
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA comprehensive HRMS methodology using LC-(ESI)-/GC-(APCI)-QTOF MS complementary platforms for wide-scope target screening of >750 pesticides in olive oil†
Sofia K.
Drakopoulou
 a,
Stefanos E.
Kokolakis
a,
Apostolos L.
Karagiannidis
a,
Marilena E.
Dasenaki
b,
Niki C.
Maragou
a and
Nikolaos S.
Thomaidis
*a
a,
Stefanos E.
Kokolakis
a,
Apostolos L.
Karagiannidis
a,
Marilena E.
Dasenaki
b,
Niki C.
Maragou
a and
Nikolaos S.
Thomaidis
*a
aLaboratory of Analytical Chemistry, Department of Chemistry, National and Kapodistrian University of Athens, Panepistimiopolis Zografou, 15771 Athens, Greece. E-mail: ntho@chem.uoa.gr; Tel: +30 210 7274430
bLaboratory of Food Chemistry, Department of Chemistry, National and Kapodistrian University of Athens, Panepistimiopolis Zografou, 15771 Athens, Greece
First published on 9th April 2024
Abstract
This study presents the development and validation of a comprehensive high-resolution mass spectrometry (HRMS) methodology for the detection of 771 pesticides in olive oil, using liquid chromatography with electrospray ionization, operating in positive and negative mode, and gas chromatography with atmospheric-pressure chemical ionization in positive mode, both coupled to quadrupole-time-of-flight mass spectrometry (LC-(ESI)-/GC-(APCI)-QTOF MS). Special reference is made to the post-acquisition evaluation step, in which all LC/GC-HRMS analytical evidence (i.e. mass accuracy, retention time, isotopic pattern, MS/MS fragmentation) is taken into account in order to successfully identify the compounds. The sample preparation of the method involves a QuEChERS-based protocol, common for both techniques, differentiated only on the reconstitution step, making the method highly applicable in routine analysis. A smart evaluation of method's performance was carried out, with 65 representative analytes comprising the validation set. The method was validated in terms of linearity, accuracy, matrix effect and precision, while the limits of detection and quantification of the method were estimated. Finally, twenty Greek olive oil samples were analysed in both analytical platforms and the findings included the pesticides lambda-cyhalothrin, chlorpyrifos, phosphamidon, pirimiphos-methyl and esprocarb at low ng g−1 level.
1 Introduction
Pesticides consist a powerful tool in the agriculture field in order to satisfy the worldwide need for food, taking into consideration the fast-occurring changes, such as overpopulation,1 climate change2 and intensive farming.3 However, the extensive use of these chemicals and the non-compliance to good agricultural practises may endanger human health safety.Extra virgin olive oil (EVOO) is one of the most essential Mediterranean diet components, being distinguished for its unique taste and nutritional value, while its consumption has been associated with longevity and good health,4,5 as well as lower risk of cardiovascular6 or neurodegenerative diseases, such as Alzheimer.7,8 In order to protect the precious crops of olive trees from insecticides, farmers often apply pesticides and the produced olive oil is a food product that is monitored for pesticides' residues.
In this framework maximum residue levels (MRLs) are established by Regulation (EC) No. 396/2005 (ref. 9) in order to ensure the lowest possible consumer exposure to pesticide residues in treated crops. In most cases the MRLs are set for the foods in their raw, unprocessed form. These MRLs can be applied to processed foods using appropriate processing factors which are based on studies which take into account the effect of processing on the food as traded. Based on processing studies EFSA has created an EU database of processing factors for pesticide residues,10 including ‘olives for oil production’ as raw primary commodity and ‘native oil’ as processed commodity.
Considering that more than 1000 pesticides are listed in the EU pesticides database,11 it is of paramount importance to develop powerful and holistic analytical methodologies that can detect a wide range of pesticides at very low concentrations. According to the European Guidance Document on Pesticide Analytical Methods for Risk Assessment and Post-approval Control and Monitoring Purposes,12 the analytical methods used for pesticides screening must meet common criteria of performance, with the confirmatory techniques of gas chromatography-mass spectrometry (GC-MS) and liquid chromatography-mass spectrometry (LC-MS) being the most prevailed ones due to their reliability and widespread application.13,14 In several studies, both LC and GC combined with MS are applied,15–19 as complementary, orthogonal methods. GC-MS is mostly used for semi-volatile compounds, whereas LC-MS is favourable for polar and thermo-labile pesticides. Therefore, the combination of these two techniques leads to a significant increase of the analytical coverage that enables the wide-scope screening of pesticides.
Over the past decades, different approaches to trace-level determination of pesticides have been investigated aiming at developing fit-for-purpose methods.20 Most of them exploit the capability of chromatographic techniques coupled to triple quadrupole mass spectrometer (LC-QqQ, GC-QqQ), based mainly at its exceptional sensitivity.16,21–23 However, relying on recent breakthroughs in the field of high-resolution mass spectrometry (HRMS), chromatographic techniques combined with HRMS (LC-QTOF, GC-QTOF) are gradually introduced in pesticide routine analysis as a very promising alternative.24–26 Specifically, HRMS-based methodologies are presented as one of the most reliable analytical platforms for pesticides analysis, mainly due to their potential of wide-scope screening and retrospective analysis, as well as due to the high confidence in identification achieved.27 Regarding the latter, especially when coupled to chromatographic techniques, the confirmation of positive findings in HRMS workflow is relied on several identification criteria that consider all the analytical evidence available (i.e., retention time, mass accuracy, isotope fitting, fragmentation pattern), thus significantly enhancing identification confidence.28
Additionally, several studies have been carried out to investigate which ionization method is more suitable and effective, with atmospheric-pressure chemical ionization (APCI) being recently highlighted as an emerging source.29 APCI is a low energy ionization mechanism (soft), which results to reduced fragmentation and high-abundance occurrence of the molecular ion, ultimately favouring the wide-range detection of the targeted compounds.30 Another advantage of APCI source is its ability to be interfaced with both GC and LC instruments.30,31 This fact adds versatility and extends analytical capabilities providing flexibility to determine volatile and semi-volatile compounds of low and intermediate polarity, traditionally analysed by dedicated vacuum GC-MS instruments.32 In recent studies, GC-APCI has been coupled to MS to address different demanding issues, not only in the case of pesticide analysis,33,34 but also in the field of food authenticity, such as characterization and classification.35,36 Furthermore, in some methodologies, different analytical approaches are combined using either electrospray ionization (ESI) or APCI (e.g. LC-ESI/APCI, GC-APCI) coupled to MS, in order to fully exploit each platforms' capabilities and increase the analytical coverage. Such approaches have been already introduced to olive oil discrimination studies, using both platforms coupled to low resolution mass spectrometry, LRMS,17 or HRMS,18,19 leading to notable results.
In the present work, a comprehensive methodology using LC-ESI and GC-APCI coupled to HRMS was developed, enabling the detection of 771 pesticides. A QuEChERS-based protocol was followed for the extraction of pesticides from olive oil matrix. The sample preparation was common for both techniques, differentiated only on the reconstitution step, thus making the method highly applicable in routine analysis. Taking advantage of HRMS potential, a strong post-acquisition evaluation of the data is being discussed considering all criteria available through LC/GC-HRMS analysis, aiming at high-confidence identification. To facilitate data evaluation, in-house databases of LC-ESI and GC-APCI-HRMS were implemented to confirm positive findings. The databases included information regarding retention time, MS and MS/MS ions, and were built after injecting standards for all the target compounds. The proposed methodology was validated based on smart evaluation of its performance through the validation of 65 selected analytes. Finally, the method was applied to olive oil samples from Greece. To the best of our knowledge, this is the first study than includes such a wide range of pesticides' detection, applied for the first time in olive oil matrix. Moreover, thanks to HRMS main capabilities, retrospective analysis is also enabled. Previous data can be mined for newly emerged compounds of concern to determine if these pesticides have been previously detected. Hence, the proposed methodology is an important tool towards pesticides control and health protection, providing a comprehensive picture of the exposure to pesticides over time.
2 Experimental
2.1. Chemicals and reagents
All solvents were of special purity, pesticide grade for residue analysis. For UPLC-ESI-QTOF system all solvents were UPLC-MS grade. Methanol (MeOH) hypergrade for LC-MS was purchased from Sigma-Aldrich (Steinheim, Germany), acetonitrile (ACN) from Honeywell (New Jersey, USA), whereas 2-propanol and ethyl acetate (EtoAC) of LC-MS grade was acquired from Fisher Scientific (Geel, Belgium). Distilled water was provided by a Milli-Q purification apparatus (Millipore Direct-Q UV, Bedford, MA, USA). Regenerated cellulose syringe filters (RC, pore size 0.2 μm, diameter 15 mm) were purchased from Phenomenex (Torrance, CA, USA). Ammonium acetate and sodium formate of 99% purity were purchased by Sigma-Aldrich (Steinheim, Germany). For GC-APCI-QTOF system, hexane for pesticide residue analysis was purchased from Honeywell (New Jersey, USA) and acetone pestipure from Carlo Erba (Barcelona, Spain).Regarding the experimental procedure, standards of hexachlorobutadiene, dichlorvos, alpha-HCH, hexachlorobenzene, beta-HCH, lindane, delta-HCH, heptachlor, aldrin, dicofol, isodrin, alpha-endosulfan, dieldrin, endrin, 4,4′-DDT, 4,4′-DDD, 4,4′-DDE, 2,4′-DDT, endosulfan-sulfate, (>99% purity) were purchased from Fluka-Sigma-Aldrich (Steinheim, Germany). Standard stock solutions were also prepared for these pesticides at a concentration of 1000 mg L−1 in hexane and stored at −20 °C. For all the other pesticides used in the study (Table S1, ESI†), standard stock solutions of individual pesticides at a concentration of 1000 mg L−1 were obtained from Bruker Daltonics GmbH (Bremen, Germany). A volume of 10 μL of the standard stock solutions were transferred into a 1 mL vial and diluted with the appropriate solvent (MeOH, EtoAc, hexane) depending on compounds' solubility, to prepare working solutions of 1000 ng L−1. Working solutions were also stored at −20 °C. Acetic acid (HAc) ≥ 99% used was purchased from Honeywell (New Jersey, USA), magnesium sulphate (MgSO4) anhydrous from Mallinckrodt (New York, USA), while sodium acetate (NaAc), PSA silica and DSC-18 (C18) were acquired from Supelco (Bellefonte, USA).
2.2. Samples and sample preparation
Olive oil samples were used to evaluate the applicability of the method. In total, 20 olive oil samples were collected from different regions of Greece (Crete, Lesvos, Peloponnese, Samos, Chios), belonging to three different varieties, namely Koroneiki, Kolovi and Throumba (Table S2, ESI†). All samples were stored at room temperature for no more than 15 days before analysis.The QuEChERS procedure was applied for sample preparation and was based on the European Standard EN 15662.37 The sample preparation protocol can be summarized as follows: 2 g of olive oil were transferred into a 50 mL centrifuge tube; 10 mL of acetonitrile were added and vortex-mixed for 1 min; centrifugation was carried out for 3 min at 4000 rpm; an aliquot of 6 mL of the acetonitrile phase (upper layer) was transferred into a 20 mL screw capped vial; 150 mg PSA and 150 mg C18 were added and vortex-mixed for 30 s; centrifugation was carried out for 3 min at 4000 rpm; an aliquot of 2 mL of the supernatant was taken for each analysis (2 mL for LC-HRMS and 2 mL for GC-HRMS); both aliquots were evaporated till dryness; reconstitution was performed with 200 μL MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O 50:50 and 5% HCOOH (LC-HRMS analysis) and with 200 μL n-hexane (GC-HRMS analysis); the extracts were vortex-mixed for 30 s each and transferred into auto-sampler vials to be used for gas- and liquid chromatographic analyses, after filtration with regenerated cellulose filters (RC 0.20 μm, 15 mm). The schematic workflow of the sample preparation followed is depicted in Fig. 1.
:H2O 50:50 and 5% HCOOH (LC-HRMS analysis) and with 200 μL n-hexane (GC-HRMS analysis); the extracts were vortex-mixed for 30 s each and transferred into auto-sampler vials to be used for gas- and liquid chromatographic analyses, after filtration with regenerated cellulose filters (RC 0.20 μm, 15 mm). The schematic workflow of the sample preparation followed is depicted in Fig. 1.
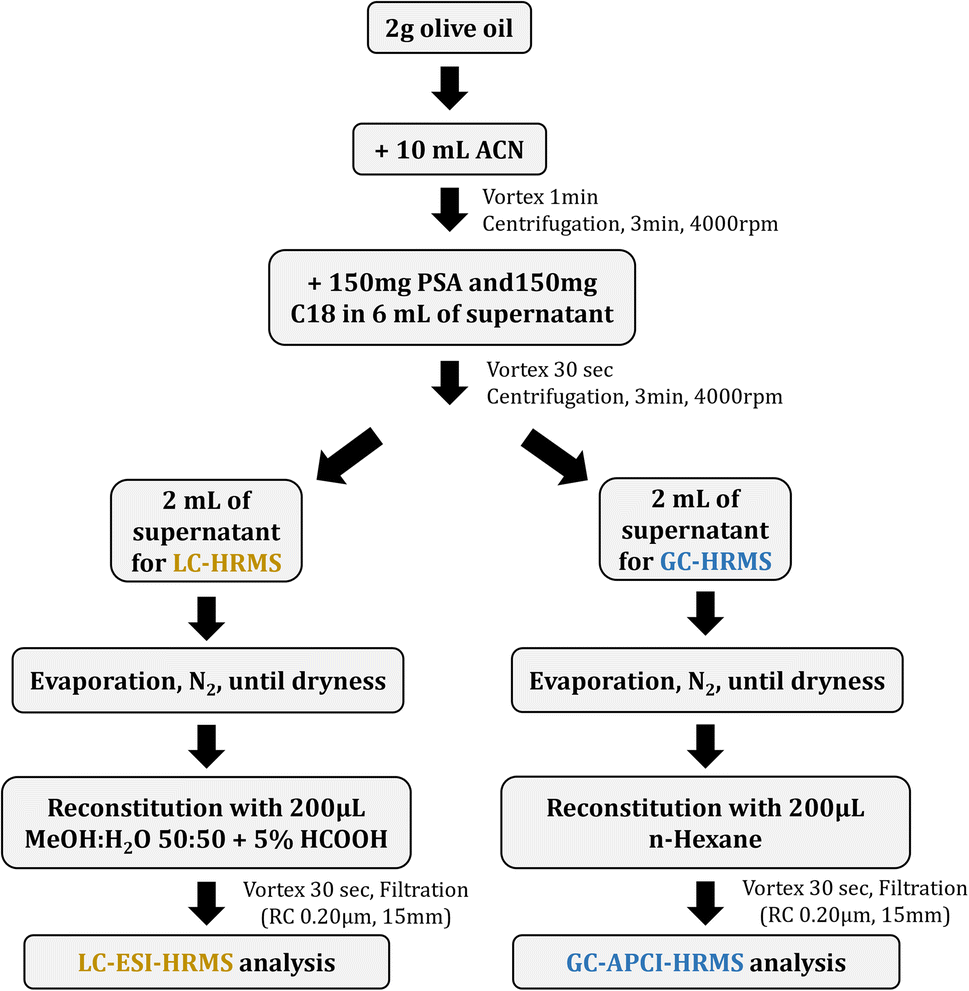 | ||
| Fig. 1 Schematic workflow of sample preparation. | ||
2.3. HRMS analysis
HRMS analysis was performed using a quadrupole time-of-flight (QTOF) mass spectrometer (Maxis Impact, Bruker Daltonics, Bremen, Germany). Two different analyses were performed for the needs of the study, with LC-(ESI)- and GC-(APCI) being coupled to the same MS arrangement (QTOF) in both cases.The QTOF-MS system was equipped with an electrospray ionization interface (ESI) with the following operation parameters: capillary voltage 2500 V (PI) and 3500 V (NI); end plate offset 500 V; nebulizer pressure 2 bar; drying gas 8 L min−1; and gas temperature 200 °C. The QTOF-MS system was operated in data independent acquisition (DIA) mode, Bruker's broadband collision-induced dissociation, bbCID mode, wherein all the precursor ions within a time cycle were subjected to fragmentation in the mass spectrometer and recorded spectra over the range of m/z 50–1000, with a scan rate of 2 Hz. A QTOF-MS calibration was performed daily with the manufacturer's solution in the beginning of the sequence and in the beginning of every injection. The instrument provided a typical mass resolving power of 36000–40000 during calibration (39274 at m/z 226.1593, 36923 at m/z 430.9137, and 36274 at m/z 702.8636).
The QTOF-MS was interfaced with an APCI source operating in positive ionization mode. Mass calibration was automatically performed with perfluorotributylamine (FC43) in the beginning of the sequence and prior to each injection. MS/MS spectra were received using DIA mode (Bruker's bbCID), scanning between 30 and 1000 Da (m/z range) with scan frequency of 8 Hz.
2.4. Data analysis and screening strategy
Data acquired from HRMS analysis were subjected to target screening using DataAnalysis 5.2 and TASQ 2.1 software (Bruker Daltonics). Target screening was performed using LC-(ESI)- and GC-(APCI)-HRMS developed databases.28,38 The databases included all information required for the identification, namely compounds' name, formula, retention time and mass-to-charge ratios (m/z) of the precursor and the qualifier ions (i.e. adducts, fragments). The LC-(ESI)-HRMS database was comprised of 663 pesticides, including compounds ionized in both positive and negative modes. As shown in the Venn diagram of Fig. 2A, 420 out of the 663 compounds of the database are being detected in positive mode and 243 in negative, with 225 compounds being detected in both modes. | ||
| Fig. 2 Venn diagrams depicting the number of pesticides detected using LC-(ESI)-HRMS analytical platform operating in positive and negative ionization modes (A) and using LC-(ESI)-, and GC-(APCI)-HRMS analytical platforms (B). | ||
In the case of GC-(APCI)-HRMS, the respective database included 270 compounds corresponding to positive ionization. The two HRMS databases were used complement to each other, to increase the depth of coverage and detect a wide range of pesticides. Therefore, 771 different pesticides constituted the overall target screening list of our study, as 162 compounds are common and can be detected in both LC-(ESI)- and GC-(APCI)-HRMS (Fig. 2B). These 771 pesticides are thoroughly presented in Table S3 in the ESI,† characterized by their name and molecular formula. The HRMS analytical platform in which they are detected along with the ionization mode are also provided.
Identification was performed on the basis of retention time, mass accuracy, isotopic fitting and MS/MS fragments. The screening parameters, previously optimized by our group for the needs of wide-scope target screening,28 were formed as follows: retention time tolerance < 0.1 min; mass accuracy < 5 m Da for at least 2 ions (molecular ion and qualifier fragment), with the extracted ions chromatograms fully overlapping; isotopic fitting between the measured and theoretical molecular formulae < 100 msigma; minimum peak area threshold at 800 and minimum intensity threshold at 200.
Taking a step forward, the identification criteria were adjusted to each platform's special attributes. Thus, in the case of GC-(APCI)-HRMS, in which we receive sharp peaks and narrow full-width half maximum (FWHM), retention time tolerance was significantly lowered (ΔRT = 0.05 min compared to LC's, ΔRT = 0.1 min) in order to reduce false positive incidence. In Fig. 3, typical extracted ions chromatograms (EICs) of pesticides analyzed in LC-(ESI)-QTOF MS (Fig. 3A) and GC-(APCI)-QTOF MS (Fig. 3B) are depicted, along with the resolving power of each platform, expressed via FWHM metric. The screening parameters were applied for the evaluation of both standard solutions and spiked samples, achieving in all cases successful identification.
 | ||
| Fig. 3 Extracted ions chromatograms (EICs) and FWHM recorded in each platform in the cases of LC-(ESI)-QTOF MS (A), and GC-(APCI)-QTOF MS (B). | ||
2.5. Method validation
A selection of a validation sub-dataset was considered necessary due to the great number of target analytes. Thus, representative compounds of each category were selected to validate the method in both analytical platforms and for both ionization modes. The compounds that constituted the validation set were of different chemical classes, to cover a broad range of pesticides, and were widely distributed in terms of retention time, too (Table S1, ESI†). A smart evaluation of method's performance was implemented, according to the validation protocol of wide-scope target screening studies presented in Gago-Ferrero et al.28 Overall, 65 pesticides were used for method validation, detected with the proposed methodology. Its performance characteristics were evaluated in terms of linearity, matrix effect, accuracy and precision, while the limits of detection and quantification of the method were estimated.The linear dynamic range of the method (based on regression coefficients) was studied in standard solutions at five different concentration levels (4, 10, 20, 50, 100 ng mL−1). The standards were made in the same solvent used in our methodology during the reconstitution step, in the case of olive oil samples (Section 2.2). Therefore, standard samples were prepared in MeOH:H2O (50:50) for LC-ESI-QTOF analysis, while in the case of GC-APCI-QTOF hexane was used as solvent. Recovery experiments were performed to study the accuracy of the method in five different concentrations of spiked olive oil samples (2, 5, 10, 25 and 50 ng g−1). The recovery R (%) of each analyte was calculated by dividing the area of spiked samples to that of the matrix-matched standard. To evaluate the matrix effect of our methodology, matrix matched standards were prepared and analyzed in each platform, using the extracts of the blank olive oil sample. Matrix matched calibration curves were made at concentrations of 2, 5, 10, 25 and 50 ng mL−1 (referring to the experimental concentrations and not the ones finally measured considering the 2-fold preconcentration). Method's limits of detection (MLOD) and quantification (MLOQ) values were estimated as the concentration of the target compound in spiked samples corresponding to signal-to-noise ratio that equals to 3.3 and 10, respectively. Procedural blank samples were also analyzed for the evaluation and subtraction of potential laboratory introduced contamination. Finally, the method's precision was evaluated through the metric of repeatability, expressed in % RSD. It was estimated from the analysis of three replicates at a spiked sample of 10 ng g−1. The validation results are presented in detail, in Table S4 of the ESI.†
3 Results and discussion
3.1. Wide-scope target screening and identification criteria
The target screening workflow, thoroughly discussed in Section 2.4, was implemented in HRMS data retrieved from both LC-(ESI)- and GC-(APCI)-HRMS analysis. Fig. 4A exemplifies the identification workflow followed in the case of malaoxon pesticide, detected using LC-(ESI)-QTOF MS methodology. The identification criteria are aligned with their limits of acceptance set per case, in terms of retention time, mass accuracy, isotope fitting, and MS/MS fragmentation. All the abovementioned parameters met the standards and were within the acceptable thresholds, compared to the respective theoretical values included in the database (resulting from standard analysis), thus leading to successful identification of the compound.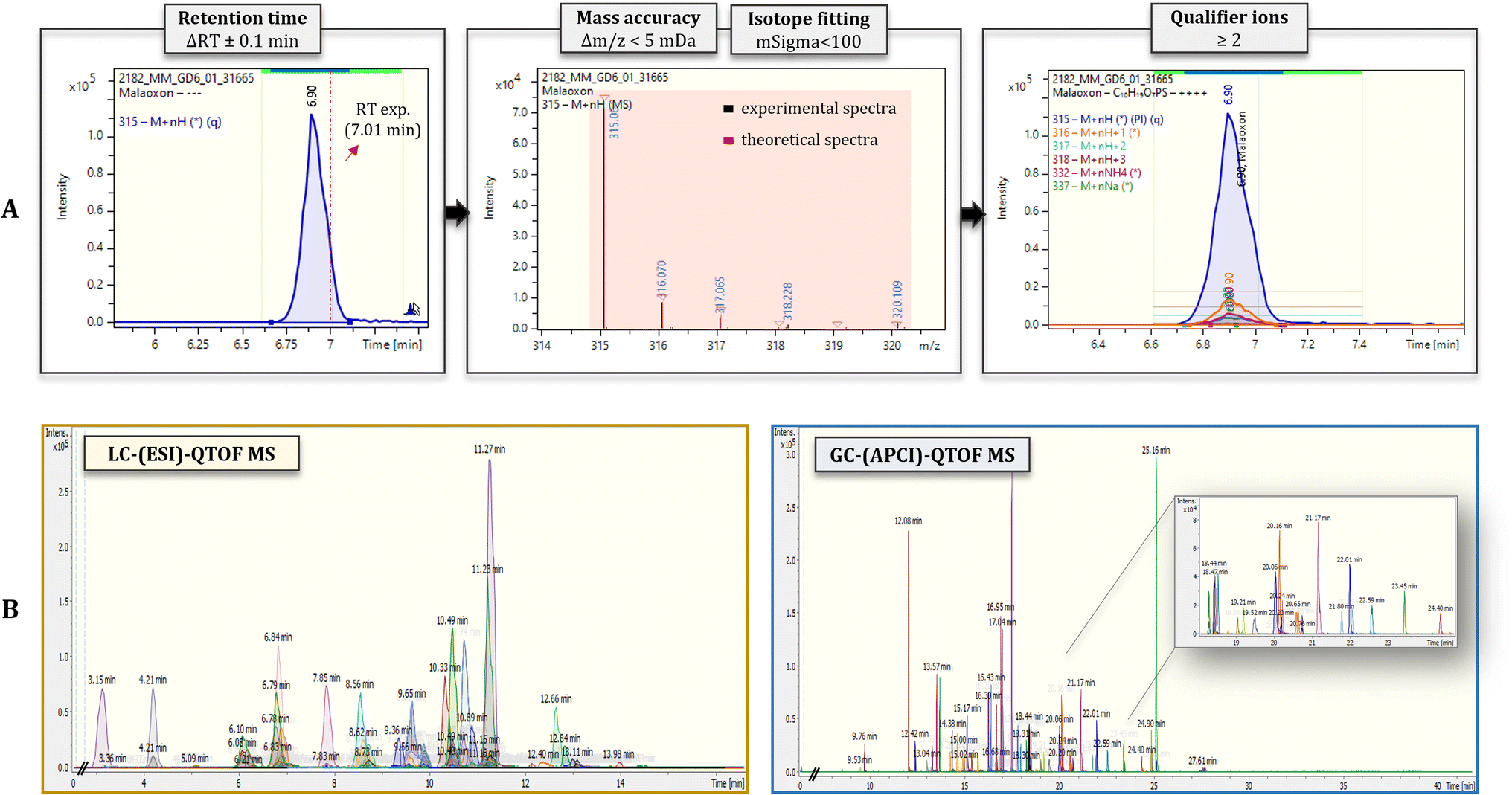 | ||
| Fig. 4 Identification workflow of malaoxon, detected in LC-(ESI)-QTOF MS. All analytical information retrieved from analysis is considered, with specific criteria and limits of acceptance being set regarding mass accuracy, retention time, isotopic pattern and MS/MS fragmentation (A). Extracted ion chromatograms (EICs) of the 65 analytes constituting the validation set, detected in positive ionization mode, in the cases of LC-(ESI)-QTOF MS, and GC-(APCI)-QTOF MS (B). | ||
3.2. Validation results
Parameters including method linearity, matrix effect, accuracy, precision, MLOD and MLOQ, were evaluated for both LC- and GC-QTOF MS analysis. The validation set was comprised of 65 pesticides, distributed across the chromatographic range, in an extensive analytical coverage, as shown in Fig. 4B.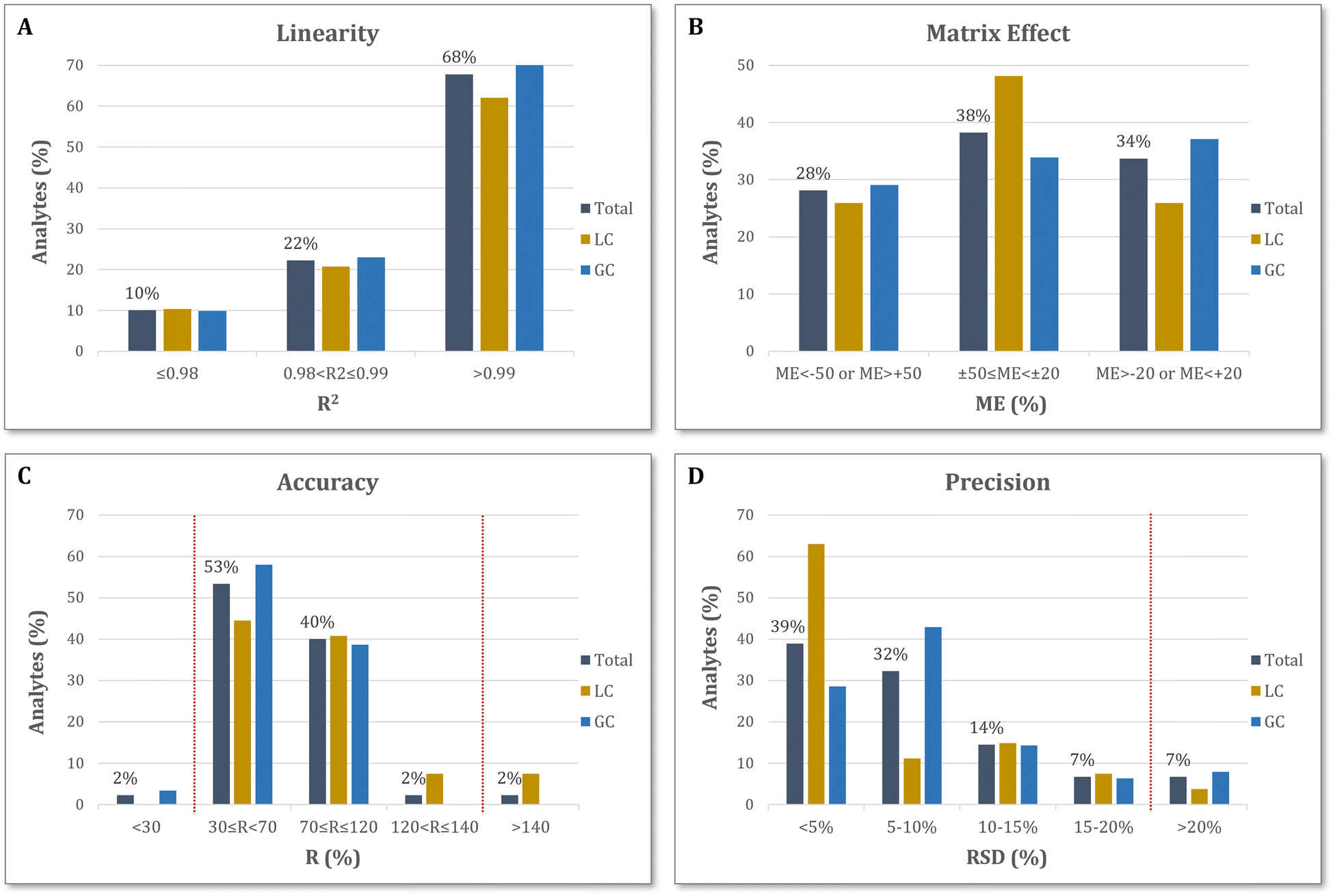 | ||
| Fig. 5 Validation results of the methodology in terms of linearity (A), matrix effect (B), accuracy (C), and precision (D). | ||
:H2O at 50:50 ratio), which is less volatile than n-hexane (used for GC analysis), thus more stable during repeatability experiments.
3.3. Application of the method
The validated methodology was applied for the analysis of 20 olive oil samples. The samples were analysed in both LC-(ESI)- and GC-(APCI)-HRMS platforms and subsequently screened for the 771 target pesticides available in our databases.Analysis was performed separately for each platform and retrieved data were subjected to wide-scope target screening using the databases of LC-(ESI)- and GC-(APCI)-HRMS accordingly. Data were processed according to the screening workflow, discussed in Section 3, with positive findings meeting all identification criteria. The screening led to the identification of 5 pesticides in the total of 20 olive oil samples. More specifically, 3 compounds were identified through LC-(ESI)-HRMS workflow (esprocarb, phosphamidon, pirimiphos-methyl) and 2 through GC-(APCI)-HRMS (chlorpyrifos, lambda-cyhalothrin). In a consequent step, for analytes detected in the olive oil samples but not included in the validation set (i.e. esprocarb, chlorpyrifos ethyl), additional analysis was performed using a spiked sample at 50 ng g−1 to confirm identification (Fig. S3†), and also perform quantification of the pesticides. The extracted ion chromatograms (EICs) of the analytes detected are presented in Fig. S4.†
The identified pesticides and their concentrations are presented in Table S5† along with MLOD and MLOQ, the recovery per analyte, the EU MRLs that correspond to the olives for oil production, the corresponding regulation and the processing factors where available. The detected compounds include one thiocarbamate herbicide (esprocarb) and four insecticides, one organophosphate (phosphamidon), two aryl organothiophosphates (pirimiphos-methyl and chlorpyrifos) and one pyrethroid (lambda-cyhalothrin).
Lambda-cyhalothrin was determined in one sample at 12 ng g−1, a concentration far below the calculated MRL for olive oil which is 500 ng g−1, taking into consideration the processing factor 1. Lambda-cyhalothrin is officially associated with olive cultivation in Greece with registered plant protection products containing this active substance as insecticide for the control of olive fruit fly Bactrocera oleae.39 Similar concentrations of lambda-cyhalothrin in Greek olive oil samples have been reported again ranging between 10 and 24 ng g−1.40
Chlorpyrifos was identified in one sample but below the quantification limit of 4.4 ng g−1. Chlorpyrifos has been identified and quantified in Greek olive oil samples with concentrations ranging between 15 and 17 ng g−1,40 and in virgin olive oils produced in Chile at average concentration of 84 ng g−1.41 Pirimiphos-methyl was also identified in one sample but below the quantification limit of 2.0 ng g−1. Phosphamidon, also known as Dimecron, was detected in four samples at concentrations between 2 and 2.5 ng g−1. This active substance is not currently approved under Regulation EC 1107/2009 however it has been reported to be tested in the past on the control of the olive fly Dacus oleae Gmel.42
It is noted that although chlorpyrifos, phosphamidon and pirimiphos-methyl are insecticides, not authorized in EU for the control of olive fruit fly in olive trees, their presence in olive oil could be attributed to pesticide applications in nearby cultivations or due to other exogenous factors such as transport of pollutants.3 These active substances are listed in the EU database and MRLs are set for olives for oil production. According to Regulation (EU) 2018/55, if no specific oil processing factor is available for virgin olive oil, a default factor of 5 may be applied for fat-soluble substances, taking into account an olive oil production standard yield of 20% of the olive harvest and a default oil processing factor of 1 may be used for non-fat-soluble substances.43 Based on this, in the strict case scenario with a processing factor 1, the calculated MRLs for olive oil are equal to the MRLs for olives for oil production which is 10 ng g−1 for the three active substances. The generated results show that the determined concentrations are in all cases below this limit. It is also noted that chlorpyrifos, lambda-cyhalothrin and pirimiphos-methyl are among the pesticides detected in virgin olive oil samples analysed during the annual EU-coordinated control programme on pesticides residues in food of year 2021.44
Regarding the herbicide esprocarb, this substance was determined in seventeen samples with concentrations ranging between 2.1 and 6.3 ng g−1. This substance is a thiocarbamate herbicide reported to be used in rice cultivations. Although it is not registered in EU as pesticide, it is included in the REACH list of pre-registered substances.45 Esprocarb has been detected in water sources in Japan and correlated with paddy rice cultivation.46 Its presence requires further investigation.
4 Conclusions
The detection of pesticides in food matrices is of paramount importance, as their presence indicates an imminent risk to human health. In the present work, a comprehensive methodology for the analysis of 771 pesticides in olive oil was developed, based on LC-(ESI)- and GC-(APCI)-HRMS. Data retrieved from HRMS analysis were subjected to wide-scope target screening, utilizing the in-house databases, developed for each analytical platform. Taking advantage of HRMS full potential, identification was performed considering all analytical evidence available from LC/GC-HRMS analysis (i.e. retention time, isotopic pattern, MS and MS/MS ions information), thus significantly enhancing identification confidence.Sample preparation was based on a QuEChERS-based protocol, common for both techniques, differentiated only on the reconstitution step, which makes the method highly applicable in routine analysis. Due to the large number of total analytes, a validation set was selected of about 10% of target compounds, including representative pesticides per category. The method was validated in terms of linearity, accuracy, matrix effect and precision, while the limits of detection and quantification of the method were also estimated. The method was then applied in olive oil samples. In total 5 pesticides were detected, with 3 compounds being identified through LC-(ESI)-HRMS workflow and 2 in GC-(APCI)-HRMS. Therefore, as indicated from the results, an increased analytical coverage was achieved, with pesticides being detected in both platforms. Finally, quantification of the analytes was performed, and the concentrations were found to be below the available MRLs considering a processing factor 1.
In conclusion, this study mainly highlights the importance of HRMS approaches in combination with enriched databases towards wide-scope target screening. To the best of our knowledge, this is the widest method developed so far in terms of number of pesticides, applied for the first time in olive oil. It is also worth mentioning that thanks to HRMS, retrospective analysis of the samples is also enabled. Namely, pesticides not yet evaluated and authorised in EU, or unknown pesticides' degradation products and metabolites could be detected in the already analysed samples, thus constituting an important asset towards pesticide control.
Author contributions
S. K. D.: data curation, investigation, visualization, writing – original draft; S. E. K.: data curation, investigation; A. L. K.: investigation, writing – original draft; M. E. D.: conceptualization, supervision, writing – review & editing; N. C. M.: writing – review & editing; N. S. T.: conceptualization, funding acquisition, project administration, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research work was financed by North Aegean Region through the program “Novel wide-scope research for the promotion of N. Aegean olive oil and olive products through the designation of their unique characteristics and bioactive content”.References
- F. P. Carvalho, Environ. Sci. Pol., 2006, 9, 685–692 CrossRef.
- R. A. Duchenne-Moutien and H. Neetoo, J. Food Prot., 2021, 84, 1884–1897 CrossRef CAS PubMed.
- M. Tudi, H. D. Ruan, L. Wang, J. Lyu, R. Sadler, D. Connell, C. Chu and D. T. Phung, Int. J. Environ. Res. Public Health, 2021, 18, 1112 CrossRef CAS PubMed.
- G. Buckland and C. A. Gonzalez, Br. J. Nutr., 2015, 113, S94–S101 CrossRef CAS PubMed.
- A. Foscolou, E. Critselis and D. Panagiotakos, Maturitas, 2018, 118, 60–66 CrossRef CAS PubMed.
- M. Xia, Y. Zhong, Y. Peng and C. Qian, Front. Nutr., 2022, 9, 1041203 CrossRef PubMed.
- L. Rita, N. R. Neumann, I. Laponogov, G. Gonzalez, D. Veselkov, D. Pratico, R. Aalizadeh, N. S. Thomaidis, D. C. Thompson, V. Vasiliou and K. Veselkov, Hum. Genom., 2023, 17, 57 CrossRef CAS PubMed.
- G. C. Román, R. E. Jackson, J. Reis, A. N. Román, J. B. Toledo and E. Toledo, Rev. Neurol., 2019, 175, 705–723 CrossRef PubMed.
- Regulation (EC) No. 396/2005, OJ L 70, 16.3 2005, pp. 1–3857 Search PubMed.
- F. Zincke, A. Fischer, A. Kittelmann, C. Kraus, R. Scholz and B. Michalski, First update of the EU database of processing factors for pesticide residues, EFSA supporting publication, 2022, p. EN-7453, DOI:10.2903/sp.efsa.2022.EN-7453.
- European Commission, https://ec.europa.eu/food/plant/pesticides/eu-pesticides-database/start/screen/active-substances, last access on 02/4/ 2024.
- SANTE/2020/12830, Rev. 1, European Union, 24 February 2021 Search PubMed.
- H. V. Botitsi, S. D. Garbis, A. Economou and D. F. Tsipi, Mass Spectrom. Rev., 2011, 30, 907–939 CrossRef CAS PubMed.
- M. Hernández-Mesa and D. Moreno-González, Separations, 2022, 9, 148 CrossRef.
- P. Stefanelli, T. Generali, D. A. Barbini and S. Girolimetti, Curr. Trends Anal. Bioanal. Chem., 2018, 3(1), 95–107 Search PubMed.
- K. Takim and M. E. Aydemir, Toxics, 2022, 11, 34 CrossRef.
- A. Bajoub, T. Pacchiarotta, E. Hurtado-Fernández, L. Olmo-García, R. García-Villalba, A. Fernández-Gutiérrez, O. A. Mayboroda and A. Carrasco-Pancorbo, J. Chromatogr. A, 2016, 1428, 267–279 CrossRef CAS PubMed.
- L. Olmo-García, N. Kessler, H. Neuweger, K. Wendt, J. Olmo-Peinado, A. Fernández-Gutiérrez, C. Baessmann and A. Carrasco-Pancorbo, Molecules, 2018, 23, 2419 CrossRef PubMed.
- L. Olmo-García, K. Wendt, N. Kessler, A. Bajoub, A. Fernández-Gutiérrez, C. Baessmann and A. Carrasco-Pancorbo, Eur. J. Lipid Sci. Technol., 2019, 121, 1800336 CrossRef.
- M. D. H. Prodhan, S. N. Alam and M. Jalal Uddin, in Pesticide Residue in Foods, ed. M. S. Khan and M. S. Rahman, Springer International Publishing, Cham, 2017, pp. 135–145 Search PubMed.
- S. Barrek, O. Paisse and M.-F. Grenier-Loustalot, Anal. Bioanal. Chem., 2003, 376, 355–359 CrossRef CAS PubMed.
- N. Chamkasem and T. Harmon, J. Regul. Sci., 2015, 3, 16–35 Search PubMed.
- J. Robles-Molina, B. Gilbert-López, J. F. García-Reyes and A. Molina-Díaz, J. Chromatogr. A, 2017, 1517, 108–116 CrossRef CAS PubMed.
- A. Agüera, A. B. Martínez-Piernas and M. C. Campos-Mañas, in Applications in High Resolution Mass Spectrometry, Elsevier, 2017, pp. 59–82 Search PubMed.
- N. Rousis, M. Denardou, N. Alygizakis, A. Galani, A. Bletsou, D. Damalas, N. Maragou, K. Thomas and N. Thomaidis, Toxics, 2021, 9, 260 CrossRef CAS.
- Z. He, Y. Xu, L. Wang, Y. Peng, M. Luo, H. Cheng and X. Liu, Food Chem., 2016, 196, 1248–1255 CrossRef CAS PubMed.
- E. L. Schymanski, J. Jeon, R. Gulde, K. Fenner, M. Ruff, H. P. Singer and J. Hollender, Environ. Sci. Technol., 2014, 48, 2097–2098 CrossRef CAS PubMed.
- P. Gago-Ferrero, A. A. Bletsou, D. E. Damalas, R. Aalizadeh, N. A. Alygizakis, H. P. Singer, J. Hollender and N. S. Thomaidis, J. Hazard. Mater., 2020, 387, 121712 CrossRef CAS.
- D.-X. Li, L. Gan, A. Bronja and O. J. Schmitz, Anal. Chim. Acta, 2015, 891, 43–61 CrossRef CAS PubMed.
- J. Fang, H. Zhao, Y. Zhang, M. Lu and Z. Cai, Trends Environ. Anal. Chem., 2020, 25, e00076 CrossRef CAS.
- R. D. O. Silva, M. G. G. De Menezes, R. C. De Castro, C. D. A. Nobre, M. A. L. Milhome and R. F. Do Nascimento, Food Chem., 2019, 297, 124934 CrossRef PubMed.
- T. Portolés, J. V. Sancho, F. Hernández, A. Newton and P. Hancock, J. Mass Spectrom., 2010, 45, 926–936 CrossRef PubMed.
- M. L. Gómez-Pérez, P. Plaza-Bolaños, R. Romero-González, J. L. Martínez Vidal and A. Garrido Frenich, J. Am. Soc. Mass Spectrom., 2014, 25, 899–902 CrossRef PubMed.
- P.-T. Jiang, K.-L. Wu, H.-T. Wang and S.-F. Chen, J. Food Drug Anal., 2022, 30, 163–171 CrossRef PubMed.
- L. Olmo-García, A. Bajoub, A. Fernández-Gutiérrez and A. Carrasco-Pancorbo, Talanta, 2016, 150, 355–366 CrossRef.
- C. Sales, M. I. Cervera, R. Gil, T. Portolés, E. Pitarch and J. Beltran, Food Chem., 2017, 216, 365–373 CrossRef CAS PubMed.
- CEN, Foods of Plant Origin—Determination of Pesticide Residues Using GC-MS and/or LC-MS/MS Following Acetonitrile Extraction/Partitioning and Clean-Up by Dispersive SPE—QuEChERS-Method; European Standard EN 15662:2008 Search PubMed.
- D. E. Damalas, S. Kokolakis, A. Karagiannidis, N. Alygizakis and N. Thomaidis, S65 | UATHTARGETSGC | University of Athens GC-APCI-HRMS Target List, Zenodo, 2020, DOI:10.5281/zenodo.3753372.
- Hellenic Republic, Ministry of Rural Development and Food, https://1click.minagric.gr/oneClickUI/frmFytoPro.zul, last access on 02/04/ 2024.
- K. Varikou, K. M. Kasiotis, E. Bempelou, E. Manea-Karga, C. Anagnostopoulos, A. Charalampous, N. Garantonakis, A. Birouraki, F. Hatjina and K. Machera, Insects, 2020, 11, 855 CrossRef.
- E. Fuentes, M. E. Báez and J. Díaz, Food Addit. Contam.: Part B, 2010, 3, 101–107 CrossRef CAS PubMed.
- A. P. Economopoulos, N. Avtzis, G. Zervas, J. Tsitsipis, G. Haniotakis, G. Tsiropoulos and A. Manoukas, Z. Angew. Entomol., 1977, 83, 201–215 CrossRef.
- Commission implementing regulation (EU) 2018/555 of 9 April 2018 concerning a coordinated multiannual control programme of the Union for 2019, 2020 and 2021 to ensure compliance with maximum residue levels of pesticides and to assess the consumer exposure to pesticide residues in and on food of plant and animal origin, Official Journal of the European Union, p. L 92/6-18 Search PubMed.
- European Food Safety Authority (EFSA), L. C. Cabrera, G. Di Piazza, B. Dujardin and P. M. Pastor, EFSA J., 2023, 21(4), 7939 Search PubMed.
- European Chemical Agency, https://echa.europa.eu/substance-information/-/substanceinfo/100.111.286, last access on 02/4/ 2024.
- M. Kamata, Y. Matsui and M. Asami, Sci. Total Environ., 2020, 744, 140930 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ay00181h |
| This journal is © The Royal Society of Chemistry 2024 |