
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDryfilm-ATR-FTIR analysis of urinary profiles as a point-of-care tool to evaluate aerobic exercise†
Jaume
Béjar-Grimalt
a,
Ángel
Sánchez-Illana
 *a,
Miguel de la
Guardia
a,
Salvador
Garrigues
a,
Ignacio
Catalá-Vilaplana
b,
Jose Luis
Bermejo-Ruiz
c,
Jose Ignacio
Priego-Quesada
*bd and
David
Pérez-Guaita
a
*a,
Miguel de la
Guardia
a,
Salvador
Garrigues
a,
Ignacio
Catalá-Vilaplana
b,
Jose Luis
Bermejo-Ruiz
c,
Jose Ignacio
Priego-Quesada
*bd and
David
Pérez-Guaita
a
aDepartment of Analytical Chemistry, University of Valencia, Burjassot, Spain. E-mail: angel.illana@uv.es
bResearch Group in Sports Biomechanics (GIBD), Department of Physical Education and Sports, Universitat de València, Valencia, Spain. E-mail: j.ignacio.priego@uv.es
cDepartment of Physical Education and Sports, Universitat de València, Valencia, Spain
dResearch Group in Medical Physics (GIFIME), Department of Physiology, Universitat de València, Valencia, Spain
First published on 13th August 2024
Abstract
The understanding of metabolic alterations triggered by intense exercise can provide a biological basis for the development of new training and recovery methods. One popular way to monitor these changes is the non-invasive analysis of the composition of urine. This work evaluates the use of attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR) and multivariate analysis as a rapid and cost-effective way to investigate changes in urine composition after intense exercise. The urine FTIR spectra of 21 volunteers (14 going through aerobic exercise and 7 controls) were measured before and immediately, 2, 5, 11, and 24 h after running 10 km. Principal component analysis (PCA) and partial least squares analysis (PLS) regression were used to investigate the changes in the spectra as a function of the recovery time. PLS models obtained for the prediction of the time points in the exercise group were deemed significant (p < 0.05, rand t-test permutation testing in cross-validation), showing changes in the urine composition after the exercise, reaching a maximum after 11 hours as opposed to the control group which did not show any significant relationship with the recovery time. In a second step, spectra of the protean extract isolated from urines at significant timepoints (before, immediately after, and 11 hours after exercise) were measured. The PCA of the protein spectra showed clear differences in the spectra obtained at the separation between the recovery time points, especially after the end of the exercise, where the protein profile was significantly different from the other times. Results indicate that the technique was able to find differences in the urine after physical exertion and holds strong potential for an easy-to-use and simple screening metabolic evaluation of recovery methods.
1. Introduction
Monitoring the stress suffered during physical exercise through the use of chemical biomarkers provides insights into how the body responds to training, including adaptations, fatigued states and/or recovery processes.1,2 By integrating these biomarkers with factors such as strength, heart rate, blood pressure, and recovery from fatigue, trainers and medical professionals can gain enhanced insight into the requirements of individuals engaging in physical activity. This promotes the improvement and optimization of training regimens, resulting in the prevention of injuries3 and cardiovascular pathologies4 as well as the discovery of hidden genetic conditions.5In the aforementioned context, the changes in metabolism associated with intense exercise have been studied in various biofluids, including blood,6,7 saliva8,9 and urine.10–12 Urine is an ideal sample in the sports context due to its non-invasive nature, making it easily and rapidly accessible during both exercise and recovery. Furthermore, urine contains a multitude of biomolecules derived from the glomerular filtration of plasma, which are associated with the physical integrity of the individual. For instance, urine components such as creatinine, creatine kinase (CK), catecholamines, and myoglobin serve as markers of muscle stress and breakdown.13 Simultaneously, changes in oxidative stress markers, such as lipid peroxidation14 and phenylalanine oxidation products,15 can provide information about oxidative damage. Additionally, alterations in hormone levels induced by exercise, including cortisol and adrenocorticotropic hormone (ACTH), could serve as indicators of the body's stress response to training.12,16,17 Another advantage of urine is that it presents a simpler matrix compared to plasma or tissue, characterized by lower concentrations of lipids and peptides.11
To study changes in the metabolome associated with exercise and recovery, urine has been analyzed with different methodologies based on gas chromatogram-mass spectrometry (GC-MS), liquid chromatography-mass spectrometry (LC-MS) or nuclear magnetic resonance (NMR).18 Although these techniques can identify a wide range of metabolites with sensitivity and specificity, they are time-consuming, require intensive sample processing and must be performed in expensive and non-portable instrumentation. To investigate both exercise-induced stress and recovery, sport science studies also need techniques that can be employed in situ and offer direct and rapid analysis of several samples at different time points, all at a reasonable cost. In this context, Fourier-transform infrared spectroscopy (FTIR) coupled with attenuated total reflectance (ATR) enables fast and cost-effective measurement with minimum or no sample treatment.19 ATR-FTIR is progressively garnering attention in clinical analysis, being the analysis of biofluids a significant portion of the field with a wide range of potential diagnostic application.19–22 In this sense, it has been demonstrated in field studies that can be used in remote locations for obtaining information about clinical samples23 and has been proposed for the identification of discrete biomarkers in urine.24–26
Despite the important benefits of ATR-FTIR, its sensitivity is lower than GC-MS or LC-MS techniques for different reasons. For example, the inherent water content of urine often masks significant biological information and limits the range of markers that can be detected. For this reason, the analysis of urine as a dry film is preferable.27 Since the spectra contain the biological information encrypted in a complex set of overlapped bands, cluster analysis, chemometric and machine learning algorithms are employed to extract potential biological markers from the infrared (IR) variables.20 However, the incorporation of simple urine sample processing steps such as centrifugation, liquid–liquid microextraction, and ultrafiltration have demonstrated improvements of the detection limit of certain molecules and the simplification of the IR spectra by eliminating potential interferents.24,26,28
In this context, this study aimed to evaluate the capability of ATR-FTIR to identify spectral markers associated with both, intense exercise, and recovery in the IR spectra. To that end, spectra of urines from different volunteers before intense training and at different recovery times were obtained and compared to similar data from control volunteers using chemometric methods. The spectra of untreated dry films of urine were evaluated for all the timepoints of the study, while the spectra of protean extracts of urine using ultrafiltration was evaluated for significant timepoints (before, immediately after, and 11 hours after exercise).
2. Material and methods
2.1. Study participants and sample collection
Urines used for this study were obtained from 14 physically active participants, considered as the experimental group (6 male and 8 female, with an age between 33 ± 9 years old, a height of 170 ± 7 cm, a body mass of 69 ± 10 kg and a body fat percentage of 21 ± 7%) and 7 participants included as controls who did not perform any exercise during the study (4 male and 3 female, with an age between 34 ± 5 years old, a height of 173 ± 7 cm, a body mass of 73 ± 13 kg and a body fat percentage of 22 ± 10%). Some instructions were given to participants to control factors that may affect the study intervention: to avoid high-intensity physical activity in the 24 h before the assessments, to avoid smoking, drinking alcohol, caffeine, or other stimulating beverages in the 12 hours before the assessments, and to avoid copious meals and other exercise that stipulated by the study during the day of the measurements.The experimental group ran 10 km at a moderate rate of perceived exertion (11 points using the 20-point Borg scale) on an outdoor running track. The time spent for the 10 km was 51 ± 9 min. The Control group did not perform any exercise. Urine samples were obtained before exercise between 7 am and 9 am (pre), immediately after finishing exercise (post; only for the experimental group), 2 hours after exercise (post 2), 5 hours after exercise (post 5) 11 hours after exercise (post 11), and finally 24 hours after exercise (post 24). Timepoints were selected to have a good sampling of the responses over time, without excessively compromising the participants' routine (for example, avoiding measuring after 8 pm and during sleeping hours). Overall fatigue perception was measured at pre and post 24 using a 150 mm visual analogue scale, labelled from the left as “absence of fatigue” to the right as “highest fatigue imaginable”. This research protocol was in accordance with the Declaration of Helsinki and was approved by the Ethics Committee for Research in Human Beings of the University of Valencia.
2.2. Sample preprocessing and ATR-FTIR analysis
Two ATR-dry film analytical approaches were undertaken. One involved the direct analysis of centrifuged urine samples and was carried out for all the different timepoints. The other one incorporated a minimal preprocessing to isolate and concentrate proteins in urine using ultrafiltration and was only carried out for 3 significant timepoints: before, immediately after, and 11 hours after exercise.ATR-FTIR spectra of the dry films were acquired using a Tensor 27 FTIR spectrometer from Bruker (Karlsruhe, Germany) equipped with a DLaTGS detector using a DuraSamplIR II ATR module with a three-reflection diamond/ZnSe DuraDisk plate, from Smith Detection Inc. (Warrington, UK). Acquisitions were performed between 550 cm−1 and 4000 by accumulation of 50 scans per spectrum and 4 cm−1 nominal resolution. The background spectrum was performed by measuring the clean ATR crystal before each sample measurement. OPUS program (version 6.5) from Bruker was employed for instrumental and measurement control. All the spectra were acquired as OPUS “ATR spectra” and thus, an internal correction of the pathlength was automatically applied (i.e., signal scaled as absorbance × wavenumber/1000).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 150 g for 10 minutes, resulting in approximately 50 μL of protein-rich extract. Then, employing the same centrifugation parameters, the extract was washed per triplicate with 450 μL ultrapure water obtained from a Labaqua BIO ultrapure water purification system from Biosan (Riga, Latvia). All filters employed were pre-rinsed with 500 mL of ultrapure water before the sample loading to remove the trace amounts of glycerine.
150 g for 10 minutes, resulting in approximately 50 μL of protein-rich extract. Then, employing the same centrifugation parameters, the extract was washed per triplicate with 450 μL ultrapure water obtained from a Labaqua BIO ultrapure water purification system from Biosan (Riga, Latvia). All filters employed were pre-rinsed with 500 mL of ultrapure water before the sample loading to remove the trace amounts of glycerine.
Finally, 1 μL of the protein-rich extract was deposited onto the ATR crystal, dried, and measured as a dry film. ATR-FTIR spectra of the protein extract were obtained using a Spectrum Two FT-IR spectrometer from PerkinElmer (Waltham, MA, USA) equipped with the UATR accessory (i.e. 1 reflexion diamond crystal), coadding 10 scans at 4 cm−1 spectral resolution over the range 4000–500 cm−1. The background spectrum was performed by measuring the clean ATR crystal before each sample measurement. Spectrum (version 10.03.06) from PerkinElmer was employed for the instrumental and measurement control and the atmospheric contribution was corrected using the CO2/H2O correction provided by the software. Prior to analysis, spectra were pre-processed employing the same algorithm for ATR correction used in OPUS.
2.3. Data analysis
ATR-FTIR spectra were analyzed using MATLAB R2022a from Mathworks (Natick, MA) employing the PLS_Toolbox functions from Eigenvector (Manson, WA) and in-home written functions and scripts. All the spectra were plotted, visually inspected and the main IR bands were annotated according to the literature. After the visual inspection, different chemometric models were built using the 550–1800 cm−1 spectral region with a common preprocessing as follows: (1) normal variate scaling normalization (SNV), (2) Savitzky–Golay smoothing and second order derivative computation (polynomial order: 2 window width: 15), and (3) mean centering.First, a PCA model was built using the spectral dataset obtained measuring the dry films of the centrifuged urine. The number of significant principal components (PC) was selected based on the minimum value in the root mean square error of cross validation (RMSECV) versus the principal component number. Cross validation was performed by the random subsets algorithm using 10 splits and 20 iterations.
In order to identify the spectral features related to the study times, an orthogonalized PLS (o-PLS) model was performed for the centrifuged urine spectra being the response variable (Y) the study times. Separate models were made for the experimental and control samples. These models were cross-validated employing the random subsets algorithm and the RMSECV was used for selecting the optimal number of latent variables. The significance of the models was studied by permutation testing29 and the regression vectors were inspected to assign the spectral features to potential molecular biomarkers.
For the protein extraction dry films, a visual quality control (QC) was carried out prior to the further chemometric analysis. This QC was based on the presence of the amide I and amide II bands and their well-known spectral profile. A PCA model was built following the same number of PC selections as above.
All the raw data generated in this work together with a MATLAB livescript containing the data analysis workflow are available as a dataset in the Zenodo repository.30
3. Results and discussion
3.1. Exploratory analysis of the spectral data
The exploratory analysis confirmed the correct formation of urine dry films. As it is shown in Fig. 1, which compares the average spectra of training and control volunteers, the spectra showed typical bands of dry films of urine obtained in previous works.24,25 Second derivatives of the spectra are available at the ESI for further inspection (see Fig. SM1†). Spectra were dominated by the signals of urea and creatinine, which are the major components of urine, found as bands at 1450, 1605 and 3340 cm−1, assigned to the νas(CN), δ(NH) and ν(NH) modes of urea, respectively.31 These bands are also overlapped with νas(CN) and δ(NH) modes from creatinine,32 and additional bands are found in the urine spectra, including overlapped bands at 1655, 1150 and 1070 cm−1 which can be assigned to a variety of urine compounds, including salts such as CaCl2, NaCl, MgSO4, NaH2PO4 or NaHCO3, or even glucose and uric acid, which are some of the major components that make up the urine.33 | ||
| Fig. 1 Average IR spectra (lines) and standard deviation (shade) of the urine dry-films for the control (blue) and the experimental (red) samples acquired with three-reflection Smiths ATR accessory. Spectra have been shifted along the Y-axis in order to provide a better comparison. | ||
On the other hand, the spectral profiles of the protein extracts (see Fig. 2) were dominated by typical protein bands, namely the amide I (1644 cm−1), II (1545 cm−1) and III (1400 cm−1) bands, which are characteristic of isolated proteins as reported elsewhere.24,34,35 However, it was noticed that some samples showed signs of urea contamination (strong band at 1605 cm−1 of compared with the amide bands) indicating that for those samples the isolation was not effective enough (see ESI, Fig. SM2†). Therefore, a visual quality control (QC) was conducted based on the presence and integrity of the amide I and amide II bands to ensure they were not overshadowed by the urea band at 1605 cm−1. Additionally, the urea band at 1467–1450 cm−1 was used as an additional check for urea presence. The spectra with non-clear amide bands were discarded resulting in the exclusion of 14 samples. Fig. 2, which represents the average of the spectra from the three classes considered that passed the QC, also shows strong bands at the 1000–1200 cm−1 region, which are assigned to the C–O from alcohols found in glycoproteins such as the uromodulin, which is the main protein component of urine in healthy individuals.36 Second derivative of the spectra are available in Fig. SM1. From the visual inspection of the average spectra, there are differences among the 3 classes, being the ratio of the amide I (1644 cm−1) to amide II (1545 cm−1) larger for the pre and post 11 samples. This can be indicative of changes in the protean composition produced immediately after exercise.
 | ||
| Fig. 2 Mean (lines) and interquartile range (shade) of the IR spectra range 1800–890 cm−1 of the urinary protein extracts dry-films acquired with single-reflection PerkinElmer ATR accessory for the pre (blue) post (red) and post 11 (green) samples. | ||
3.2. Urine dry film chemometric analysis
The PCA built for the centrifuged urine dry films did not indicate clear clustering for control and experimental groups, suggesting that the exercise was not a primary source of variation (see ESI, Fig. SM3a†). Notably, even though PC5 captured only 4.85% of the variance, a discernible trend emerges when examining the relationship between the score values of PC5 and the sampling times (see ESI, Fig. SM3b†). This temporal trend, despite its limited contribution to the variance, could be indicative of a dynamic process, a gradual shift, or an evolving state within the study.To better study the relationship between the spectra and the recovery time, we conducted an o-PLS regression with the same dataset employing the time points as response variables in Y vector. The t = 0 was assigned to the sample spectra immediately obtained after finishing the training, and because of that, this time was absent for the control group. It was also assumed that samples obtained before the physical activity are like those obtained the next day after full rest and thus, t = −1 hour was assigned to samples obtained before (pre) and 24 hour after the training.
For the training group, the o-PLS was built with 10 LVs, selected according to the RMSECV (see ESI, Fig. SM4†). The model suggested a modest quantification performance, with an apparently linear relationship between the CV-predicted time and the measured time (see Fig. 3(a)). There is a lot of variation among the samples for the same timepoints, probably caused by the strong biological and nutritional differences among the different volunteers. The RMSECV value obtained was 3.22 hours, suggesting that despite the presence of other confounding factors, a relationship between the IR variables and the recovery time could be observed.
 | ||
| Fig. 3 PLS model of the urine dry film extracts for the experimental samples (a) and the controls (b), and permutation test result of 500 permutations for the experimental samples (c) and the controls (d). | ||
To ensure that the obtained relationship was statistically significant, a permutation test was performed. The real error obtained with the actual timepoints was compared to the null distribution of errors obtained from 500 models constructed with the permuted timepoints. A p-value of 0.005 for the rand t-test was obtained for the cross-validation prediction of the experimental model, meaning that it is significant at the 95% confidence level. As it can see in Fig. 3(c), when representing the results of the permutation test for the experimental samples, the unpermuted results are significantly away from the bulk of the corresponding permuted results when looking at the standardized fractional sum squared Y captured (SSQ Y), which are relatively close to each other meaning that the model is robust.
On the other hand, in the o-PLS analysis of the control samples data, the optimum number of latent variables (LVs) was 2, because as employing additional latent variables noticeably increased the RMSECV. In this case, the predictive performance was significantly inferior to that of the training group, reflected in an RMSECV value of 4.08 hours (R2 = 0.123). Fig. 3(b) clearly shows the lack of linearity between the CV predicted time and the real time for the control group. A permutation test constructed in the same conditions as those employed for the training group also revealed that the model obtained was not statistically significant from the permuted classes (p-value = 0.283). As it can be seen in Fig. 3(d), the bulk is split away and closer to the unpermuted data indicating a likely overfit of the model.
In summary, the lack of a significant prediction in the control group suggested that, without training, the urine spectra remained unaffected by the passage of time over a 1 day cycle. In contrast, the spectra from the training group held significant information related to the recovery time, enabling its prediction from the IR variables. Values of overall fatigue for the experimental group were 0.9 ± 1.5 mm at pre and 2 ± 2 mm at post 24 (p = 0.071 between moments), and for the control group were 1.6 ± 3.0 mm at pre and 1.2 ± 2.2 mm at post 24 (p = 0.638). Therefore, these results are in agreement with the idea that a minimum fatigue was observed 1 day after exercise. To identify the spectral features responsible for the quantification, the loading of the LV1 vector of the o-PLS was inspected. The first o-PLS loading (i.e., ‘component loading’) captures the covariance with Y (i.e., sample time) being orthogonal to any confounding variation.37Fig. 4 shows local maxima and minima at different regions related to the main constituents of urine reported elsewhere such as urea, creatinine, uric acid, sulfate, and phosphate25 (For the second derivative of the spectra check the Fig. SM5†). This includes the strong bands located at the 1700–1600 cm−1 region, that can be assigned to urea and creatinine (see Fig. 1). Most interestingly, other spectral markers or band shifts cannot be assigned to typical bands from major constituents. For example, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching located at 1710–1740 cm−1 can be associated with urinary ketones or metabolites related to glycolysis (e.g., lactate, pyruvate) and tricarboxylic acid cycle (e.g., cis-aconitate, malate). In addition, the band at 1735 cm−1 may be associated with the increase of specific carbonyl groups in the post urine due to the oxidative stress.38 Also, the C–H bending vibrations 1420 cm−1 and 1000 cm−1 are also consistent with the reported changes on the purines (e.g., hypoxanthine) or amino acids like alanine. Changes in the concentration of these compounds have already been linked to intense exercise in recent metabolomics studies.39 The bands at low wavenumbers between 617 cm−1 and 551 cm−1 were tentatively assigned to the X–H wag of hydrogen bonded water, amines, amides, and alcohols. Since hydrogen bonding affects these bands substantially, they are difficult to be interpreted and associated with certain molecules.40 Interestingly, these bands show a high value for the component loading but a low value for the regression vector (see ESI, Fig. SM6†). This means that, despite having covariance with the study time, these bands are not as important for the prediction as the other marker bands.
O stretching located at 1710–1740 cm−1 can be associated with urinary ketones or metabolites related to glycolysis (e.g., lactate, pyruvate) and tricarboxylic acid cycle (e.g., cis-aconitate, malate). In addition, the band at 1735 cm−1 may be associated with the increase of specific carbonyl groups in the post urine due to the oxidative stress.38 Also, the C–H bending vibrations 1420 cm−1 and 1000 cm−1 are also consistent with the reported changes on the purines (e.g., hypoxanthine) or amino acids like alanine. Changes in the concentration of these compounds have already been linked to intense exercise in recent metabolomics studies.39 The bands at low wavenumbers between 617 cm−1 and 551 cm−1 were tentatively assigned to the X–H wag of hydrogen bonded water, amines, amides, and alcohols. Since hydrogen bonding affects these bands substantially, they are difficult to be interpreted and associated with certain molecules.40 Interestingly, these bands show a high value for the component loading but a low value for the regression vector (see ESI, Fig. SM6†). This means that, despite having covariance with the study time, these bands are not as important for the prediction as the other marker bands.
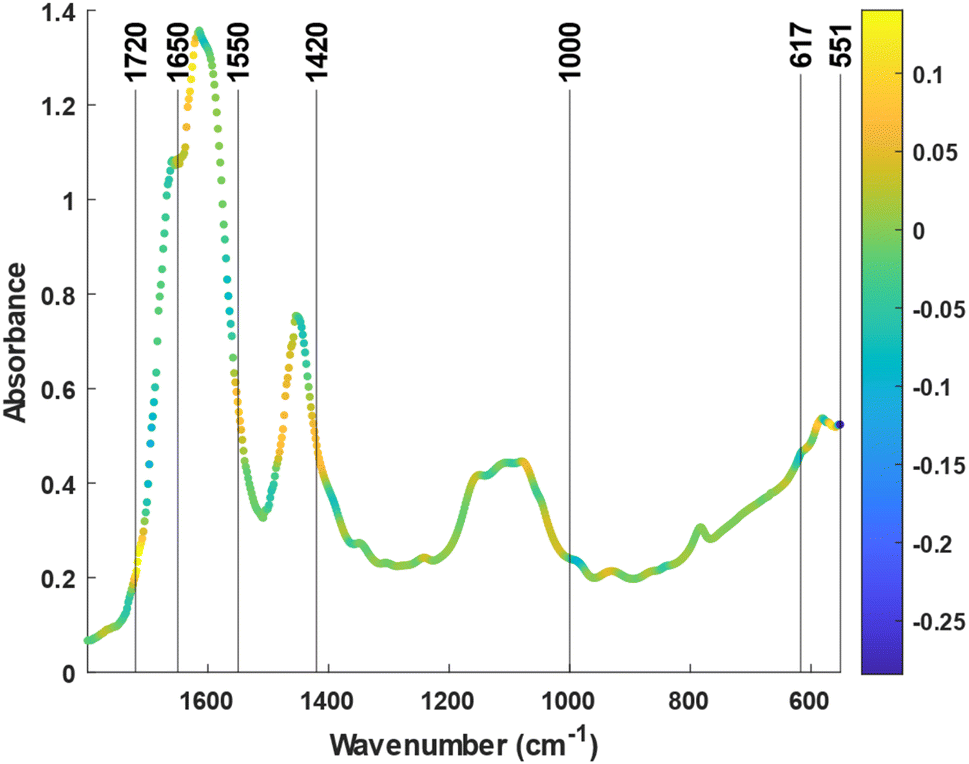 | ||
| Fig. 4 Average spectrum of the dry films of urine. The color scale of the points indicates the values of the “component loading” (LV1 in the o-PLS). | ||
3.3. Protein extraction dry-film data analysis
A PCA was built with the dry film spectra of the protein fractions of urine. Fig. 5(a) shows the scores from the PC1, that explained a significant proportion of the variance (29.6%). The boxplot shows that the values of the scores for the samples obtained immediately after training (“post”) are significantly lower than those for the samples obtained before training (p < 0.001, two-sided Wilcoxon rank sum test) and after 11 hours of rest (p < 0.01). Moreover, samples obtained before training and after resting 11 hours showed a more similar distribution of score values, and two-sided Wilcoxon rank sum test indicated that there were not statistically significant differences (p > 0.39). This suggests that contrary to the results obtained with the raw urine, where the changes gradually augmented until the 11 hours mark and then returned to normal precedent values, the changes in the protein profile in urine extract are found just after the intense exercise is done. This fact has been previously observed in previous works that studied the proteome of urine before and immediately after physical activity, which evidenced changes in the protein profile as well as an increase in protein content in urine after training.41 | ||
| Fig. 5 (a) Boxplots indicating the distribution of the PC1 scores for the 3 classes considered in the analysis of protein extracts: pre (before training), post (immediately after training) and post 11 (11 hours after training) and the p-values resulting of the Wilcoxon rank sum test. (b) Average spectrum of the protean extracts. The color scale of the points represents the values of the PC1 loading. | ||
Loadings associated with PC1 were examined to identify spectral makers indicative of compositional changes in urinary proteins after training. Fig. 5(b) shows the average spectrum of the protein extracts, with the points coloured as a function of the PC1 loading 1. Second derivatives of the spectra with the same colouring are available at the ESI (see Fig. SM7†). Taking into account that a 2nd derivative was included in the preprocessing and that the samples of the “post” class showed negative values of the PC score, bands with positive values in the PC1 loading should be more intense in samples obtained immediately after the training. The two high intense values of the loading at 1657 cm−1 and 1584 cm−1 are assigned to less presence of alpha-helical or beta-sheet structures in the proteins on the pre-urine. The two high intense values of the loading at 1540 cm−1 and 1650 cm−1 are assigned to the amide II and I bands, respectively, and show an increase of the protein content in urines obtained immediately after training. Furthermore, the amide I band shows asymmetric values of the PC1 loading, being larger than those located on the left side of the band (1650–1670 cm−1) and those located on the right side (1650–1630 cm−1). This is indicative of changes in the secondary structure of urine proteins in the “post” samples (e.g. increasing of proteins with a higher proportion of alpha-helical structures42) caused by changes in the proteome induced by the exercise. This finding is in agreement with previous proteomic MS studies, where the presence of collagen alpha-1 chains showed a positive correlation with the mean heart rate, indicator of physical condition of the participants, during high intensity simulated firefighting tasks, highlighting its potential significance in monitoring protein changes associated to intense exercise.41
The results of this work have important practical applications. In the study of recovery after exercise, some blood markers are used, such as serum creatine kinase levels, as well as physical tests (for example the countermovement jump) and pain/fatigue perception scales.43–45 The problem with all of these measurements is that some of them are invasive (some people are apprehensive about drawing blood by puncture, even if it is minimal), economically costly or with high interindividual variability.44,46 Moreover, physical tests have also the associated inconvenience of performing a motor action in moments of fatigue or muscle damage. The ATR-FTIR method has the benefits that is easy to perform and low-cost, and it can interfere to a lesser extent with the athlete's routine and recovery process. Another advantage of the proposed technique is that the IR spectrum also contains information about the hydration levels (e.g. concentration of urinary creatine). Nevertheless, it must be noted that in our study design it was not monitored the volunteer's hydration level by reference methods limiting the acquisition of information about changes in hydration states of participants.
4. Conclusions
In this study, we have evidenced the utility of dry film ATR-FTIR as a point-of-care tool for assessing physical stress following intense exercise using urine samples and urine protein extracts. The employed approach based on o-PLS modelling enabled the detection of spectral features that predicted the recovery time elapsed since the physical activity was conducted. These results indicate that the spectra detected a linear change in urine composition over the 11 hours following the exercise, with the baseline recovering after 24 hours of rest. Thus, ATR-FTIR analysis of urine was able to follow up the rest phase, with spectral markers that included typical bands of urine such as urea and creatinine as well as minor components.In contrast, the spectra of the protein fractions showed intense differences between the samples obtained before and immediately after the exercise, thus evidencing that the extract of proteins was able to capture immediate markers of the effort. Compared to the dry films of untreated urine, these changes were more evident, as they were featured in the first PC of a PCA. An analysis of the loadings showed changes in the protein composition as well as an increase in protein concentration in urine obtained just after the exercise, which was in good agreement with previous studies. In summary, ATR-FTIR shows substantial potential for becoming an innovative means of monitoring training progress in the exercise context, but future investigations utilizing metabolomic and proteomic techniques are essential to validate the band assignments.
Data availability
Data for this article, including IR raw data and MATLAB scripts are available at Zenodo at https://zenodo.org/doi/10.5281/zenodo.8355736.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We would like to thank the participants for their voluntary participation in this study. J. B. acknowledges financial support from the INVESTIGO program (INVEST/2022/95) funded by the Next Generation EU. Á. S. I. acknowledges the support of Margarita Salas grant (UP2021-044-MS21-084) from the Ministry of Universities of the Government of Spain, financed by the European Union, NextGeneration EU and grant JDC2022-049354-I funded by MCIN/AEI/10.13039/501100011033 by the “European Union NextGenerationEU/PRTR”. D. P-G. acknowledges the financial support from the projects RYC2019-026556-I and RPID2020-119326RA-I00 funded by MCIN/AEI/10.13039/501100011033. J. P. Q acknowledges the “Grupos Emergentes” funding by Conselleria de Innovación, Universidades, Ciencia y Sociedad Digital de la Generalitat Valenciana (GV/2020/050).References
- L. Djaoui, M. Haddad, K. Chamari and A. Dellal, Physiol. Behav., 2017, 181, 86–94 CrossRef CAS PubMed.
- S. L. Hooper, L. T. Mackinnon, A. Howard, R. D. Gordon and A. W. Bachmann, Med. Sci. Sports Exercise, 1995, 27, 106–112 CrossRef CAS.
- K.-A. Shin, K. D. Park, J. Ahn, Y. Park and Y.-J. Kim, Medicine, 2016, 95, e3657 CrossRef CAS PubMed.
- G. Iaccarino and B. Trimarco, BMC Med., 2019, 17, 166 CrossRef PubMed.
- F. Girolami, G. Frisso, M. Benelli, L. Crotti, M. Iascone, R. Mango, C. Mazzaccara, K. Pilichou, E. Arbustini, B. Tomberli, G. Limongelli, C. Basso and I. Olivotto, J. Cardiovasc. Med., 2018, 19, 1–11 CrossRef PubMed.
- L. A. Conlay, R. J. Wurtman, I. L. G. Coviella, J. K. Blusztajn, C. A. Vacanti, M. Logue, M. During, B. Caballero, T. J. Maher and G. Evoniuk, J. Neural Transm., 1989, 76, 65–71 CrossRef CAS PubMed.
- J. Ollesch, M. Heinze, H. M. Heise, T. Behrens, T. Brüning and K. Gerwert, J. Biophotonics, 2014, 7, 210–221 CrossRef CAS PubMed.
- E. Papacosta and G. P. Nassis, J. Sci. Med. Sport, 2011, 14, 424–434 CrossRef PubMed.
- J. M. Yoshizawa, C. A. Schafer, J. J. Schafer, J. J. Farrell, B. J. Paster and D. T. W. Wong, Clin. Microbiol. Rev., 2013, 26, 781–791 CrossRef CAS PubMed.
- T. G. Mundie and B. Hare, Biol. Trace Elem. Res., 2001, 79, 23–28 CrossRef CAS PubMed.
- T. Pisitkun, R. Johnstone and M. A. Knepper, Mol. Cell. Proteomics, 2006, 5, 1760–1771 CrossRef CAS PubMed.
- T. Anderson, A. R. Lane and A. C. Hackney, Eur. J. Appl. Physiol., 2016, 116, 1503–1509 CrossRef CAS PubMed.
- P. Brancaccio, N. Maffulli and F. M. Limongelli, Br. Med. Bull., 2007, 81–82, 209–230 CrossRef PubMed.
- S. K. Powers and M. J. Jackson, Physiol. Rev., 2008, 88, 1243–1276 CrossRef CAS PubMed.
- J. I. Priego-Quesada, A. Pérez-Guarner, A. Gandia-Soriano, F. Oficial-Casado, C. Galindo, R. M. C. O. de Anda, J. D. Piñeiro-Ramos, Á. Sánchez-Illana, J. Kuligowski, M. A. G. Barbosa, M. Vento and R. S. Palmer, Int. J. Sports Physiol. Perform., 2020, 15, 1467–1475 Search PubMed.
- T. Anderson and L. Wideman, Sports Med., 2017, 3, 37 Search PubMed.
- R. Pero, M. Brancaccio, C. Mennitti, L. Gentile, S. Arpino, R. De Falco, E. Leggiero, A. Ranieri, C. Pagliuca, R. Colicchio, P. Salvatore, G. D'Alicandro, G. Frisso, B. Lombardo, C. Mazzaccara, R. Faraonio and O. Scudiero, Int. J. Environ. Res. Public Health, 2020, 17, 6065 CrossRef CAS PubMed.
- K. Khoramipour, Ø. Sandbakk, A. H. Keshteli, A. A. Gaeini, D. S. Wishart and K. Chamari, Sports Med., 2022, 52, 547–583 CrossRef PubMed.
- V. Balan, C.-T. Mihai, F.-D. Cojocaru, C.-M. Uritu, G. Dodi, D. Botezat and I. Gardikiotis, Materials, 2019, 12, 2884 CrossRef CAS PubMed.
- D. Perez-Guaita, S. Garrigues and M. De La Guardia, TrAC, Trends Anal. Chem., 2014, 62, 93–105 CrossRef CAS.
- E. L. Callery and A. W. Rowbottom, Appl. Spectrosc. Rev., 2022, 57, 411–440 CrossRef CAS.
- S. Roy, D. Perez-Guaita, D. W. Andrew, J. S. Richards, D. McNaughton, P. Heraud and B. R. Wood, Anal. Chem., 2017, 89, 5238–5245 CrossRef CAS PubMed.
- P. Heraud, P. Chatchawal, M. Wongwattanakul, P. Tippayawat, C. Doerig, P. Jearanaikoon, D. Perez-Guaita and B. R. Wood, Malar. J., 2019, 18, 348 CrossRef PubMed.
- D. Perez-Guaita, Z. Richardson, P. Heraud and B. Wood, Anal. Chem., 2020, 92, 2409–2416 CrossRef CAS PubMed.
- N. Sarigul, İ. Kurultak, A. U. Gökceoğlu and F. Korkmaz, J. Biophotonics, 2021, 14, e202100009 CrossRef CAS PubMed.
- Z. Richardson, A. Kincses, E. Ekinci, D. Perez-Guaita, K. Jandeleit-Dahm and B. R. Wood, Analysis Sensing, 2023, 3, e202200094 CrossRef CAS.
- M. J. Baker, J. Trevisan, P. Bassan, R. Bhargava, H. J. Butler, K. M. Dorling, P. R. Fielden, S. W. Fogarty, N. J. Fullwood, K. A. Heys, C. Hughes, P. Lasch, P. L. Martin-Hirsch, B. Obinaju, G. D. Sockalingum, J. Sulé-Suso, R. J. Strong, M. J. Walsh, B. R. Wood, P. Gardner and F. L. Martin, Nat. Protoc., 2014, 9, 1771–1791 CrossRef CAS PubMed.
- D. Pérez-Guaita, Á. Sánchez-Illana, S. Garrigues and M. de la Guardia, Microchem. J., 2015, 121, 178–183 CrossRef.
- D. Pérez-Guaita, J. Kuligowski, S. Garrigues, G. Quintás and B. R. Wood, Analyst, 2015, 140, 2422–2427 RSC.
- J. Béjar-Grimalt, Á. Sánchez Illana, M. de la Guardia, S. Garrigues, I. Catalá-Vilaplana, J. L. Bermejo Ruiz, J. I. Priego-Quesada and D. Pérez-Guaita, Zenodo, 2023, DOI:10.5281/zenodo.8355736.
- J. Grdadolnik and Y. Maréchal, J. Mol. Struct., 2002, 615, 177–189 CrossRef CAS.
- K. V. Oliver, A. Maréchal and P. R. Rich, Appl. Spectrosc., 2016, 70, 983–994 CrossRef CAS PubMed.
- N. Sarigul, F. Korkmaz and İ. Kurultak, Sci. Rep., 2019, 9, 20159 CrossRef CAS PubMed.
- S. Aitekenov, A. Gaipov and R. Bukasov, Talanta, 2021, 223, 121718 CrossRef CAS PubMed.
- Á. I. López-Lorente and B. Mizaikoff, Anal. Bioanal. Chem., 2016, 408, 2875–2889 CrossRef PubMed.
- F. Serafini-Cessi, N. Malagolini and D. Cavallone, Am. J. Kidney Dis., 2003, 42, 658–676 CrossRef CAS PubMed.
- H. Stenlund, E. Johansson, J. Gottfries and J. Trygg, Anal. Chem., 2009, 81, 203–209 CrossRef CAS PubMed.
- J. Bujok, M. Gąsior-Głogowska, M. Marszałek, N. Trochanowska-Pauk, F. Zigo, A. Pavľak, M. Komorowska and T. Walski, BioMed Res. Int., 2019, 2019, e2181370 Search PubMed.
- S. Kistner, C. I. Mack, M. J. Rist, R. Krüger, B. Egert, N. Biniaminov, A. K. Engelbert, S. Seifert, C. Dörr, P. G. Ferrario, R. Neumann, S. Altmann and A. Bub, Front. Physiol., 2023, 14, 1028643 CrossRef PubMed.
- P. J. Larkin, in Infrared and Raman Spectroscopy, ed. P. J. Larkin, Elsevier, 2nd edn, 2018, pp. 135–151 Search PubMed.
- T. Zhu, Y. Hu, J. Hwang, D. Zhao, L. Huang, L. Qiao, A. Wei and X. Xu, Int. J. Environ. Res. Public Health, 2021, 18, 10618 CrossRef CAS PubMed.
- A. Terzi, E. Storelli, S. Bettini, T. Sibillano, D. Altamura, L. Salvatore, M. Madaghiele, A. Romano, D. Siliqi, M. Ladisa, L. De Caro, A. Quattrini, L. Valli, A. Sannino and C. Giannini, Sci. Rep., 2018, 8, 1–13 CAS.
- G. Banfi, A. Colombini, G. Lombardi and A. Lubkowska, Adv. Clin. Chem., 2012, 56, 1–54 CAS.
- S. L. Halson, Sports Med., 2014, 44, 139–147 CrossRef PubMed.
- A. E. Saw, L. C. Main and P. B. Gastin, Br. J. Sports Med., 2016, 50(5), 281–291 CrossRef PubMed.
- M. Clarkson, H. Priscilla and J. Monica, Am. J. Phys. Med., 2002, 81(11), S52–S69 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ay00913d |
| This journal is © The Royal Society of Chemistry 2024 |