DOI:
10.1039/D4AY00403E
(Paper)
Anal. Methods, 2024,
16, 2983-2996
Determining monolignol oxifunctionalization by direct infusion electrospray ionization tandem mass spectrometry†
Received
4th March 2024
, Accepted 22nd April 2024
First published on 8th May 2024
Abstract
We have successfully developed a validated high-throughput analysis method for the identification and quantification of native and oxifunctionalized monolignols using direct infusion electrospray ionization tandem mass spectrometry (DI-ESI-MS/MS). Oxifunctionalized monolignols generated through unspecific peroxygenase catalysis present a sustainable alternative to fossil aromatic hydrocarbons. This study emphasizes a sustainable analytical approach for these renewable biocatalytic precursors, addressing challenges such as matrix effects, accuracy, precision, and sensitivity of the method. Our findings demonstrate the potential of overcoming quantification difficulties using DI-ESI-MS. Notably, this analytical methodology represents a novel utilization of DI-ESI-MS/MS in examining monolignols and their functionalization, thereby advancing the exploration of lignin as a valuable and sustainable bioresource.
Introduction
Lignin is the most abundant aromatic polymer in nature, serving as an essential element of protection and structural support in plant cell walls. Accordingly, lignin holds the potential to serve as a sustainable substitute for fossil aromatic hydrocarbons. However, the utilization of this biopolymer is currently limited due to the high cost and energy requirement for lignin depolymerization as a consequence of lignin's resistance to decomposition.1 Sustainable lignin valorization relies on effective biorefinery of intricate mixtures of lignin-derived compounds, into valuable and reactive depolymerized lignin (Fig. 1). One example is reductive catalytic fractionating (RCF) of lignocellulose, a promising strategy in lignin-first biorefinery. RCF yields extensively depolymerized lignin and, depending on the conditions, nearly theoretical quantities of lignin monomers such as 4-propylphenol (4PP), 4-propylguaiacol (4PG), and 4-propylsyringol (4PS), here termed monolignols.2 The value of such lignin monomers is enhanced by oxifunctionalization, which involves the introduction of functional groups through, i.e., selective hydroxylation.3 Functional groups such as hydroxyl and carbonyl groups serve as versatile intermediates for target-oriented synthesis. Oxifunctionalized monolignols hence serve as alternative intermediates for a sustainable green industry (Fig. 2A). Targets for oxifunctionalization include the aromatic ring and the propyl chain of monolignols. An oxifunctionalized benzene ring is used to produce epoxy thermoplastics,4–9 agrochemicals,10 pharmaceuticals,10 and cosmetics.11 The oxidation of the monolignol propyl chain facilitates aromatic ring opening12 as well as ketone production. Such ketones find application in pharmaceuticals12 and cosmetics.13 An oxifunctionalized propyl chain is also attractive as an intermediate in industries such as pharmaceuticals14,15 and bio-based benzoxazine production.14,15 Achieving the targeted oxifunctionalization of organic compounds stands as a challenge within synthetic chemistry, where the utilization of chemical catalysts raises environmental concerns and substantial expenses. Moreover, especially for propyl chain hydroxylation, control over regioselectivity is a concern. To address these challenges and expand the scope of versatile intermediates, our attention shifted toward biotransformations using fungal unspecific peroxygenases (UPO, E.C. 1.11.2). UPOs (Fig. 2B) are biocatalysts that hold a potential for catalyzing the oxifunctionalization of the monolignol structure. A strong indication for this was given by the O-demethylation and cleavage of various non-phenolic lignin model compounds catalyzed by AaeUPO using H2O2.16 UPOs represent a rather young class of iron-dependent oxidative enzymes first described by Ullrich et al. in 2004.17 Similar to cytochrome P450 monooxygenases (P450s), these enzymes are capable of complex oxyfunctionalization chemistry catalyzing regio- and stereoselective oxidization of C–H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, and C–C bonds. However, in contrast to the complex cytochrome P450 system, UPOs are self-sufficient, require only H2O2 as a co-substrate, and are naturally secreted soluble proteins.18 Additionally, they are known for their outstanding substrate spectrum (over 400 different substrates have been tested) and ability to catalyze one-as well as two-electron transfers. This catalytic peculiarity suggests that they are related to both cytochrome P450 monooxygenases and chloroperoxidases and represent the “missing link” between these two classes of enzymes.19 Phylogenetically, there are two classes of UPOs, “short” and “long” family UPOs. They differ not only in their average molecular mass – around 29 kDa for short and 44 kDa for long type UPOs – but also their conserved protein motifs and catalytic residues, with -PCP-EHD-E- and a histidine or -PCP-EGD-R-E- and an arginine as the charge stabilizer in “short” and “long” UPOs, respectively.20 The broad substrate scope, catalytic versatility, and fungal origin of UPOs suggest involvement in various environmental detoxification reactions e.g. plant phytoalexins, microbial toxins, and xenobiotics, as well as in lignin and humus degradation. However, due to their structural characteristics – narrow substrate channel and internal active center – UPOs are presumed to be active on small lignin fragments rather than on the lignin polymer.18,21
C, and C–C bonds. However, in contrast to the complex cytochrome P450 system, UPOs are self-sufficient, require only H2O2 as a co-substrate, and are naturally secreted soluble proteins.18 Additionally, they are known for their outstanding substrate spectrum (over 400 different substrates have been tested) and ability to catalyze one-as well as two-electron transfers. This catalytic peculiarity suggests that they are related to both cytochrome P450 monooxygenases and chloroperoxidases and represent the “missing link” between these two classes of enzymes.19 Phylogenetically, there are two classes of UPOs, “short” and “long” family UPOs. They differ not only in their average molecular mass – around 29 kDa for short and 44 kDa for long type UPOs – but also their conserved protein motifs and catalytic residues, with -PCP-EHD-E- and a histidine or -PCP-EGD-R-E- and an arginine as the charge stabilizer in “short” and “long” UPOs, respectively.20 The broad substrate scope, catalytic versatility, and fungal origin of UPOs suggest involvement in various environmental detoxification reactions e.g. plant phytoalexins, microbial toxins, and xenobiotics, as well as in lignin and humus degradation. However, due to their structural characteristics – narrow substrate channel and internal active center – UPOs are presumed to be active on small lignin fragments rather than on the lignin polymer.18,21
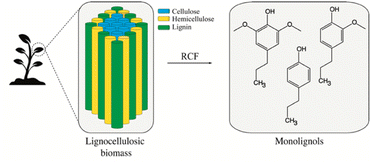 |
| | Fig. 1 Schematic presentation of monolignol production. Lignin is obtained from lignocellulosic biomass and undergoes reductive catalytic fractionation (RCF) as part of the processing. This process generates monolignols. | |
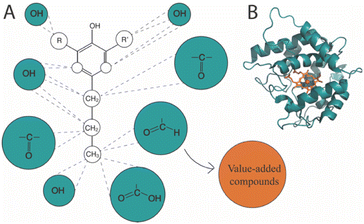 |
| | Fig. 2 Oxifunctionalization of the monolignol propyl chain. (A) Oxifunctionalization of the monolignol structure produces versatile intermediates suitable for generating value-added compounds. Proposed precursor monolignols are 4-propylphenol (R = R′ = H), 4-propylguaiacol (R = H, R′ = –OCH3), and 4-propylsyringol (R = R′ = –OCH3). (B) Unspecific peroxygenases have demonstrated catalytic activity towards aromatic substrates. We propose leveraging this property for the monolignol activation in the context of the proposed DI-ESI-MS/MS method. The presented crystalline structure is HspUPO (PDB: 7O2G). | |
We consider an optimized enzymatically catalyzed oxifunctionalization of the monolignols to be a specific, possibly cost-effective, and environmentally favorable process for monolignol functionalization and would represent a breakthrough in lignin valorization. To achieve this, it is crucial to establish a viable analytical method for efficient, comprehensive, and sustainable screening concerning the nature and quantity of the derived products. Commonly used screening techniques include colorimetric and spectrophotometric assays, and more specific and comprehensive analytics such as high-performance liquid chromatography (HPLC) and gas chromatography (GC) in combination with mass spectrometry (MS). Both analytics come with their own set of advantages and disadvantages. Colorimetric and spectrophotometric assays are regarded as straightforward and fast. Still, information on the regioselectivity of the oxifunctionalization is often elusive. The use of HPLC-MS and GC-MS provides qualitative and quantitative information about the products, even for beyond-model substrates. GC-MS has been recognized as the golden standard for structure elucidation. The downside is the time consumed for analysis, specialized equipment, and extensive sample preparation. However, the use of liquid tandem mass spectrometry (LC-MS/MS) has increased as an analytical tool as this technique requires less analytical time and sample preparation compared with conventional GC-MS. LC-MS/MS holds the potential for structure elucidation of a broad range of compounds especially due to the soft ionization techniques such as atmospheric pressure ionization mass spectrometry (API-MS) and electrospray ionization mass spectrometry (ESI-MS). Examples using LC-MS/MS include the structure elucidation of lignin oligomers.22 ESI-MS exhibits selectivity towards analytes that are either acidic or basic making this an interesting ionization technique for the weak acidic phenols found in the monolignols. ESI has been utilized for the characterization of analogous molecules; e.g. model compounds of processed lignin described by Haupert et al.23 However, so far ESI has not been a traditional choice of ionization for the structural elucidation of lignin monomers. Lignin monomers are traditionally ionized by electron ionization (EI) or chemical ionization (CI), where the lignin monomers are identified based on pyrolysis- or reductive cleavage GC.24,25
This paper introduces a method that aims to strike a balance between the straightforwardness of a plate assay and the precision and diversity of the time-consuming chromatography. We present a flow injection analysis (FIA) method designed for high-throughput analysis of both native and oxifunctionalized monolignols using direct infusion electrospray ionization tandem mass spectrometry (DI-ESI-MS/MS), offering advantages such as minimal reagent usage, straightforward sample preparation, and a rapid analytical time without compromising the analytical accuracy. Our focus is on the validation and application of DI-ESI analytics for the identification and quantification of 4PP, 4PG, 4PS, and various oxifunctionalized monolignols. Moreover, we demonstrate the utility of DI-ESI-MS for qualitative and quantitative analyses of oxifunctionalized monolignols using two separate UPO-based catalysts, one “short” (HspUPO) and one “long” (CmaUPO-I). To our knowledge, this analytical method marks the first instance of the identification and quantification of monolignols using DI-ESI-MS/MS.
Results and discussion
Identification of native and oxifunctionalized monolignols
Full scan DI-ESI-MS.
Oxifunctionalization of monolignols may result in a plethora of products. For this reason, incorporating a full scan into our analytical approach for monolignol identification was essential. The full scan mass spectra in Fig. 3 provided a comprehensive overview of the standard solutions by scanning a wide range of mass-to-charge (m/z) ratios showing deprotonated molecular ions, [M − H]−, within a brief run time of 24 seconds. DI-ESI-MS/MS was used to validate the monolignols present in full scan mode. Consistently detected anions were observed at m/z 167, 197, and 227 for the monolignols derived from 4PP (Fig. 3E), 4PG (Fig. 3H), and 4PS (Fig. 3K), respectively. We posited that these peaks represented water adducts formed by the monolingol aldehydes. Our hypothesis was confirmed through isotopic labeling experiments using heavy water, D2O (Fig. S1†). The aldehyde-water adduct ion may be used as a diagnostic indicator for Cγ oxidation of the monolignol propyl chain, as isomeric compounds generated through Cα and Cβ oxidation do not produce these adduct ions (Fig. 3C, D, F, G, I and J). Full scan mode also provided insight into how the presence of the individual analytes impacted the ionization of all the analytes. The relative signal intensity derived from the deprotonated molecular ions observed during full scan mode differed based on the type of compound. Monolignols with a carboxylic acid functional group generally exhibited higher ion intensity compared with monolignols with a hydroxyl or carbonyl functional group. This observation can be attributed to the relatively low pKa values of the carboxylic acid functional groups compared to the phenolic functional groups. Compounds with low pKa values have a low energy barrier for proton loss in a basic environment, rendering these compounds readily ionizable. Moreover, compounds with high acidity yield increased ion currents, potentially causing ion suppression. Removing carboxylic acid functionalized monolignols from the standard solutions had a minimal impact on the ion current (Fig. S2†). The same trend was observed for the standard solution comprising the Cα and Cβ oxidized monolignols, which were devoid of any propanoic acids (Fig. 3C, D, F, G, I and J). These provided support for the absence of ion suppression.
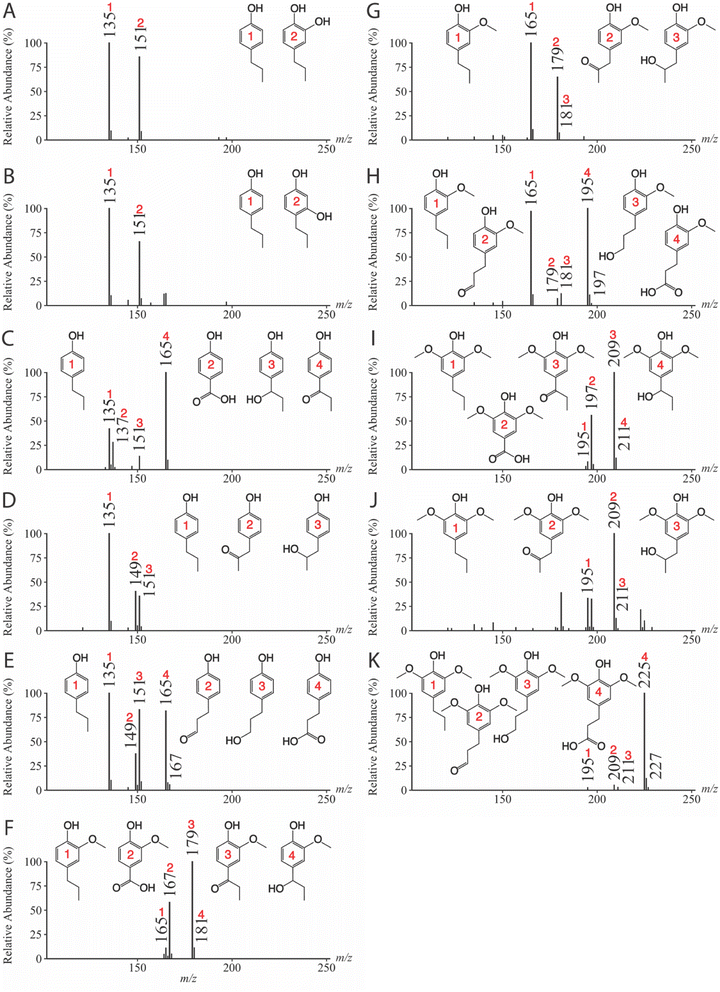 |
| | Fig. 3 Full scan mass spectra. Monolignol standard solutions were subjected to negative mode ESI, generating [M − H]− ions detected in the full scan between m/z 70–2000. The relative [M − H]− abundance (%) is presented. Ions generating a 2% or lower signal relative to the base peak are not shown. NL refers to the base peak abundance. (A) 4PP and one ring-oxidized monolignol. NL: 1.88 × 106. (B) 4PP and one ring-oxidized monolignol. NL: 7.41 × 105. (C) 4PP and Cα oxidized monolignols. NL: 3.20 × 106. (D) 4PP and Cβ oxidized monolignols. NL: 1.67 × 106. (E) 4PP and Cγ oxidized monolignols. NL: 1.33 × 106. (F) 4PG and Cα oxidized monolignols. NL: 3.12 × 106. (G) 4PG and Cβ oxidized monolignols. NL: 1.11 × 104. (H) 4PG and Cγ oxidized monolignols. NL: 1.34 × 106. (I) 4PS and Cα oxidized monolignols. NL: 3.67 × 106. (J) 4PS and Cβ oxidized monolignols. NL: 5.86 × 105. (K) 4PS and Cγ oxidized monolignols. NL: 3.92 × 106. | |
DI-ESI-MS/MS.
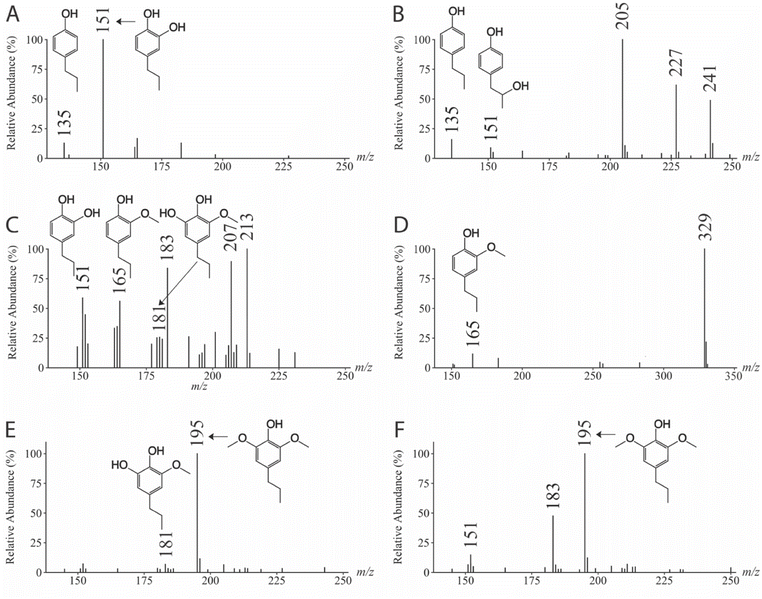
The utilization of tandem mass spectrometry in our analytical approach for monolignol identification, with a run time of 24 seconds, contributed to enhanced information on the specificity of the oxifuntionalization and provided valuable structural information. We would like to highlight that prior MS/MS analyses have been conducted on analogous compounds, including some compounds characterized in this study, i.e., demonstrated by Owen et al.26 and Mattonai et al.27 Anions generated through ESI resulted from monolignol deprotonation. Deprotonation is hypothesized to occur at various sites within the molecule, creating anions with the charge located at different atoms. Both the hydroxyl group in the propyl chain and the phenol hydroxyl group were prone to deprotonation, where removal of the hydrogen from the phenol group induced resonance stabilization of the resulting anion. Utilizing the monolignol collision-induced dissociation (CID) pattern enabled the deduction of the protons lost during ionization, as deprotonation centers the energy within the compound, essential for subsequent fragmentation. The proposed fragmentation mechanisms are illustrated for 4PP and the corresponding oxifunctionalized products (Fig. 4). 4PG and 4PS with the respective oxifunctionalized products exhibited similar fragmentation patterns (Fig. S3†). We propose deprotonation of the hydroxyl group of 4PP producing the deprotonated molecular ions with m/z 135 (Fig. 4A). The negative charge is most likely delocalized through resonance stabilization. CID of the 4PP ions led to the loss of C2H4, yielding a product ion found at m/z 106. The deprotonated molecular ions of 4-propylbenzene-1,2-diol at m/z 151 (Fig. 4B) are proposed to undergo a similar fragmentation pathway, where the base peak (m/z 122) corresponds to the loss of C2H4 from the deprotonated molecular ions. In contrast, we suggest deprotonation of the Cα, Cβ or Cγ position for the 3-(4-hydroxyphenyl)propanal (Fig. 4C). CID of m/z 149 yielded the base peak at m/z 107, corresponding to the loss of the radical C2H3O˙ likely due to homolytic bond cleavage. The deprotonated molecular ions of 4-hydroxybenzoic acid (Fig. 4D) at m/z 137 and 3-(4-hydroxyphenyl) propanoic acid (Fig. 4E) at m/z 165 had their charge centered on the carbonyl groups, leading to CID-induced decarboxylation. The neutral losses yielded radical anions at m/z 93 and 121 for 4-hydroxybenzoic acid and 3-(4-hydroxyphenyl) propanoic acid respectively. We propose that the radical anion at m/z 121 could fragment producing an anion at m/z 93. For both 4-(1-hydroxypropyl) phenol (Fig. 4F) and 4-(2 hydroxypropyl) phenol (Fig. 4G), we posit a resonance scenario between two deprotonated molecular ions, wherein the negative charge was stabilized through resonance between the oxygen in the propyl chain and the phenol. This assertion was substantiated by the numerous fragments produced through potential rearrangement reactions. Upon CID of these alcohols, we observed dehydration resulting from 1,2-elimination of water, giving rise to the base peak for each compound (m/z 133) that signified their structural analogs. These ions denoted extensive aromatic systems exhibiting resonance stabilization, providing an explanation for their role as base peaks. CID of 4-(3-hydroxypropyl)phenol (Fig. 4H) yields an intriguing monolignol fingerprint, where α elimination of CH2O˙ from the deprotonated molecular ions resulted in an anion (m/z 121) hypothesized to exist in equilibrium with the unstable tropylium anion. Fragmentation of the tropylium anion was suspected to generate the detected anion at m/z 95 and 106. 3-(4-Hydroxyphenyl)propane-1-one (Fig. 4I) and 3-(4-hydroxyphenyl)propane-2-one (Fig. 4J) were both hypothesized to experience α elimination of the propyl chain as a result of CID, leading to the formation of structural isomeric fragment ions at m/z 134. This fragment ion of 3-(4-hydroxyphenyl)propane-1-one was proposed to undergo another α elimination, yielding the base peak at m/z 121 where the compound demonstrates significant stability due to a high degree of resonance. Finally, we hypothesize that the energy of 4-propylbenzene-1,3-diol was located at the Cα (Fig. 4K), where CID-induced α elimination yielded the fragment ions at m/z 122. The monolignol fragmentation was not only helpful for locating the deprotonation of the molecule but also for identification purposes (Table 1). Most monolignol compounds fragmented differently by the experimentally determined optimal CE, enabling us to differentiate between structural isomers. However, certain monolignol isomers, such as 1-(4-hydroxy-3-methoxyphenyl)propane-1-one and 1-(4-hydroxy-3-methoxy phenyl)propane-2-one (product ion: m/z 164), or 1-(4-hydroxy-3,5-methoxyphenyl)propan-1-one and 1-(4-hydroxy-3,5-methoxyphenyl)propan-2-one (product ion: m/z 194), exhibited similar fragmentation patterns. Nevertheless, we may still distinguish between the production of such monolignol isomers during biotransformation by considering potential biotransformation in perspective. Suppose the monolignol propyl chain is suspect to oxifunctionalization. In that case, one such reaction occurs in multiple steps (Fig. 5). Initially, hydroxylation initiates the formation of the alcoholic monolignol, succeeded by either oxidizing Cα or Cβ to produce the corresponding ketones or oxidizing Cγ to yield the corresponding aldehyde, eventually resulting in the carboxylic acid. The presence of all reaction products combined may be used in the monolignol structure elucidation in addition to the fragmentation patterns.
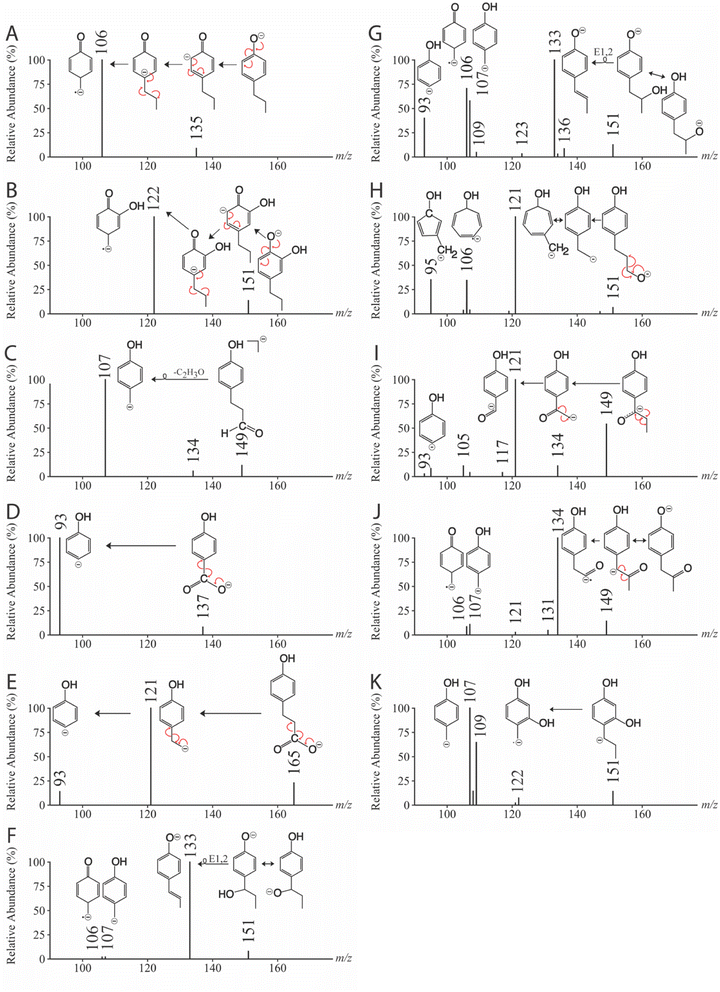 |
| | Fig. 4 MS/MS mass spectra. The proposed reaction mechanisms occurring during CID of the [M − H]− ions. The product ions were detected in the range of m/z 70–2000. Product ions generating a signal of 2% or lower relative to the base peak are not shown. NL denotes the magnitude of the base peak. (A) 4-Propylphenol [M − H]− 135 (NL = 1.12 × 105). (B) 4-Propylbenzene-1,2-diol [M − H]− 151 (NL = 1.30 × 105). (C) 3-(4-Hydroxyphenyl)propanal [M − H]− 149 (NL = 2.14 × 104). (D) 4-Hydroxybenzoic acid [M − H]− 137 (NL = 6.05 × 105). (E) 3-(4-Hydroxyphenyl) propanoic acid [M − H]− 165 (NL = 2.59 × 106). (F) 4-(1-Hydroxypropyl)phenol [M − H]− 151 (NL = 1.62 × 103). (G) 4-(2-Hydroxypropyl)phenol [M − H]− 151 (NL = 1.89 × 103). (H) 4-(3-Hydroxypropyl)phenol [M − H]− 151 (NL = 2.24 × 104). (I) 3-(4-Hydroxyphenyl) propan-1-one [M − H]− 149 (NL = 7.16 × 101). (J) 3-(4-Hydroxyphenyl)propan-2-one [M − H]− 149 (NL = 9.56 × 103). (K) 4-Propylbenzene-1,3-diol [M − H]− 151 (NL = 1.76 × 105). | |
Table 1 Monolignol fragmentation pattern. The monolignols were analyzed at the optimal collision energy (CE, eV) for the generation of a precursor ion with 10% intensity compared to the base peak. CEs were determined experimentally and used to generate the fragmentation fragments consistent across multiple replicates. Product ions generating a signal of 2% or lower relative to the base peak are not listed. Representative examples of fragmentation patterns can be found in the corresponding reference figures
| Monolignol |
CE (eV) |
Precursor ion (m/z) |
Product ions (m/z) |
Reference |
| 4-Propylphenol |
34 |
135 |
106 |
Fig. 4A
|
| 4-Propylbenzene-1,2-diol |
34 |
151 |
122 |
Fig. 4B
|
| 3-(4-Hydroxyphenyl)propanal |
27 |
149 |
107 |
Fig. 4C
|
| 4-Hydroxybenzoic acid |
28 |
137 |
93 |
Fig. 4D
|
| 3-(4-Hydroxyphenyl)propanoic acid |
26 |
165 |
93, 121 |
Fig. 4E
|
| 4-(1-Hydroxypropyl)phenol |
30 |
151 |
107, 122 |
Fig. 4F
|
| 4-(2-Hydroxypropyl)phenol |
36 |
151 |
107, 133 |
Fig. 4G
|
| 4-(3-Hydroxypropyl)phenol |
34 |
151 |
95, 106, 121 |
Fig. 4H
|
| 3-(4-Hydroxyphenyl)propane-1-one |
27 |
149 |
93, 95, 105, 107, 117, 121, 134 |
Fig. 4I
|
| 3-(4-Hydroxyphenyl)propane-2-one |
31 |
149 |
106, 107, 121, 131, 134 |
Fig. 4J
|
| 4-Propylbenzene-1,3-diol |
34 |
151 |
107, 108, 109, 121, 122 |
Fig. 4K
|
| 4-Propylguaiacol |
26 |
165 |
150 |
Fig. S3A |
| 1-(4-Hydroxy-3-methoxyphenyl)propane-1-one |
24 |
179 |
164 |
Fig. S3B |
| 4-Hydroxy-3-methoxybenzoic acid |
24 |
167 |
123, 152 |
Fig. S3C |
| 1-(4-Hydroxy-3-methoxyphenyl)propane-2-one |
25 |
179 |
164 |
Fig. S3D |
| 4-(3-Hydroxypropyl)-2-methoxyphenol |
24 |
181 |
166 |
Fig. S3E |
| 3-(4-Hydroxy-3-methoxyphenyl)propanal |
27 |
179 |
137, 164 |
Fig. S3F |
| 3-(4-Hydroxy-3-methoxyphenyl)propanoic acid |
23 |
195 |
119, 123, 136, 151, 177, 180 |
Fig. S3G |
| 4-Propylsyringol |
24 |
195 |
180 |
Fig. S3H |
| 1-(4-Hydroxy-3,5-methoxyphenyl)propane-1-one |
23 |
209 |
194 |
Fig. S3I |
| 4-Hydroxy-3,5-dimethoxybenzoic acid |
22 |
197 |
120, 153, 182 |
Fig. S3J |
| 1-(4-Hydroxy-3,5-methoxyphenyl)propane-2-one |
24 |
209 |
194 |
Fig. S3K |
| 4-(3-Hydroxypropyl)-2,6-dimethoxyphenol |
22 |
211 |
196 |
Fig. S3L |
| 3-(4-Hydroxy-3,5-dimethoxyphenyl)propanal |
25 |
209 |
167, 194 |
Fig. S3M |
| 3-(4-Hydroxy-3,5-dimethoxyphenyl propanoic acid) |
23 |
225 |
149, 153, 166, 181, 207, 210 |
Fig. S3N |
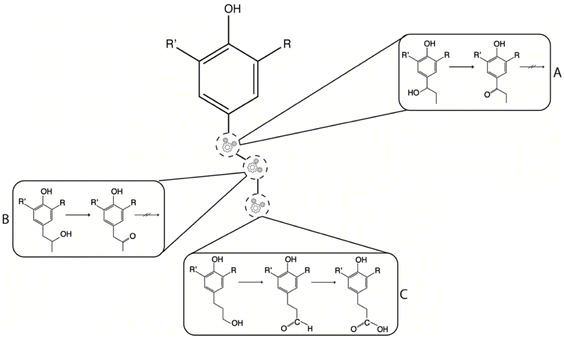 |
| | Fig. 5 Proposed reaction scheme for monolignol oxifunctionalization of the propyl chain. Detecting various oxifunctionalized monolignols can be employed to infer the specific carbon atom undergoing biotransformation. The oxifunctionalized monolignols are derived from 4-propylphenol (R = R′ = H), 4-propylguaiacol (R = H, R′ = –OCH3), and 4-propylsyringol (R = R′ = –OCH3). (A) Cα oxifunctionalization will potentially lead to a two-step oxidation with alcohol and ketone as functionalized products. The ketone is the final product. (B) An alcohol and ketone may be generated in a two-step oxidation of the Cβ, where the ketone is the end compound. (C) Cγ oxidation may yield an alcohol, aldehyde, and carboxylic acid through a multiple-step oxidation reaction if biotransformation occurs. | |
Quantification of native and oxidized monolignols
Selected ion monitoring (SIM) mode measured the intensity of the deprotonated molecular ions for quantification purposes within a run time of 24 seconds. This targeted approach is particularly advantageous for achieving high sensitivity and selectivity and minimizing interference from background noise in complex sample matrices.
Linear ranges.
DI-ESI-MS quantification is acknowledged to be challenging due to the matrix effects and ion suppression as mentioned above. Nevertheless, our findings strengthen our confidence in overcoming these difficulties. The signal intensity and the known monolignol concentration were used to generate monolignol calibration curves for quantification purposes (Table S1†). Following this, the monolignol linear range was established, ranging from 1–200 μg mL−1 (Fig. 6A). The lower and upper limits of quantification (LLOQ and ULOQ) were defined as the extremes of the estimated linear range for each monolignol, with LLOQ representing the lower limit and ULOQ representing the upper limit. The monolignols exhibited a progressively expanding linear range as the pKa value of the compounds increased, corresponding to a positive correlation between the linear range of the monolignols and the acidity of the compound. The variation in intensity predicted from the concentration (R2) ranged from 77–100%, showing that the monolignol concentration has a significant influence on the signals' intensity variation. This establishes confidence in overcoming the difficulties associated with quantification using ESI as an ionization source.
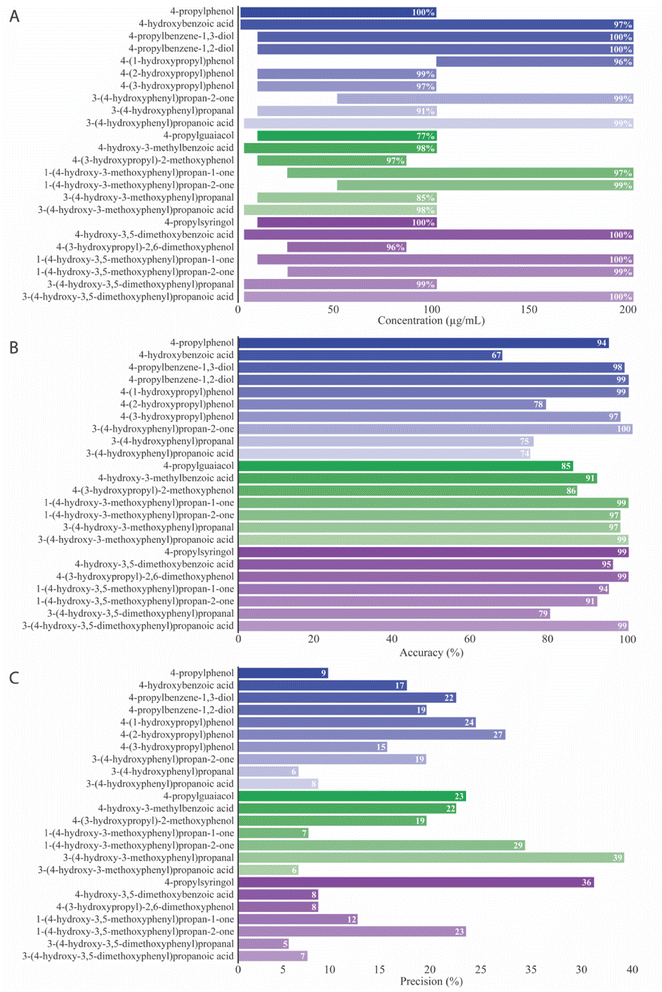 |
| | Fig. 6 Validation of monolignol quantification. The blue-, green-, and purple-coloured bars depict 4-propylphenol, 4-propylguaiacol, and 4-propylsyringol with their corresponding oxidized products, respectively. (A) Monolignol linear concentration range. The linear concentration range was estimated for the monolignols' concentration estimation. Each linear range is presented with the calculated coefficient of determination (R2, %), providing the quality of the fit for the linear regression model. The model was generated using the standard concentration and signal intensity as the independent and dependent variables, respectively. (B) Accuracy of monolignol quantification (%). Accuracy was evaluated by comparing the concentration estimated derived from the corresponding linear regression model with the established concentration of the respective monolignol (10–150 μg mL−1). (C) Precision of monolignol quantification (%). The average relative standard deviation computed based on the intensity across various dilutions of the mixed standard solutions was applied to estimate the quantification's precision. | |
Accuracy.
71% of the quantification assessments were documented to have an accuracy surpassing 90% (Fig. 6B). The estimated accuracy for each monolignol calibration curve was not correlated with the monolignol functional group, reinforcing the idea that ion suppression did not impact the accuracy of quantification estimates.
Precision.
Quantification utilizing DI-ESI-MS is excessively responsive due to high sensitivity. However, the matrix effect mentioned earlier could introduce variability in the results. To address this, we used a high number of replicates and injection parallels. The precision of the monolignol calibration curves was based on the relative standard deviation of the measured intensities, and spanned from 5–39%, with 38% of the monolignols achieving precision levels below 10% (Fig. 6C). Merely 8% of the monolignols attained precision levels exceeding 30%. No distinct correlation between the quantification precision and the monolignol functional group was evident, supporting that matrix effects did not affect the precision of quantification estimates.
Sensitivity.
In the examination of the sensitivity metrics inherent to our methodology, we investigated its capacity to discriminate minute variations and detect trace amounts. This involves quantifying both the limit of detection (LOD) and the limit of blank (LOB). LOB, representing the background, denotes the highest measurable signal intensity in the absence of the analyte and ranged with an average signal intensity from 136 ± 49 to 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 840 ± 1006 for the different monolignols (Fig. 7A). Compounds exhibiting the least acidity seem to achieve the lowest LOB. LOD, the lowest quantifiable analyte concentration discernible from background noise, was found to range from 0.3–25 μg mL−1 (Fig. 7B). Compounds such as 4-(1-hydroxypropyl)phenol (25 μg mL−1), 3-(4-hydroxyphenyl)propan-2-one (10 μg mL−1), and 4-(3-hydroxypropyl)-2,6-dimethoxyphenol (10 μg mL−1) exhibited the highest LOD and were the compounds with lowest acidity.
840 ± 1006 for the different monolignols (Fig. 7A). Compounds exhibiting the least acidity seem to achieve the lowest LOB. LOD, the lowest quantifiable analyte concentration discernible from background noise, was found to range from 0.3–25 μg mL−1 (Fig. 7B). Compounds such as 4-(1-hydroxypropyl)phenol (25 μg mL−1), 3-(4-hydroxyphenyl)propan-2-one (10 μg mL−1), and 4-(3-hydroxypropyl)-2,6-dimethoxyphenol (10 μg mL−1) exhibited the highest LOD and were the compounds with lowest acidity.
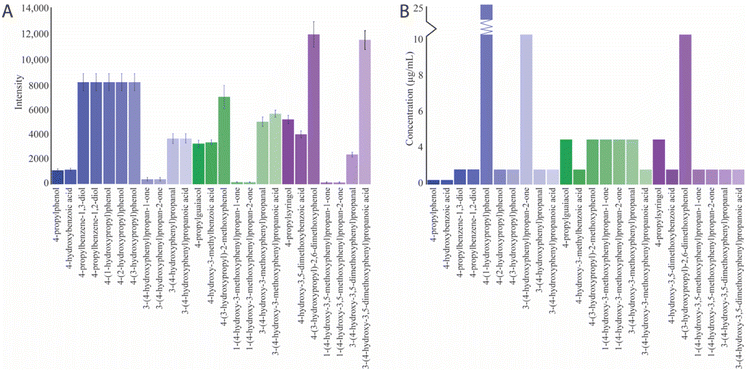 |
| | Fig. 7 Sensitivity analysis. The bars are distinguished in blue, green, and purple to illustrate the estimated analytic sensitivities pertaining to 4-propylphenol, 4-propylguaiacol, and 4-propylsyringol, along with their respective oxidized products. (A) The intensities present in a blank sample represent the LOB. (B) LOD is shown as the minimum detectable concentration for the monolignols. | |
Detection of monolignols generated by UPO-catalyzed biotransformation
Having established the DI-ESI-MS method, we turned to the nature (Fig. 8) and quantity of products (Table 2) resulting from the oxifunctionalization of monolignols catalyzed by UPOs. Incubating 4PP, 4PG, and 4PS with HspUPO and CmaUPO-I in the presence of H2O2 revealed varied oxidation specificity, as evidenced by comparing the established monolignol fragmentation patterns with CID scans from the UPO-catalyzed reactions (Fig. S4†). These variations are attributed to differences in the enzyme composition and the methoxy groups of the monolignols, potentially influencing the interaction within the enzyme–substrate complex and variations in the transition state energy.
 |
| | Fig. 8 Full scan DI-ESI-MS of UPO-catalyzed biotransformations. Deprotonated molecular ions were detected in the m/z 70–2000 range. [M − H]− ions generating a signal of 2% or lower relative to the base peak are excluded for clarity. NL is a shorthand representation of the intensity level of the base peak. The star annotates a peak with familiar molecular mass (m/z 135); however, the CID spectra did not match the established CID spectra of 4PP. (A) In the HspUPO-catalyzed oxifunctionalization of 4PP, the main product was 4-propylbenzene-1,2-diol. NL: 1.04 × 106. (B) CmaUPO-I catalyzed oxifunctionalization of 4PP, producing 4-(2-hydroxypropyl)phenol. NL: 1.67 × 105. (C) HspUPO catalyzed the demethylation and hydroxylation of 4PG, yielding mainly 4-propylbenzene-1,2-diol and possibly 3-methoxy-5-propylbenzene-1,2-diol. NL: 5.23 × 105. (D) Dimerization of 4PG, possibly resulting from phenol radicals in the CmaUPO-I catalyzed biotransformation. 4-Propylbenzene-1,2-diol was also produced in this biocatalysis. NL: 3.67 × 106. (E) Demethylation of 4PS by HspUPO suggested the production of 3-methoxy-5-propylbenzene-1,2-diol. NL: 5.29 × 105. (F) CmaUPO-I catalyzed the biotransformation of 4PS, but we have not confirmed the generated products. NL: 4.63 × 105. | |
Table 2 Quantification of Oxifunctionalized Monolignols. The FIA-based method was employed to emphasize the amount (μg mL−1) of monolignol degradation as well as the production of oxifunctionalized monolignols in UPO catalyzes. Each UPO-catalyzed reaction can be visualized in full scan mode found in the corresponding reference figures
| UPO |
Native monolignol |
Oxifunctionalized monolignols |
| Substrate |
Concentration prior to reaction (μg mL−1) |
Concentration after reaction (μg mL−1) |
Products |
Concentration (μg mL−1) |
Reference |
|
The absence of the standard introduces uncertainty, and the assumption made is a tentative inference relying on the observed CID spectra, mass calculations, and reaction pathways. Hence, the final concentration remains non-estimated.
|
|
HspUPO |
4-Propylphenol |
1362 ± 100 |
341 ± 46 |
4-Propylbenzene-1,2-diol |
1081 ± 123 |
Fig. 8A
|
| 4-Propylguaiacol |
1662 ± 278 |
83 ± 45 |
4-Propylbenzene-1,2-diol 3-methoxy-5-propylbenzene-1,2-diola |
307 ± 49 |
Fig. 8C
|
| — |
| 4-Propylsyringol |
1962 ± 230 |
357 ± 141 |
3-Methoxy-5-propylbenzene-1,2-diola |
— |
Fig. 8E
|
|
CmaUPO-I |
4-Propylphenol |
1362 ± 99 |
5 ± 1 |
4-(2-Hydroxypropyl)phenol |
66 ± 1 |
Fig. 8B
|
| 4-Propylguaiacol |
1938 ± 336 |
433 ± 160 |
Dimerizationa |
— |
Fig. 8D
|
| 4-Propylsyringol |
1962 ± 230 |
568 ± 170 |
Unknowna |
— |
Fig. 8F
|
For the enzymatically oxyfunctionalization of 4PP (start concentration 1362 ± 99 μg mL−1), HspUPO catalyzed an electrophilic attack on the π system to yield 4-propylbenzene-1,2-diol (1081 ± 123 μg mL−1, m/z 151, Fig. 8A) as the main product. At the end of the reaction, 341 ± 46 μg mL−1 4PP remained after biotransformation, which is not unexpected as there is a 1:1 molar ratio between 4PP and H2O2. In contrast, CmaUPO-I targeted mainly the Cβ position of the propyl chain, producing 4-(2-hydroxypropyl)phenol (66 ± 1 μg mL−1, m/z 151, Fig. 8B). This reaction is likely the result of the hydroxylation of Cβ–H bond by compound I. Upon completion of the reaction, 5 ± 1 μg mL−1 of 4PP remained. Given that there is a discrepancy between the amount of substrate and the hydroxylated product, there is likely a competition between a peroxygenase and a peroxidase mechanism. The latter will likely have a dimerization product formation initially (see more below). Moreover, inspection of the mass spectrum shows products with m/z values deviating what is expected of what is obtained by only insertion of one oxygen in a C–H bond. Poor chemo selectivity in alkane oxidation is a known problem leading to low yields of the desired products.28 B3LYP-D3 calculations indicate the relative activation energies of the phenol H, Cα–H, Cβ–H, and Cγ–H of 4PP to be 20.5, 20.5, 27.2, and 41.8 kJ mol−1, respectively. Similar trends were observed for 4-propylguaiacol and 4-propylsyringol. This shows that, kinetically, the Cγ–H is the most difficult to oxifunctionalize. Still, the key parameter for using enzymes for regioselectivity is the positioning of the substrate in the active site concerning which C–H will be in the closest proximity to the activated oxygen species.
When 4PG (start concentration 1662 ± 278 μg mL−1) was the substrate for HspUPO, 83 ± 45 μg mL−1 remained upon completion of the reaction. Again, hydroxylation of the aromatic ring (m/z 181, Fig. 8C) was observed. Interestingly, HspUPO catalyzed a demethylation reaction to yield 4-propylbenzene-1,2-diol (307 ± 49 μg mL−1, m/z 151, Fig. 8C) as the primary product. The addition of ascorbate enhanced the yield of reaction products by preventing phenol radical formation. CmaUPO-I catalyzed mainly a peroxidase reaction resulting in the potential dimerization of 4PG (m/z 329, Fig. 8D). Here, the 4PG concentration decreased to 433 ± 160 μg mL−1. The same biotransformation was tested in the presence of ascorbate to see if this would favor the peroxygenase reaction. Now, the dimerization of 4PG was restricted, yet there was no significant substrate degradation.
HspUPO catalyzed demethylation of 4PS (start concentration 1962 ± 230 μg mL−1) that produced ions at m/z 181 (Fig. 8E). Upon completion of the reaction, the 4PS concentration was 357 ± 141 μg mL−1. Although the proposed identity of 3-methoxy-5-propylbenzene-1,2-diol could not be verified due to a lack of a suitable standard, the fact that the same peak with comparable fragmentation patterns was observed in the HspUPO-catalyzed biotransformation of 4PG, we express confidence that the compounds at m/z 181 produced from 4PG and 4PS are the same, likely 3-methoxy-5-propylbenzene-1,2-diol. CmaUPO-I catalyzed the biotransformation of 4PS resulting in substrate degradation to a concentration of 568 ± 170 μg mL−1. However, the nature of the products could not be determined (Fig. 8F).
It is essential to emphasize that although all experiments were conducted under identical conditions, altering parameters such as temperature, component concentrations, or buffers could significantly impact the biotransformations. We repeated the biotransformation of 4PS catalyzed by CmaUPO-I at 37 °C to explore potential product formation, as the nature of the products was unclear at 25 °C. Subsequently, we identified a formed product at m/z 211, indicating a potential isomeric mixture of hydroxylated 4PS (Fig. S5†). Additionally, a product emerged at m/z 209, indicating second-step oxidation of the Cα, Cβ, and Cγ of the hydroxylated products. Notably, this resulted in an isomeric mixture of ketones and an aldehyde (Fig. S5†). Quantification of the isomeric mixtures was not attempted; nevertheless, the intensities of the deprotonated molecular ions at m/z 209 and m/z 211 were significantly higher compared to the controls.
Materials and methods
Standard solution preparation
Monolignol standard stock solutions (1.0 mg mL−1) were prepared separately in 50% ACN (LC-MS grade) and 50% Milli-Q water with 150 mM NH4OH and filtered (0.2 μm, Pall Corporation, Port Washington, New York, USA). The stock solutions comprised the following compounds, each purchased from their respective suppliers: 4-propylphenol, 4-hydroxy benzoic acid, 4-(3-hydroxypropyl)phenol, 3-(4 hydroxyphenyl) propanoic acid, 4-propylguaiacol, 4-hydroxy-3-methoxy-benzoic acid, 3-(4-hydroxy-3-methoxyphenyl)propanoic, 4-hydroxy-3,5-methoxy-benzoic acid, and 3-(4-hydroxy-3-methoxyphenyl)propanal (Sigma-Aldrich, Burlington, Massachusettes, USA), 4-(3-hydroxypropyl)-2-methoxy phenol, 3-(4-hydroxyphenyl) propanal, 1-(4-hydroxyphenyl)propane-1-one, 1-(4-hydroxy-3-methoxy-phenol)propane-1-one, 1-(4-hydroxy-3-methoxy-phenol)propane-2-one, and 1-(4-hydroxy-3,5-dimethoxy-phenol)propane-2-one (BLD Pharmatech Ltd, Shanghai, China), 4-propylsyringol, 4-(2-hydroxypropyl) phenol, 4-(3-hydroxypropyl)-2,6-dimethoxy phenol, and 1-(4-hydroxy-3,5-dimethoxy-phenol)propane-1-one (Enamine, Kyiv, Ukraine), 3-(4-hydroxy-3,5-dimethoxyphenyl) propanal (ChemSpace, Riga, Latvia), 3-(4-hydroxy-3,5-dimethoxyphenyl) propanoic acid (Accela, San Diego, California, USA), 4-(1-hydroxypropyl) phenol (LGC Standards Ltd, Teddington, Middlesex, UK), and 1-(4-hydroxyphenyl)propane-2-one (Tokyo Chemical Industry, Tokyo, Japan).
Instrumentation and conditions
MS analyses were performed using a Dionex Ultimate 3000 RS autosampler (Thermo Fisher Scientific, Waltham, MA, USA) coupled to a linear ion trap (LTQ XL Thermo Fisher Scientific, Waltham, MA, USA) equipped with an ESI source. Instrument control, data acquisition, and processing were carried out using LTQ Tune software and Xcalibur 2.2 SP1.48 (Thermo Fisher Scientific, Waltham, MA, USA). All injections were performed as three replicates of 10 μL with three injection parallels. The transfer capillary was set at 275 °C and comprised the sheath, aux, and sweep gas flow of 12, 2, and 0 arb. units, respectively. 50% ACN with NH4OH (pH 10) was the mobile phase that directed the monolignol standard solutions to the ESI source with a flow of 0.3 mL min−1. The MS operated in negative mode with an electrospray voltage of 3.0 kV, providing optimal ionization conditions for these phenolic compounds.
Identification of native and oxidized monolignols
Standard Preparation.
Monolignol standard solution dilutions were prepared at 50 μg mL−1 as separate standards (25 separate standard solution dilutions) and as a mixture of the standards. The standard mixtures (Fig. 9) combined 4PP (mix I–V), 4PG (mix VI–VIII), and 4PS (mix IX–XI) with their corresponding oxidized monolignols (eleven mixed standard solution dilutions). By analyzing the monolignols both separately and in a mixture, one could evaluate whether the presence of other compounds would affect the generated signal of an individual compound.
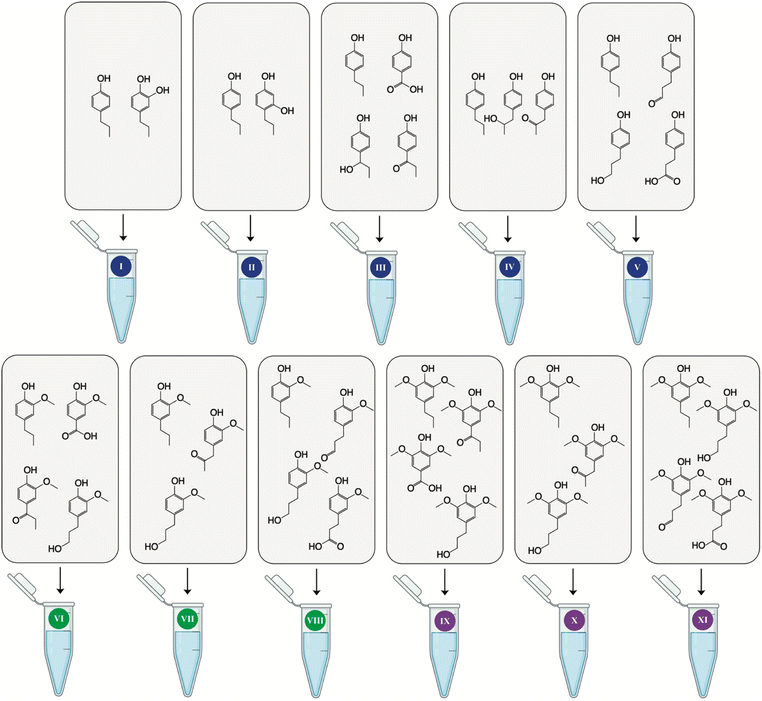 |
| | Fig. 9 Mixed standard solutions. Each mix was prepared at 50 μg mL−1 dissolved in 50% ACN with 150 mM NH4OH. The labeled reaction tubes are numerically arranged to indicate the various mixtures. The color-coded scheme employs blue, yellow, and green to signify the mixtures comprising 4-propylphenol, 4-propylguaiacol, and 4-propylsyringol with their corresponding oxidized products, respectively. Notably, the lack of commercially available standards for the hydroxyl group at the Cα or Cβ position of the propyl chain derived from 4-propylguaiacol and 4-propylsyringol led to their replacement with the Cγ hydroxyl compound, simulating the potential ion suppression from a phenolic alcohol. | |
Full scan DI-ESI-MS.
The ESI-generated deprotonated molecular ions [M−H−] in the standard solution dilutions were detected using the full scan mode employed in a m/z range of 70 to 2000. For method validation, separate and mixed standard solution dilutions were subjected to full scan analysis on different days.
DI-ESI-MS/MS.
Native and oxidized monolignols were identified using tandem MS in product ion scan mode. The deprotonated molecular ions of each monolignol were detected and selected within a range of ±m/z 0.5. CID was then utilized to fragment the selected ions by colliding them with He gas. The operated collision energy (CE) was determined experimentally as the optimal CE for each compound. Various CE were tested (20–40 eV), and the CE that generated a precursor ion intensity of approximately 10% of the base peak was chosen as the optimal CE for that specific compound. The product ions generated from CID at the optimal CE were used as the fingerprint for the identification of monolignols.
Quantification of native and oxidized monolignols
Calibration curves.
The eleven mixed standard solution dilutions were employed for quantification, as they provided a more representative sample estimate than single standard solutions. Calibration curves were constructed within the linear range of each compound by measuring the signal intensity of five monolignol deprotonated molecular ions of known concentration using three replicates and three injection parallels in SIM mode.
Method validation.
Method validation encompassed the determination of the LOB, LOD, LLOQ, and ULOQ, as well as the assessment of the method's precision and accuracy. Every validation step was performed in three replicates with three injection parallels. The signal intensity generated from a sample comprising solvent was the LOB estimate. LOD, LLOQ, and ULOQ were established based on the three and ten signal-to-noise (S/N) ratios, respectively. Accuracy was assessed by comparing the known concentration of each monolignol in the mixed standard solution dilutions (10–150 μg mL−1) with the estimated concentration obtained using the regression line of the respective calibration curve. The method's precision was assessed by calculating the relative standard deviation (RSD) of the intensities measured from the mixed standard solution dilutions.
Unspecific peroxygenase activity assays and analyzation of monolignol oxifunctionalization
Reaction setup.
The analytical method and enzymatically catalyzed monolignol oxifunctionalization presented in this study were evaluated using a generic aromatically active enzymatic system, specifically UPOs (HspUPO and CmaUPO-I). Enzyme selection was performed by screening a wide range of different UPOs produced by bisy GmbH (Wuenschendorf, Hofstaetten a. d. Raab, AUT) (ESI†). HspUPO and CmaUPO-I were expressed and purified as described previously.29,30 Biotransformations were conducted in 500 μL volume in 1.5 mL reaction tubes at 25 °C and 750 rpm in triplicates, comprising 1 μM UPO, 10 mM substrate, and 10 mM H2O2 in 100 mM ammonium bicarbonate buffer pH 6.0. The HspUPO catalyzed reaction also comprised 20 mM AscA to reduce transiently formed phenoxy radicals. All biotransformations were quenched with 50% ACN with 150 mM NH4OH after 10 minutes and extracted with 200 μL ethyl acetate. Ethyl acetate was removed from the samples prior to injection and the samples were redissolved in 50% ACN with 150 mM NH4OH. The negative control samples were (1) solvent only, (2) without H2O2, (3) without UPO, and (4) without H2O2 or UPO. Another control included subjecting intermediates (e.g., an alcohol or aldehyde derived from one of the native monolignols) to incubation with H2O2 in the absence of an enzyme. This control aimed to ascertain whether H2O2 could induce the oxifunctionalization of monolignols independently of enzyme catalysis.
DI-ESI-MS analysis.
The full scan provided an overview of the remaining substrates and the generated oxifunctionalized monolignols after the UPO-catalyzed biotransformations. Subsequently, the tandem MS protocol was employed to identify oxifunctionalized monolignols in the samples. SIM mode was used for quantification, as the intensity of the generated signal generated by the deprotonated molecular ion was proportional to the monolignol concentration. Substrate conversion was calculated based on substrate depletion, where the control comprising substrate only was set to 100%.
Conclusions
This study addresses the challenges associated with lignin valorization by focusing on the oxifunctionalization of lignin monomers, particularly 4-propylphenol, 4-propylguaiacol, and 4-propylsyringol. We emphasize the potential of lignin monomers as valuable intermediates for a sustainable green industry. Oxifunctionalization enhances the value of the monomers that can be achieved by using UPO catalysts. Our research introduces a novel, validated, and efficient method using DI-ESI-MS in full scan, SIM, and MS/MS mode for efficient and sustainable identification and quantification of native and oxifunctionalized monolignols. Each analysis segment is completed within a concise run time of 24 seconds. The proposed fragmentation mechanisms provide valuable insights into the deprotonation and identification of these compounds. Additionally, we present the quantification of monolignols, addressing challenges associated with matrix effects, accuracy, precision, and sensitivity. Our findings demonstrate the potential of overcoming quantification difficulties using DI-ESI-MS. This screening method overcomes the limitations of existing techniques by providing a balance between the straightforwardness of plate assays and the precision of chromatography and represents an analytical breakthrough. We illustrate the method's utility in both qualitative and quantitative analyses of oxifunctionalized monolignol products derived from different UPO catalyzes. The varied oxidation specificity observed with different UPOs highlights the potential for tailoring enzymatic processes for specific applications. Notably, this analytical method represents a versatile application of DI-ESI-MS/MS for the analysis of monolignols, contributing to the exploration of lignin as a valuable and sustainable bioresource.
Author contributions
K. E. expressed HspUPO and CmaUPO-I. R. S. purified HspUPO and CmaUPO-I, performed the formal analysis and investigation, ensured experiment reproducibility, and project administration, and authored the manuscript including visualization and data presentation. D. E. and H. D. provided supervision, investigation, and manuscript review. M. S. was responsible for conceptualization, resources, and funding acquisition and contributed to manuscript review.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge funding from the European Innovation Council (EIC) Pathfinder program under grant agreement no. 101046815. We also acknowledge Professor Tomasz Borowski for the computation of the activation parameters of the monolignols and Dr Gabriela Schröder for helpful discussions. The PhD thesis of K. E. was funded by the Eureka project PUMLA (FFG Basisprogramm).
Notes and references
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef PubMed.
- S. Van den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S. F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Energy Environ. Sci., 2015, 8, 1748–1763 RSC.
- Y. Y. Wang, X. Meng, Y. Pu and A. J. Ragauskas, Polymers, 2020, 12(10), 2277 CrossRef CAS PubMed.
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- J. F. Zhang, Y. Jiang, L. F. Easterling, A. Anstner, W. R. Li, K. Z. Alzarieni, X. M. Dong, J. Bozell and H. I. Kenttamaa, Green Chem., 2021, 23, 983–1000 RSC.
- C. K. Zhao, Z. H. Hu, L. L. Shi, C. Wang, F. X. Yue, S. X. Li, H. Zhang and F. C. Lu, Green Chem., 2020, 22, 7366–7375 RSC.
- A. De Santi, M. V. Galkin, C. W. Lahive, P. J. Deuss and K. Barta, Chemsuschem, 2020, 13, 4468–4477 CrossRef CAS PubMed.
- A. Kumar and B. Thallada, Sustainable Energy Fuels, 2021, 5, 3802–3817 RSC.
- J. Zhu, C. Yan, X. Zhang, C. Yang, M. Jiang and X. Zhang, Prog. Energy Combust. Sci., 2020, 76, 100788 CrossRef.
- J. Bomon, M. Bal, T. K. Achar, S. Sergeyev, X. Wu, B. Wambacq, F. Lemière, B. F. Sels and B. U. W. Maes, Green Chem., 2021, 23, 1995–2009 RSC.
- J.-M. Gaudin and J.-Y. de Saint Laumer, Eur. J. Org Chem., 2015, 2015, 1437–1447 CrossRef CAS.
- E. Erickson, A. Bleem, E. Kuatsjah, A. Werner, J. Dubois, J. McGeehan, L. Eltis and G. Beckham, Nat. Catal., 2022, 5, 86–98 CrossRef CAS.
- J. Beekwilder, I. M. van der Meer, O. Sibbesen, M. Broekgaarden, I. Qvist, J. D. Mikkelsen and R. D. Hall, Biotechnol. J., 2007, 2, 1270–1279 CrossRef CAS PubMed.
- A. Piazzon, U. Vrhovsek, D. Masuero, F. Mattivi, F. Mandoj and M. Nardini, J. Agric. Food Chem., 2012, 60, 12312–12323 CrossRef CAS PubMed.
- M. W. Hackl, M. Lakemeyer, M. Dahmen, M. Glaser, A. Pahl, K. Lorenz-Baath, T. Menzel, S. Sievers, T. Böttcher, I. Antes, H. Waldmann and S. A. Sieber, J. Am. Chem. Soc., 2015, 137, 8475–8483 CrossRef CAS.
- M. Kinne, M. Poraj-Kobielska, S. A. Ralph, R. Ullrich, M. Hofrichter and K. E. Hammel, J. Biol. Chem., 2009, 284, 29343–29349 CrossRef CAS PubMed.
- R. Ullrich, J. Nüske, K. Scheibner, J. Spantzel and M. Hofrichter, Appl. Environ. Microbiol., 2004, 70, 4575–4581 CrossRef CAS PubMed.
-
M. Hofrichter, H. Kellner, M. J. Pecyna and R. Ullrich, Monooxygenase, Peroxidase and Peroxygenase Properties and Mechanisms of Cytochrome P450, 2015, vol. 851, pp. 341–368 Search PubMed.
-
M. Hofrichter, H. Kellner, R. Herzog, A. Karich, C. Liers, K. Scheibner, V. W. Kimani and R. Ullrich, in Grand Challenges in Fungal Biotechnology, ed. H. Nevalainen, Springer International Publishing, Cham, 2020, pp. 369–403, DOI: DOI:10.1007/978-3-030-29541-7_14.
- M. Faiza, S. Huang, D. Lan and Y. Wang, BMC Evol. Biol., 2019, 19, 76 CrossRef PubMed.
- M. Hofrichter, H. Kellner, R. Herzog, A. Karich, J. Kiebist, K. Scheibner and R. Ullrich, Antioxidants, 2022, 11, 163 CrossRef CAS PubMed.
- R. Seraglia, L. Molin, I. Isak and P. Traldi, Eur. J. Mass Spectrom., 2012, 18, 195–203 CrossRef CAS PubMed.
- L. J. Haupert, B. C. Owen, C. L. Marcum, T. M. Jarrell, C. J. Pulliam, L. M. Amundson, P. Narra, M. S. Aqueel, T. H. Parsell, M. M. Abu-Omar and H. I. Kenttämaa, Fuel, 2012, 95, 634–641 CrossRef CAS.
- J. MacKay and D. Dimmel, J. Wood Chem. Technol., 2001, 21, 1–17 CrossRef.
- F. Lu and J. Ralph, J. Agric. Food Chem., 1997, 45, 2590–2592 CrossRef CAS.
- B. C. Owen, L. J. Haupert, T. M. Jarrell, C. L. Marcum, T. H. Parsell, M. M. Abu-Omar, J. J. Bozell, S. K. Black and H. I. Kenttämaa, Anal. Chem., 2012, 84, 6000–6007 CrossRef CAS.
- M. Mattonai, E. Parri, D. Querci, I. Degano and E. Ribechini, Microchem. J., 2016, 126, 220–229 CrossRef CAS.
-
J. A. Davies, Selective Hydrocarbon Activation: Principles and Progress, VCH Publishers, 1990 Search PubMed.
- L. Rotilio, A. Swoboda, K. Ebner, C. Rinnofner, A. Glieder, W. Kroutil and A. Mattevi, ACS Catal., 2021, 11, 11511–11525 CrossRef CAS PubMed.
- K. Ebner, L. J. Pfeifenberger, C. Rinnofner, V. Schusterbauer, A. Glieder and M. Winkler, Catalysts, 2023, 13, 206 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Hanne
Devle
*a,
Hanne
Devle








