the run time. These improvements make the new method more efficient and economical, suitable for routine analysis in various practical applications.
4.2. Method validation
Validation studies demonstrated the reliability and suitability of the method for the quantitative analysis of TVB. Parameters including precision, linearity, accuracy, specificity, and robustness were evaluated according to guidelines.
4.2.1. Specificity and forced degradation.
The FD studies of TVB were conducted to evaluate its stability profile under different stress conditions (Table 2). This analysis aims to clarify possible degradation pathways and assess the drug's susceptibility to chemical, physical, and photolytic degradation.32,33 Under chemical degradation conditions, TVB showed degradation when exposed to an acidic (0.5 mL 0.5 N HCl), basic (0.5 mL 0.5 N NaOH), and peroxide (0.5 mL 30% H2O2) environment. Acidic and basic conditions led to hydrolysis-mediated degradation, with 6.68% and 8.13% degradation rates, respectively. The peroxide-induced degradation, likely due to oxidative reactions, resulted in the highest degradation rate of 22.93%. Physical stress testing, such as thermal degradation (3 hours at 105 °C) and humidity degradation (95% RH for 24 hours), also affected TVB stability. Thermal degradation caused a degradation rate of 6.12%, indicating susceptibility to heat-induced degradation. Thermal degradation caused a degradation rate of 6.12%, indicating susceptibility to heat-induced degradation. However, exposure to humidity resulted in minimal degradation (1.73%), suggesting relative stability under high humidity conditions. Regarding photolytic degradation, TVB remained stable when protected from light (dark conditions covered with aluminum foil).
Table 2 Forced degradation of TVB
| Condition |
Sample treatment |
Assay (%) |
t
R (min) |
% Degradation |
Peak purity |
| Control |
No treatment |
99.31 |
6.82 |
— |
1.000000 |
| Acidic |
0.5 mL–0.5 N HCl; immediate |
92.60 |
6.81 |
6.71 |
1.000000 |
| Alkali |
0.5 mL–0.5 N NaOH; immediate |
91.18 |
6.82 |
8.14 |
1.000000 |
| Peroxide |
0.5 mL–30% H2O2; immediate |
76.47 |
6.84 |
22.84 |
1.000000 |
| Humidity |
95% RH; 24 hours |
97.52 |
6.84 |
1.79 |
1.000000 |
| Thermal |
3 hours; 105 °C |
93.19 |
6.83 |
6.12 |
1.000000 |
| Photostability control |
Dark condition UV light; 7 days; 25 °C |
95.41 |
6.87 |
— |
1.000000 |
| Photostability |
UV light; 7 days; 25 °C |
93.67 |
6.87 |
1.73 |
1.000000 |
However, exposure to light (1.2 million lux hours & 200 watt hour m−2 under open conditions) resulted in a degradation rate of 3.17%, indicating susceptibility to light-induced degradation. The forced degradation studies demonstrated that TVB remained within acceptance criteria for total degradation (below 30%). Chromatograms are illustrated in Fig. 3. The observed degradation pathways provided insights into possible degradation mechanisms and supported the development of appropriate storage and handling protocols. Furthermore, confirmation of method specificity by chromatographic analysis confirmed the reliability of the assay method for assessing TVB stability. These results contribute to the broader research to ensure safety and efficacy.
 |
| | Fig. 3 Chromatograms showing forced degradation studies of TVB under varied settings: (A) control (B) acidic; (C) basic; (D) peroxide (E) humidity (F) thermal (G) photostability control (H) photolytic degradations (I) standard chromatogram. | |
4.2.2. Evaluation of system suitability.
Six identical injections of the system suitability solution were introduced into the HPLC system to assess resolution, retention time, column efficiency and repeatability. Fig. 3(I) illustrates the standard chromatogram. Statistical analysis showed no significant deviations in peak area, retention time, theoretical plate count ( 1500 theoretical plates), or tailing factor (
1500 theoretical plates), or tailing factor ( 2). The % RSD for these parameters was found to be <2.0% (Table 3).
2). The % RSD for these parameters was found to be <2.0% (Table 3).
Table 3 Precision, linearity and accuracy of TVB
| System suitability |
| Concentration |
t
R
|
Peak area |
| 500 ppm |
6.93 |
1570.87 |
| 6.93 |
1565.23 |
| 6.94 |
1565.67 |
| 6.94 |
1565.33 |
| 6.94 |
1562.02 |
| 6.95 |
1563.24 |
| Average |
6.94 |
1565.39 |
| SD |
0.01 |
3.04 |
| RSD |
0.11 |
0.19 |
| Precision |
| Precision parameter |
System precision |
Method precision |
Intermediate precision |
| % RSD |
0.19 |
0.38 |
0.53 |
| Theoretical plate count |
8602 |
8602 |
7611 |
| Tailing factor |
1.12 |
1.12 |
1.1 |
| % Assay |
— |
98.3 |
97.3 |
| Ruggedness |
| Overall SD |
0.67 |
| Overall RSD |
0.69 |
| Linearity |
| Concentration (ppm) |
Mean area |
Observed concentration (ppm) |
| 5 |
10.790 |
3.0 |
| 50 |
159.841 |
50.46 |
| 150 |
474.033 |
150.52 |
| 250 |
787.426 |
250.33 |
| 375 |
1170.487 |
372.32 |
| 500 |
1562.972 |
497.32 |
| 625 |
1958.909 |
623.41 |
| 750 |
2353.390 |
749.04 |
| 1000 |
3143.168 |
1000.56 |
| Accuracy |
| Accuracy range (%) |
Recovery (%) |
SD |
RSD (%) |
| 1 |
100 |
0 |
0 |
| 50 |
99.73 |
0.24 |
0.25 |
| 100 |
99.32 |
0.14 |
0.15 |
| 150 |
99.34 |
0.09 |
0.09 |
| 200 |
99.81 |
0.22 |
0.22 |
4.2.3. Evaluation of precision.
The system precision results revealed that the % RSD of the TVB peak area was 0.19%, which was well within the acceptance criteria. Moreover, the theoretical plate count and tailing factor from the first injection of the standard were 8601.6 and 1.12, respectively, meeting the specified criteria. Method precision assessments showed that % assay for each preparation met the specification limit, with the % RSD of TVB content for six sample preparations observed at a commendably low 0.38%. Intermediate precision analyses revealed % assay for each preparation on different days, using different HPLC and columns, all meeting the specification limit. The % RSD of assay results across six sample preparations was 0.53%, while the overall % RSD of replicate test preparations from both method and intermediate precision was a satisfactory 0.69%. These findings demonstrated ruggedness to various variables, such as different HPLC instruments, columns, and days, confirming its precision and reliability in analytical settings (Table 3).
4.2.4. Evaluation of linearity.
The linearity of TVB response was evaluated at concentrations from approximately 1% (5 ppm) to 200% (1000 ppm) of the standard concentration range (Table 3). Standard solutions within this range were prepared and subjected to analysis using the proposed method. A correlation coefficient of  0.999 is used as a measure of acceptance. The linear equation Y = 3.14x – 1.3971 was observed, and the goodness-of-fit or correlation coefficient was found to be 1.00000. The linearity is shown graphically in Fig. 4. The detection and quantitation were calculated using the y-intercept SD and the slope of the calibration curve; the LOD and LOQ values were observed at 1.47 ppm and 4.45 ppm, respectively.
0.999 is used as a measure of acceptance. The linear equation Y = 3.14x – 1.3971 was observed, and the goodness-of-fit or correlation coefficient was found to be 1.00000. The linearity is shown graphically in Fig. 4. The detection and quantitation were calculated using the y-intercept SD and the slope of the calibration curve; the LOD and LOQ values were observed at 1.47 ppm and 4.45 ppm, respectively.
 |
| | Fig. 4 (A) Chromatogram of TVB standard solution; (B) linearity plot; and (C) overlay. | |
4.2.5. Evaluation of accuracy.
The “accuracy” parameter was evaluated by spiking the TVB standard in the diluent at about 1% (5 μg mL−1) to about 200% (1000 μg mL−1) of the sample concentration in triplicate at each level, demonstrating good recovery rates, which are within acceptable criteria. All samples exhibited complete recovery at the 1% level, with a mean recovery of 100.00% and negligible % RSD of 0.00%. Similarly, at higher concentration levels (50%, 100%, 150%, and 200%), the samples consistently showed recovery rates close to 100%, ranging from 99.20% to 100.06%. The overall mean recovery was 99.64%, with an overall % RSD of 0.31%, indicating excellent accuracy of the analytical method used to quantify TVB. The data are shown in Table 3.
4.2.6. Evaluation of robustness.
The proposed method was validated for robustness by examining the sample and the standard TVB solution, showing minor changes (Table 4). Despite intentional parameter adjustments, comparative analysis using the optimized method revealed no significant change in retention time, tailing factor, SD, % RSD, and theoretical plates. The system suitability parameters complied under each robustness condition, and the TVB peak tR remained consistent. TVB assay values were within the specified acceptance criteria. Variations in flow rate (±10%), COT (±5 °C), mobile phase composition (±2%), and the detection wavelength (±5 nm) all met the acceptance criteria, indicating the robustness of the method to pre-meditated analytical changes. Consequently, the method proves to be robust for its intended application.
Table 4 Robustness of TVB
| Parameter |
Variation level |
Theoretical plates |
Tailing factor |
% RSD |
t
R (min) |
Assay (%) |
| Normal condition |
265 nm; 75:25; 35 °C; 1 mL min−1; 3.0 |
8601.6 |
1.12 |
0.19 |
6.93 |
98.09 |
| Change in wavelength (nm) |
260 |
8520.2 |
1.12 |
0.16 |
6.95 |
99.25 |
| 270 |
8461.7 |
1.13 |
0.14 |
6.96 |
99.23 |
| Change in mobile phase composition |
73:27 |
8509.6 |
1.09 |
0.64 |
7.93 |
100.19 |
| 77:23 |
8619.3 |
1.13 |
0.29 |
6.18 |
99.94 |
| Change in temperature (°C) |
30 |
8433.3 |
1.11 |
0.07 |
7.31 |
99.01 |
| 40 |
8775.5 |
1.12 |
0.08 |
6.6 |
99.22 |
| Change in flow rate (ml min−1) |
0.9 |
8787.8 |
1.12 |
0.18 |
7.72 |
99.02 |
| 1.1 |
8328 |
1.11 |
0.23 |
6.32 |
99.43 |
| Change in pH |
2.8 |
8500.2 |
1.11 |
0.08 |
7 |
97.09 |
| 3.2 |
8499 |
1.12 |
0.18 |
6.96 |
97.26 |
4.2.7. Evaluation of stability in analytical solution.
The SIAS over different time period showed a % cumulative RSD of 0.30 and 0.18 up to 35 hours at 5 °C, respectively, for standard and sample solutions. These findings indicate that both solutions remain stable for up to 35 hours. These results are relevant to demonstrate the reliability of the HPLC method used to analyze TVB. A low % cumulative RSD value indicated good stability, vital for accurate and reproducible results. The stability of the standard and sample solutions over 35 hours at 5 °C substantiates the reliability and efficacy of the methodology, instilling confidence in the precision of the analytical findings.
5. Application of method
An RP-HPLC method, ensuring it aligns with ICH guidelines, specifically tailored to measure TVB. The approach has been characterized and confirmed to be rapid, simple, precise, accurate, and specific in its ability to quantify TVB levels accurately. Runtime of 10 minutes enables the analysis of samples in a short time, tailoring it for regular examination of TVB in formulations and ensuring that the analysis is rapid and reliable.
The methodology carefully considered factors such as cost-effectiveness, simplicity, equipment compatibility, solvent suitability, analysis speed, and adaptability. These factors are crucial for routine analysis and are effectively addressed in this approach. TVB's quantitative estimation and assay results are within acceptable limits using an optimized and validated HPLC method, demonstrating its effectiveness. Furthermore, the developed chromatographic method shows promise in the analysis of samples from accelerated stability experiments and routine formulation testing, incorporating its potential for practical use, which extends to aiding in the identification and quantification of degradation products in formulation, thus contributing to the enhancement of quality and safety measures. Stability studies conducted under different stress conditions confirm significant degradation of TVB in the oxidation medium compared to others, supporting previously reported methods of oxidative degradation of TVB. In particular, chromatograms from degradation studies reveal no interference with TVB peaks, confirming the specificity of the developed method for formulations containing TVB. This research presents a novel HPLC methodology for TVB, contributing to advancements, ensuring the safe and effective use of TVB for onychomycosis treatment, and improving the quality of formulations.34 Furthermore, the method's efficiency is confirmed by validation, making it suitable for future bioanalytical applications.
6. Conclusion
An HPLC approach was devised and validated for the TVB quantification. Validation adhered to ICH guidelines, assessing specificity, precision, linearity, detection, quantification limit, accuracy, robustness, and stability-indicating potential. Stress testing was conducted under various conditions, with all outcomes meeting the predefined acceptance criteria, demonstrating the method's suitability for sample analysis. The solution stability study showed that standard and sample solutions remained stable for up to 35 hours at 5 °C. This finding is significant as it confirms the reliability and robustness of the HPLC method over an extended period, ensuring that accurate and consistent results can be obtained during routine analysis. These comprehensive validation results highlight the method's effectiveness, accuracy, and reliability. The stability-indicating HPLC method thus provides a robust analytical tool for the quality control and development of TVB, ensuring the integrity and consistency of the drug product throughout its shelf life. This method is imperative for pharmaceutical quality control laboratories, facilitating the accurate assessment of TVB stability and potency in various formulations.
Abbreviation
| ACN | Acetonitrile |
| COT | Column oven temperature |
| D0 | Zero order |
| D1 | First-order derivative |
| FD | Forced degradation |
| H2O2 | Hydrogen peroxide |
| HCl | Hydrochloric acid |
| KH2PO4 | Potassium dihydrogen orthophosphate |
| NaOH | Sodium hydroxide |
| OPA | Orthophosphoric acid |
| PDA | Photodiode array |
| RSD | Relative standard deviation |
|
t
R
| Retention time |
| SD | Standard deviation |
| TVB | Tavaborole |
| UPLC | Ultra-performance liquid chromatography |
| USFDA | US food and drug administration |
| WL | Wavelength |
Data availability
The data that supports the findings are within the paper and additional data may be requested from the authors.
Author contributions
Conceptualization: Shiv Kumar Prajapati; data curation: Shiv Kumar Prajapati; formal analysis: Meenakshi Bajpai; methodology: Shiv Kumar Prajapati; software: Shiv Kumar Prajapati; supervision Meenakshi Bajpai & Ankit Jain; validation: Shiv Kumar Prajapati; writing – original draft: Shiv Kumar Prajapati; writing – review & editing: Shiv Kumar Prajapati, Ankit Jain, & Meenakshi Bajpai.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We would like to thank the Birla Institute of Technology and Science, Pilani, for providing generous financial support, which made it possible to publish this article as open access.
References
- A. K. Leung, J. M. Lam, K. F. Leong, K. L. Hon, B. Barankin, A. A. Leung and A. H. Wong, Recent Pat. Inflammation Allergy Drug Discovery, 2020, 14, 32–45 CAS.
- F. Yousefian, C. Smythe, H. Han, B. E. Elewski and M. Nestor, J Clin Aesthet Dermatol, 2024, 17, 24 Search PubMed.
- H. Eftekhari, Y. H. Saheli, M. T. Ashoobi, M. Mahjoob, E. K. Leyli and P. B. Toolaroud, Heliyon, 2024, 10(4), e25737 CrossRef CAS PubMed.
- G. Kuvandik, M. Çetin, G. Genctoy, M. Horoz, M. Duru, C. Akcali, S. Satar, A. A. Kiykim and H. Kaya, BMC Infect. Dis., 2007, 7, 1–5 CrossRef PubMed.
- B. E. Elewski and A. Tosti, Expert Opin. Pharmacother., 2014, 15, 1439–1448 CrossRef CAS PubMed.
- M. E. Toledo-Bahena, A. Bucko, J. Ocampo-Candiani, M. E. Herz-Ruelas, T. M. Jones, M. T. Jarratt, R. A. Pollak and L. T. Zane, J. Drugs Dermatol., 2014, 13, 1124–1132 CAS.
- A. Kuplińska and K. Rząd, Amino Acids, 2021, 53, 961–991 CrossRef PubMed.
- A. H. Indabawa, S. U. Abubakar and S. Yahaya, J. Chem. Pharm., 2021, 13, 01–09 Search PubMed.
- P. D. Harak, A. G. Zalte and V. S. Gulecha, Res. J. Pharm. Technol., 2023, 16, 1342–1346 Search PubMed.
- A. K. Gupta and D. Daigle, Food Res. Int., 2014, 9, 1243–1250 CAS.
- A. Markham, Drugs, 2014, 74, 1555–1558 CrossRef CAS PubMed.
- M. Poulakos, Y. Grace, J. D. Machin and E. Dorval, J. Pharm. Pract., 2017, 30, 245–255 CrossRef PubMed.
- S. Tampucci, E. Terreni, S. Burgalassi, P. Chetoni and D. Monti, J. AOAC Int., 2018, 101, 437–443 CrossRef CAS PubMed.
- U. Puppala, V. M. Marisetti, K. S. Srinivas, K. V. Reddy, M. Kaliyaperumal and R. Doddipalla, Biomed. Chromatogr., 2021, 35, e5070 CrossRef CAS PubMed.
- A. S. Sheela, M. M. Annapurna and R. S. Yasaswini, Res. J. Pharm. Technol., 2020, 13, 1895–1900 CrossRef.
-
ICH Q14, Analytical procedure development Q14, https://database.ich.org/sites/default/files/ICH_Q14_Document_Step2_Guideline_2022_0324.pdf.
-
ICH Q2(R2), ICH guideline Q2(R2) on validation of analytical procedures, https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-q2r2-validation-analytical-procedures-step-2b_en.pdf, accessed 21-02-2024.
- D. V. McCalley, Chem. Commun., 2023, 59, 7887–7899 RSC.
- A. Czyrski and J. Sznura, Sci. Rep., 2019, 9, 19458 CrossRef CAS PubMed.
- T. Siva Rao, V. Balireddi and T. Krishna Murthy, Eur. J. Biomed. Pharm. Sci., 2018, 5, 545–550 Search PubMed.
- A. Jain, A. Gulbake, A. Jain, S. Shilpi, P. Hurkat, S. Kashaw and S. K. Jain, J. Chromatogr. Sci., 2014, 52, 697–703 CAS.
- T. Zelesky, S. W. Baertschi, C. Foti, L. R. Allain, S. Hostyn, J. R. Franca, Y. Li, S. Marden, S. Mohan and M. Ultramari, J. Pharm. Sci., 2023, 112, 2948–2964 CrossRef CAS PubMed.
- S. Singh, M. Junwal, G. Modhe, H. Tiwari, M. Kurmi, N. Parashar and P. Sidduri, TrAC, Trends Anal. Chem., 2013, 49, 71–88 CrossRef CAS.
- M. Blessy, R. D. Patel, P. N. Prajapati and Y. Agrawal, J. Pharm. Anal., 2014, 4, 159–165 CrossRef CAS PubMed.
-
ICH Q1A (R2), ICH guideline Q1A (R2) stability testing of new drug substances and products, https://database.ich.org/sites/default/files/Q1A%28R2%29%20Guideline.pdf, accessed 21-02-2024.
-
ICH Q2B, Q2B validation of analytical procedures: methodology, https://www.fda.gov/regulatory-information/search-fda-guidance-documents/q2b-validation-analytical-procedures-methodology, accessed 21-02-2024.
- H. Darji, Z. Dedania, R. Dedania and V. Jain, Green Anal. Chem., 2024, 8, 100092 CrossRef.
- U. Nepal, V. K. Panthi, N. P. Chaudhary and S. Chaudhary, Int. J. Anal. Chem., 2022, 2022(1), 8331762 Search PubMed.
- P. Gupta, S. Sharma, A. Gupta, S. Kawish, M. Iqbal, S. Rahman, M. Aqil, K. Kohli and Y. Sultana, ACS Omega, 2024 DOI:10.1021/acsomega.3c08078.
- A. Najmi, Z. u. Rehman, H. A. Alhazmi, M. M. Albratty, N. H. Majrashi, K. M. Hakami, N. A. Najmi and A. A. Mobarki, Separations, 2023, 10, 346 CrossRef CAS.
- U. D. Sharma, L. Kumar and R. Verma, Res. J. Pharm. Technol., 2022, 15, 4325–4332 Search PubMed.
- D. C. Kulkarni, A. S. Dadhich and M. M. Annapurna, J. Drug Delivery Ther., 2024, 14, 161–169 CrossRef.
- A. Tiwari, D. Bose, P. Mishra, A. Jain and S. K. Jain, J. AOAC Int., 2022, 105, 999–1007 CrossRef PubMed.
- T. Tome, N. Žigart, Z. Časar and A. Obreza, Org. Process Res. Dev., 2019, 23, 1784–1802 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Ankit
Jain
a,
Ankit
Jain
 1500, and % RSD should not exceed 2.0%.
1500, and % RSD should not exceed 2.0%.
 0.999. LOD is the minimum detectable sample concentration but not quantifiable under specified conditions, while LOQ is the lowest determinable concentration. A statistical approach was used to establish the limits of detection and quantification. This involved the use of a calibration curve containing analyte concentrations in close proximity to the respective limits. The limits were computed using the equations for detection and quantitation limit by applying 3.3σ/s and 10σ/s, respectively.27
0.999. LOD is the minimum detectable sample concentration but not quantifiable under specified conditions, while LOQ is the lowest determinable concentration. A statistical approach was used to establish the limits of detection and quantification. This involved the use of a calibration curve containing analyte concentrations in close proximity to the respective limits. The limits were computed using the equations for detection and quantitation limit by applying 3.3σ/s and 10σ/s, respectively.27
 2) and theoretical plates (
2) and theoretical plates ( 1500) must meet the requirements under various robustness conditions. The assay value should also not deviate >±2% from the control condition.
1500) must meet the requirements under various robustness conditions. The assay value should also not deviate >±2% from the control condition.

 1500 theoretical plates), or tailing factor (
1500 theoretical plates), or tailing factor ( 2). The % RSD for these parameters was found to be <2.0% (Table 3).
2). The % RSD for these parameters was found to be <2.0% (Table 3).
 0.999 is used as a measure of acceptance. The linear equation Y = 3.14x – 1.3971 was observed, and the goodness-of-fit or correlation coefficient was found to be 1.00000. The linearity is shown graphically in Fig. 4. The detection and quantitation were calculated using the y-intercept SD and the slope of the calibration curve; the LOD and LOQ values were observed at 1.47 ppm and 4.45 ppm, respectively.
0.999 is used as a measure of acceptance. The linear equation Y = 3.14x – 1.3971 was observed, and the goodness-of-fit or correlation coefficient was found to be 1.00000. The linearity is shown graphically in Fig. 4. The detection and quantitation were calculated using the y-intercept SD and the slope of the calibration curve; the LOD and LOQ values were observed at 1.47 ppm and 4.45 ppm, respectively.
