
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of a generic sample preparation method using dispersive liquid–liquid microextraction for the monitoring of leachable compounds in hospital pharmacy-prepared prefilled drug products†
William
Bello
 abcd,
Julian
Pezzatti
a,
Serge
Rudaz
cde and
Farshid
Sadeghipour
*abcd
abcd,
Julian
Pezzatti
a,
Serge
Rudaz
cde and
Farshid
Sadeghipour
*abcd
aPharmacy Department, Lausanne University Hospital, Switzerland. E-mail: farshid.sadeghipour@unige.ch; farshid.sadeghipour@chuv.ch
bCenter for Research and Innovation in Clinical Pharmaceutical Sciences, Lausanne University Hospital, University of Lausanne, Switzerland
cSchool of Pharmaceutical Sciences, University of Geneva, CMU-Rue Michel Servet 1, 1211, Geneva 4, Switzerland
dInstitute of Pharmaceutical Sciences of Western Switzerland, University of Geneva, University of Lausanne, Switzerland
eSwiss Center of Applied Human Toxicology (SCATH), Basel, Switzerland
First published on 13th February 2024
Abstract
Performant sample preparation is mandatory in any leachable study to clean and preconcentrate analytes within the sample to offer the best possible extraction recovery as well the best precision for any given substance. The aim consists in developing a sample preparation method for hospital pharmacy-prepared drug products such as long-term storage prefilled syringes, vials and IV bags for the screening of leachable compounds. The Quality Control Laboratory of the Pharmacy of the Lausanne University Hospital (Switzerland) has developed a time- and cost-effective, highly sensitive, robust, and fast method using liquid chromatography coupled with high-resolution mass spectrometry (LC-HRMS) for the analysis of 205 plastic additives. An innovative setup, based on postcolumn infusion (PCI) using 2% ammonium hydroxide in methanol was used to boost the signal intensity of the analytes in MS detection. A database for extractable and leachable trace assessment (DELTA) was built to assist in the screening process of 205 plastic packaging-related compounds. The development of the sample preparation was based on 33 plastic additive candidates in different hospital pharmacy compounding solutions, and their extraction recovery rates as well as their relative standard deviation were taken into consideration. In conclusion, the developed DLLME was assigned with ultrasound assistance and triple extraction, which brought about extraction recovery rates between 67% and 92%, a good RSD <10%, and a preconcentration factor of 50×. Therefore, DLLME could be considered suitable for the semiquantitative screening of leachable additives in simple hospital pharmacy-prepared prefilled drug products.
Introduction
Prefilled drug products are very practical in any hospital environment. They can be timesaving and have a positive impact on the reduction of preparation errors and microbial contamination. In case of emergencies, such prefilled drug products could be given at a moment's notice, which could be beneficial for patient treatment.1–5 They come in different formats, such as syringes, IV bags and vials.5,6 These containers can be made of different polymer materials, such as polypropylene, high-density and low-density polyethylene, ethylene vinyl acetate, cyclic olefin polymer and copolymers. All these materials possess many attributes, such as physical, chemical and biological resistance, thanks to the presence of a series of different plastic additives, such as antioxidants, UV stabilizers, lubricants, and plasticizers.7–10In a hospital setting, using the best quality plastic material is essential for patient care and to comply with Good Manufacturing Practice (GMP) regulations such as FDA and EMEA standards.11–13 A good quality plastic material would be less susceptible to leaching of additives in drug solutions, as demonstrated by extractable and leachable tests carried out by industries and independent laboratories. However, leaching of polymer-related compounds (polymer additives, processing aids, material degradants, processing residuals, and their breakdown products) into the surrounding solution can nonetheless occur due to long-term storage regardless of the quality of the plastic material. Many of these compounds could significantly alter the active pharmaceutical ingredient (API) and/or excipients, with different chemical reactions or interactions, such as oxidation and hydrolysis, which could lead to therapeutic failure or toxicity.9,10 Drafts and guidelines coming from the United States Pharmacopoeia (USP), International Council for Harmonization (ICH), International Organization for Standardization (ISO) and nongovernmental organizations such as the Product Quality Research Institute (PQRI) help industries perform extractable and leachable experiments. It is a legal requirement for all industries to perform leachable studies on a drug product containing a drug matrix to evaluate content-container interactions.14–19 In a hospital pharmacy, when prefilled drug products are being prepared in situ, the screening and semiquantitation of plastic additives in drug products are not mandatory. However, for patient safety, it is of paramount importance to screen leachable compounds and assess their presumptive toxicology. Hospital pharmacy-prepared prefilled drug products for long-term storage are often compounded with inappropriate use of medical devices for a specific population that is made of frail and/or vulnerable patients such as preterm, neonates, children and even chronically ill adults. Treatments involving frequent administrations, sometimes in the presence of complex formulations, are also risk factors for the presence of potentially toxic leachates. Therefore, a screening system for the identification and semiquantitation of nonvolatile compounds was built to promote the monitoring of leachable compounds in these particular cases of prefilled drug products.10,20 To perform analyses on drug products compounded in hospital pharmacies, a generic and reliable sample preparation method is extremely important to take into consideration the influence of drug matrices and enable a considerable enrichment of leachates with the promotion of medium to high recovery rates.
Considering that the matrix of drug products is often an aqueous solution (e.g., normal saline, 5% glucose, and salt buffers at different pH values), various sample preparations exist, such as liquid–liquid extraction (LLE), solid–liquid extraction (SLE), QuEChERS and solid phase extraction (SPE). LLE is mentioned as a reference sample preparation method for leachable studies by the United States Pharmacopoeia (USP) and PQRI.9,10,15,16 Despite its ease of use, there are two main drawbacks regarding LLE, which are the high consumption of large volumes of organic solvent and manual handling causing the formation of emulsions.21 On the other hand, dispersive liquid–liquid microextraction (DLLME), a microextraction method, could be an interesting alternative to both SPE and LLE because of its simplicity, high throughput, high recovery rates and high enrichment factor.22–26 DLLME is based on the use of a suitable extraction solvent injected at high velocity, i.e., a few microlitres of a high-density organic solvent (i.e., dichloromethane, chlorobenzene, chloroform or carbon disulfide) and a dispersion solvent that is highly miscible with the aqueous phase of the sample (i.e., methanol, acetonitrile or acetone).27
In this article, the development of a generic sample preparation method via DLLME is presented and evaluated to help the user identify and estimate the concentration of plastic additives for leachable studies via a LC-HRMS screening platform and an in-house database (DELTA).10 This sample preparation was established thanks to a set of 33 representative compounds with a log P ranging from approximately 1 to 20. Eventually, the performance of the method was evaluated on different drug-related matrices (normal saline, 5% glucose, 10% glucose, pH 2.5 and 9.5 buffer solution) for extraction recovery and retention time variation caused by matrix effects. Last but not least, a long-term stored prefilled IV bag containing parenteral nutrition will be presented as a sample preparation application of DLLME.
Experimental
Reagents and materials
Diphenyl phosphate, bisphenol S, butylated hydroxyanisole, bisphenol F, 4-cumylphenol, bisphenol E, butylated hydroxytoluene, 3,5-di-tert-butyl-4-hydroxyanisole, bisphenol B, 3,5-di-tert-butyl-4-hydroxybenzoic acid, 2,4,6-tri-tert-butylphenol, 3-(3,5-di-tert-butyl-4-hydroxyphenyl)propanoic acid, bumetrizole, octabenzone, 2,2′-methylenebis(6-tert-butyl-4-methylphenol), bisphenol M, 2-(2H-benzotriazol-2-yl)-4,6-ditertpentylphenol, 4,4′-sulfanediylbis(2-tert-butyl-5-methylphenol), 2,2′-methanediylbis(6-tert-butyl-4-ethylphenol), octyl 3-(3,5-di-tert-butyl-4-hydroxyphenyl)propanoate, alpha-tocopherol, 1,2-bis(3,5-di-tert-butyl-4-hydroxyhydrocinnamoyl)hydrazine, triphenyl phosphate, oleamide, dilauryl 3,3′-thiodipropionate, 1,3,5-tris[4-hydroxy-3,5-bis(2-methyl-2-propanyl)benzyl]-1,3,5-triazinane-2,4,6-trione, 1,3,5-trimethyl-2,4,6-tris(3,5-di-tert-butyl-4-hydroxybenzyl)benzene, pentaerythritol tetrakis(3-(3,5-di-tert-butyl-4-hydroxyphenyl)propionate), bisphenol A-d16, and bis(2-ethylhexyl)phthalate-3,4,5,6-d4, 2,4-di-tert-butyl-6-(5-chloro-2H-1,2,3-benzotriazol-2-yl)phenol were obtained from Sigma Aldrich® (Buchs, Switzerland) and 3,3-bis(3-tert-butyl-4-hydroxyphenyl)ethylene butyric acid ester was obtained from the Council of Europe (EDQM, Strasbourg, France) and were used for the development of the sample preparation methods. For the mobile phase, MS-grade water and MS-grade methanol were purchased from Biosolve® (Dieuze, France). Five internal standards were used for the development of the sample preparation method, i.e., bis(2-ethylhexyl)phthalate-3,4,5,6-d4, bisphenol M, 4,4′-sulfanediylbis(2-tert-butyl-5-methylphenol), 2,4-di-tert-butyl-6-(5-chloro-2H-1,2,3-benzotriazol-2-yl)phenol and bisphenol A-d16. For more information, Table S1 is presented in the ESI† containing physical–chemical properties as well as chromatographic and mass spectral data. For the postcolumn infusion installation, 25% ammonium hydroxide (LC-MS quality) was purchased from Merck® (Gygli, Switzerland).For the development of the DLLME sample preparation, Cellstar® Greiner Bio-One polypropylene tubes (Huberlab®, Aesch, Switzerland) and Brand® glass tubes (Wertheim, Germany) were purchased for the experiment. Glass syringes with luer-lock stainless steel needles (1 and 5 mL) were procured from Hamilton® (Nevada, USA). The following solvents were used for the development/optimization of the dispersion and extraction solvents: methanol, acetonitrile and acetone from Biosolve® (Dieuze, France) and dichloromethane, chloroform, 1,2-dichloroethane, chlorobenzene, carbon tetrachloride and carbon disulfide from Sigma Aldrich® (Buchs, Switzerland). Four-millilitre glass vials were purchased from BGB Analytic (Boeckten, Switzerland). Since this experiment is dealing with leachable compounds, it is crucial to place any liquid solvent in glass containers to avoid further plastic additive contamination.
Different representative materials (matrices) were prepared for the evaluation of the sample preparation matrix effect. They were carefully fashioned to avoid any plastic additive contamination. The raw materials used were as follows: sodium chloride, glucose monohydrate, hydrochloric acid, potassium chloride, monobasic sodium phosphate, dibasic sodium phosphate, and sodium hydroxide, provided by Merck® (Gygli, Switzerland). The aqueous solutions were selected based on the drug matrices used in the drug product inventories of the pharmacy department of the Lausanne University Hospital and are as follows: sodium chloride 0.9%, glucose 5% and 10%, and pH 2.5 and pH 9.5 buffers, stored in glass containers to minimize plastic additive contamination.
Postcolumn infusion (PCI)
A Chemyx® Fusion 100T syringe pump (TX, USA) was used, along with a 10 mL glass syringe (Hamilton, Nevada, USA) containing a concentration of 2% ammonium hydroxide in methanol. The solution was then infused into the MS source via a stainless tee-piece. After performing the analysis, a cleaning step by infusing equal parts of MS-grade water and methanol was performed.Liquid chromatography and mass spectrometry conditions
A Thermo Scientific™ ultrahigh-performance liquid chromatography instrument Vanquish Horizon (Thermo Scientific™, MA, USA) was hyphenated to a Thermo Scientific™ Orbitrap Q Exactive mass spectrometer (Thermo Scientific™, MA, USA) equipped with a heated electrospray ionization (HESI-II) source. The temperature in the sample chamber was set at 10 °C during analyses, and a 10 μL volume was injected. LC experiments were performed on a Waters™ Acquity™ BEH Phenyl column (100 × 2.1 mm, 1.7 μm) (Waters™, Milford, MA, USA) and the corresponding VanGuard precolumn. The flow rate was set at 0.2 mL min−1, and the column temperature was 60 °C. Mobile phase A was pure water, and mobile phase B was absolute methanol. The gradient profile used was as follows: a linear increase from 70% B to 85% B in 6 minutes, followed by an increase to 95% B in 4 minutes and another increase to 100% B in 2 minutes, holding at 100% B for 4 minutes, before returning back at 70% B in 0.1 minutes and re-equilibrating the column for 9 minutes.For the HESI II parameters, the sheath gas flow rate and auxiliary gas flow rate were programmed at 30 and 5 arbitrary units, respectively. The capillary temperature was 275 °C, and the auxiliary heater temperature was 290 °C. Analytes were scanned at both polarities, with a positive ion spray voltage of 3 kV and a negative ion spray voltage of 2.7 kV.
Parallel-reaction monitoring (PRM) was used at a mass resolution of 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500, at an AGC target of 2 × 105, using a maximum filling time of the C-trap of 50 ms. A normalized collision energy was set at 10%. All chromatograms were obtained using an m/z tolerance of 5 ppm. An isolation window of 1 m/z was programmed without an isolation offset and without defining the maximum number of precursor ions to be multiplexed. Mass calibration was performed once a week at both polarities using a Pierce® Velos ESI Ion Calibration standard mixture (Thermo Scientific™, MA, USA). For positive ion calibration, the mix consisted of n-butylamine, caffeine, MRFA (peptide of the Met-Arg-Ala acetate salt) and Ultramark 1621, and for negative ion calibration, it contained sodium dodecyl sulfate, sodium taurocholate and Ultramark 1621. MS Tune 2.8 (Thermo Scientific™, MA, USA) was used to control the instrument, and Chromeleon™ 7.2.7 (Thermo Scientific™, MA, USA) was employed to acquire data.
500, at an AGC target of 2 × 105, using a maximum filling time of the C-trap of 50 ms. A normalized collision energy was set at 10%. All chromatograms were obtained using an m/z tolerance of 5 ppm. An isolation window of 1 m/z was programmed without an isolation offset and without defining the maximum number of precursor ions to be multiplexed. Mass calibration was performed once a week at both polarities using a Pierce® Velos ESI Ion Calibration standard mixture (Thermo Scientific™, MA, USA). For positive ion calibration, the mix consisted of n-butylamine, caffeine, MRFA (peptide of the Met-Arg-Ala acetate salt) and Ultramark 1621, and for negative ion calibration, it contained sodium dodecyl sulfate, sodium taurocholate and Ultramark 1621. MS Tune 2.8 (Thermo Scientific™, MA, USA) was used to control the instrument, and Chromeleon™ 7.2.7 (Thermo Scientific™, MA, USA) was employed to acquire data.
Standard solution preparation for the development of the sample preparation using DLLME
A stock solution containing the complete set of 33 compounds, as described in Section 2.1, at 100 μg mL−1 was first prepared by weighing 10 mg of each analyte and dissolving them in 100 mL of MeOH. The stock solution was then diluted 1000× in MeOH to reach a concentration of 100 ng mL−1 and further diluted 50× with H2O/MeOH (1:1) to reach a concentration of 2 ng mL−1. This final concentration was used for a working solution to test the performance and enrichment factor of the sample preparation.
Preparation of an internal standard cocktail mix for the official DLLME sample preparation method
A stock solution containing all five internal standards (bis(2-ethylhexyl)phthalate-3,4,5,6-d4, bisphenol M, 4,4′-sulfanediylbis(2-tert-butyl-5-methylphenol), 2,4-di-tert-butyl-6-(5-chloro-2H-1,2,3-benzotriazol-2-yl)phenol and bisphenol A-d16) at 100 μg mL−1 was first prepared by weighing 10 mg of each compound and dissolving them in 100 mL of MeOH. The stock solution was then diluted 1000× in H2O:MeOH (1:1) to reach a concentration of 100 ng mL−1. This final concentration was used as a working solution to spike samples and blanks with 0.25 mL prior to DLLME sample preparation.
Official DLLME sample preparation method
Sample preparation was performed via a DLLME method. A sample volume of 10 mL was transferred into a 15 mL glass vial. A total of 0.25 mL of the test mixture containing only the five internal standards at 100 ng mL−1 was injected into the sample and vortexed before sample preparation. A mixture of 2 mL of acetone and 0.35 mL of 1,2-dichloroethane was then rapidly injected into the sample via a 2.5 mL Hamilton® glass syringe to obtain a microemulsion. The samples were then sonicated in an ultrasonic bath for 5 min and centrifuged for 10 min at 3,500×g. After centrifugation, the sedimented phase was extracted via a 1 mL Hamilton® glass syringe into a small glass vial. A second dispersion was performed by injecting 0.35 mL of 1,2-dichloroethane again into the sample. After this, they underwent a 5 min ultrasonication followed by a 5 min centrifugation, as explained before, and then the sediment phase was extracted, which was then transferred into the same vial. A third and final dispersion and extraction was performed again with 0.35 mL of 1,2-dichloroethane. Once all microextraction samples were collected, they were evaporated under nitrogen and reconstituted in 0.2 mL of H2O:MeOH (1:1) before analysis. The whole procedure is illustrated in Fig. S1 in the ESI.†
Results & discussion
Choice of sample preparation method
The screening of leachable compounds in aqueous solutions is often impacted by very low concentrations, often below the limit of detection (LOD) of analytes. An optimal sample preparation is therefore crucial to isolate and enrich analytes to facilitate detection. Such sample preparation should also remove any unwanted interferences mainly related to polar substances such as salts and sugars. They are excipients commonly found in parenteral hospital pharmacy-prepared drug products.Numerous sample preparations were looked at as candidates for the setup of sample enrichment of plastic additives. PQRI guidelines consider liquid–liquid extraction (LLE) as the gold standard.9,10 However, there are many disadvantages of LLE, such as low to moderate extraction recoveries, high consumption of halogenated solvents and relatively long and tedious procedures, such as triple extractions.27 The collected halogenated solvents need to be evaporated to enable an appropriate sample preconcentration factor. Halogenated solvents have to be tested regarding plastic additive contamination before their LLE application. The least contaminated ones are usually selected. Numerous scientific articles have discussed other sample preparations, such as solid phase extraction (SPE), which is considered an easy, efficient, and robust sample preparation technique that enables high analytical throughput. However, plastic-based cartridges and frits as well as other accessories could be deemed inappropriate for application in plastic additive analysis due to possible plastic additive contamination with regard to screening applications and not for the absolute quantitation of a small range of plastic additives.28
In this work, dispersive liquid–liquid microextraction (DLLME) was developed, and its parameters were investigated for sample preparation. The mechanism of DLLME is similar to that of LLE. Two different organic solvents are used, i.e., a dispersion solvent, which enables the dispersion of the extraction solvent into tiny droplets, forming a microemulsion in the process. This phenomenon increases the transfer of analytes into the organic phase by increasing the surface area contact with the organic extraction phase. The main advantage of this method is the use of a microvolume of organic solvents in comparison with LLE. Moreover, extraction recovery was often found to be between moderate and high depending on the affinity of the dispersion and extraction solvent combination with the candidate compounds. This method is becoming popular for the analysis of pesticides, plastic additives and pharmaceutically active compounds in water and food samples, although it can be used for biological samples such as plasma and urine.29–34 On the other hand, two issues must be considered: the method could be tedious at some points, similar to LLE, and it could bring about a narrow extraction range for certain classes of compounds. Some advantages that could make this method suitable for plastic additive analysis are the number of plastic-based accessories that could be kept to a minimum. A thorough investigation of the dispersion and extraction solvents as well as the critical DLLME parameters, i.e., sample sonication and centrifugation will be presented in the present article.
Selection of plastic additive candidates for the development of sample preparation
The 33 representative analytes were selected in reference to the article to build a generic sample preparation strategy meant to clean and enrich leached plastic additives coming from diverse samples of different hospital pharmacy-based matrices.10 The list of compounds consists of 28 plastic additive compounds and 5 internal standards, including plasticizers, antioxidants, UV stabilizers, UV absorbers, monomers, degradants and complex compounds. These compounds were selected because they represent a wide range of plastic additive categories, with logP ranging from approximately 1 to 20, to evaluate the performances of the designed DLLME method as well as to estimate the extraction recovery of the listed compounds in the internal database (DELTA). A table containing their physical–chemical properties as well as chromatographic and MS information used in the optimization of the sample preparation can be found in Table S1 (ESI).†
Semiquantitative approach
For an absolute quantification system, an ideal range of extraction recovery of compounds would normally depend on the matrix of the sample and the application, demanding a stricter and more precise range of acceptance, as per ICH Q2 (R1) guidelines.35 However, the current analytical method used consists of a semiquantitative approach in which the aimed extraction recovery for all 33 compounds after DLLME extraction was set to be between 50 and 200%. These values are more adapted for the screening of all 205 compounds according to the internal database DELTA.10 This approach is currently being used in industrial E&L, as needed by law to perform wide untargeted screening of plastic additives during E&L studies.36,37DLLME extraction developments
To optimize DLLME performance and efficiency, one must consider a series of parameters, such as the selection of the dispersion solvent, extraction solvent, physical assistance (with/without, i.e., vortexing, ultrasonication or a combination of both) at different durations and centrifugation times and speeds. The evaluated dispersion solvents were methanol, acetonitrile and acetone. The evaluated extraction solvents were dichloromethane, dichloroethane, chloroform, carbon disulfide, chlorobenzene and carbon tetrachloride. Different volumes of dispersion and extraction solvents were investigated, i.e., dispersion: 0.25, 0.5, 1, 1.5, 2, and 2.5 mL; extraction: 0.05, 0.2, 0.35, 0.5, 1, and 2 mL for a sample volume of 10 mL. All variables and the level of design are presented below in Table 1.| Dispersion solvents | Methanol | Acetonitrile | Acetone | |||
| Volume of dispersion solvents (μL) | 0.25 | 0.5 | 1 | 1.5 | 2 | 2.5 |
| Extraction solvents | Dichloromethane | Dichloroethane | Chloroform | Chlorobenzene | Carbon tetrachloride | |
| Volume of extraction solvents (μL) | 0.05 | 0.2 | 0.35 | 0.5 | 1 | 2 |
| Physical assistance | No assistance | Vortex | Ultrasound | Vortex + ultrasound | ||
| Vortex time (minutes) | N/A | 3 | 5 | 10 | ||
| Ultrasound time (minutes) | N/A | 3 | 5 | 10 | ||
| Centrifugal time (minutes) | N/A | 3 | 5 | 10 | ||
| Centrifugal speed (rpm) | 2000 | 2500 | 3000 | 3500 | ||
A one factor at a time approach (OFAT) was applied to evaluate the best possible parameters for the development of the sample preparation. The approach was applied in a step-by-step fashion by first determining the optimal extraction recovery produced by the combination of the dispersion and extraction solvents and their volumes. Once that was determined, the physical parameters, according to Table 1, were investigated to search for further improvements in extraction recovery. The final step would be to experiment on the concept of multiple extractions to further bring about optimization.
The performance parameters considered were the preconcentration factor (PF) and the extraction recovery (ER%), obtained via the following relations:32
 | (1) |
 | (2) |
The extraction recovery value of analytes via DLLME sample preparation is equal to PF multiplied by the ratio between the reconstituted volume from the evaporation of the sedimented phase and the volume of aqueous solution, multiplied by 100.
All experiments were performed with three replicate extractions (n = 3). All variables are known except for the concentration of analyte in the sedimented phase. The latter is obtained via calculation with the point-slope equation, which is compound specific. Before starting the development, one must ensure that the material used to house the sample as well as the solvents (extraction and dispersion) do not interfere with the experiment by cross-contamination of plastic additives.
Evaluation of dispersion solvents
The selection of an appropriate dispersion solvent for the development of a DLLME sample preparation is crucial. The solvent must be miscible with both the extraction solvent and the aqueous sample. Its purpose is to disperse the extraction solvent as droplets to promote a microemulsion. This phenomenon was observed when the dispersion and extraction solvents were rapidly injected via a syringe and a blunt needle in the aqueous solution. There is a rapid transfer of analytes from the aqueous sample to the droplets containing the extraction solvent. The dispersion solvent is crucial for the transfer of analytes because it could affect the solubility of analytes in the aqueous phase, as well as the distribution coefficient of analytes.34Different dispersion solvents were selected at different volumes according to Table 1 to evaluate their influence on the extraction recovery of compounds and were calculated according to eqn (2). According to the results shown in Fig. 1, acetone had the highest extraction recovery for most of the compounds. It was observed that upon increasing the volume of acetone, the extraction efficiency also increased. Acetone enabled a better dispersion of the extraction solvent in the aqueous sample. A well-formed microemulsion was clearly observed after the dispersion of the combined proportions of organic solvents. The best volume of acetone was established at 2 mL. The majority of candidate compounds showed maximal extraction recovery (%) except for very lipophilic compounds with logP values above 10 (Irganox 3114, Hostanox O3, tocopherol, Naugard DLTDP, Irganox 1330 and Irganox 1010). This was because the combination of acetone and dichloroethane did not seem to be the best fit due to the lack of lipophilicity of the extraction solvent. The optimal combination would be acetone and tetrachloromethane, enabling a better solubility of the relevant hyperlipophilic compounds (see the “Evaluation of extraction solvents” section). However, above 2 mL of acetone, the extraction recovery appeared to decrease. As the volume of acetone increased, so did the solubility of all lipophilic analytes in the aqueous sample, affecting the extraction efficiency negatively by decreasing the distribution coefficient of the analytes. Moreover, the miscibility of extraction solvent could also increase in the aqueous part of the sample, inducing a lowering of the volume of extraction solvent at the end of the DLLME process, diminishing the concentration of analytes in the extraction phase. At a higher volume of acetone, the microemulsion was no longer obtained and was short-lived, producing larger droplets upon dispersion of the extraction solvent and causing an aggregation of droplets into a separated phase. In contrast, acetone at low volume (below 2 mL) yielded suboptimal extraction recovery for all analytes. The extraction solvent injected into the sample with a low volume of acetone formed much larger extraction solvent droplets, which aggregated easily and caused phase separation. This would decrease the extraction surface area, hindering the transfer of analytes.
 | ||
| Fig. 1 Different dispersion solvents at a fixed volume of 2 mL tested for their performance in terms of extraction recovery (%) via DLLME for all 33 compounds (fixed volume of 0.35 mL of extraction solvent 1,2-dichloroethane). | ||
For the other dispersion solvents, methanol seemed to give the least optimal results of extraction recoveries. On the other hand, acetonitrile seemed to perform better than methanol for the extraction of UV stabilizers. Finally, acetone was selected as the dispersion solvent at a volume of 2 mL, which gave the best extraction recovery for the majority of representative test compounds.
Evaluation of extraction solvents
In DLLME, extraction solvents should exhibit specific physical–chemical properties such as water immiscibility and higher density than water, i.e., once centrifuged when possible. In this case, it is more practical to obtain the sediment phase. Therefore, high-density halogenated solvents are often selected as extraction solvents. They were tested at different volumes in combination with different dispersion solvents (see the previous section). Optimal extraction recoveries were obtained from 1,2-dichloroethane. This enabled a compromised optimal extraction for the overall compounds. Chlorobenzene and carbon tetrachloride resulted in better extraction recovery for very lipophilic compounds with logP values higher than 9–10, such as Hostanox® O3, Irganox® 3114, Irganox® 1330 and Irganox® 1010. This could be due to the logP values of both extraction solvents being greater than those of the other solvents. For dichloromethane, the microemulsion was not well formed, and a sediment phase was quickly reached in combination with acetone, which negatively affected the values of the extraction recovery. This was due to dichloromethane's low lipophilicity, affecting the affinity of compounds bearing higher logP. Finally, 1,2-dichloroethane was selected as the extraction solvent at a volume of 0.35 mL, which gave optimal extraction recoveries for the majority of analyte candidates.
To evaluate the efficiency of the extraction solvent, additional experiments using 2 mL of acetone as a constant were considered. As seen in Fig. 2, 1,2-dichloroethane was observed to give optimal results for the majority of the candidate compounds, except for molecules with low logP values, such as diphenyl phosphate, and very high logP values, such as Hostanox® O3 and Irganox® 1010. Consequently, the optimal DLLME extraction solvent was a mixture of acetone as the dispersion solvent and 1,2-dichloroethane as the extraction solvent at proportions of 2 mL and 0.35 mL, respectively, with recovery rates of 9–79% and a relative standard deviation (RSD) <10%. However, with the current DLLME solvent combination, the overall compounds still did not show the best possible results. More extraction recovery improvements will be discussed in the next section.
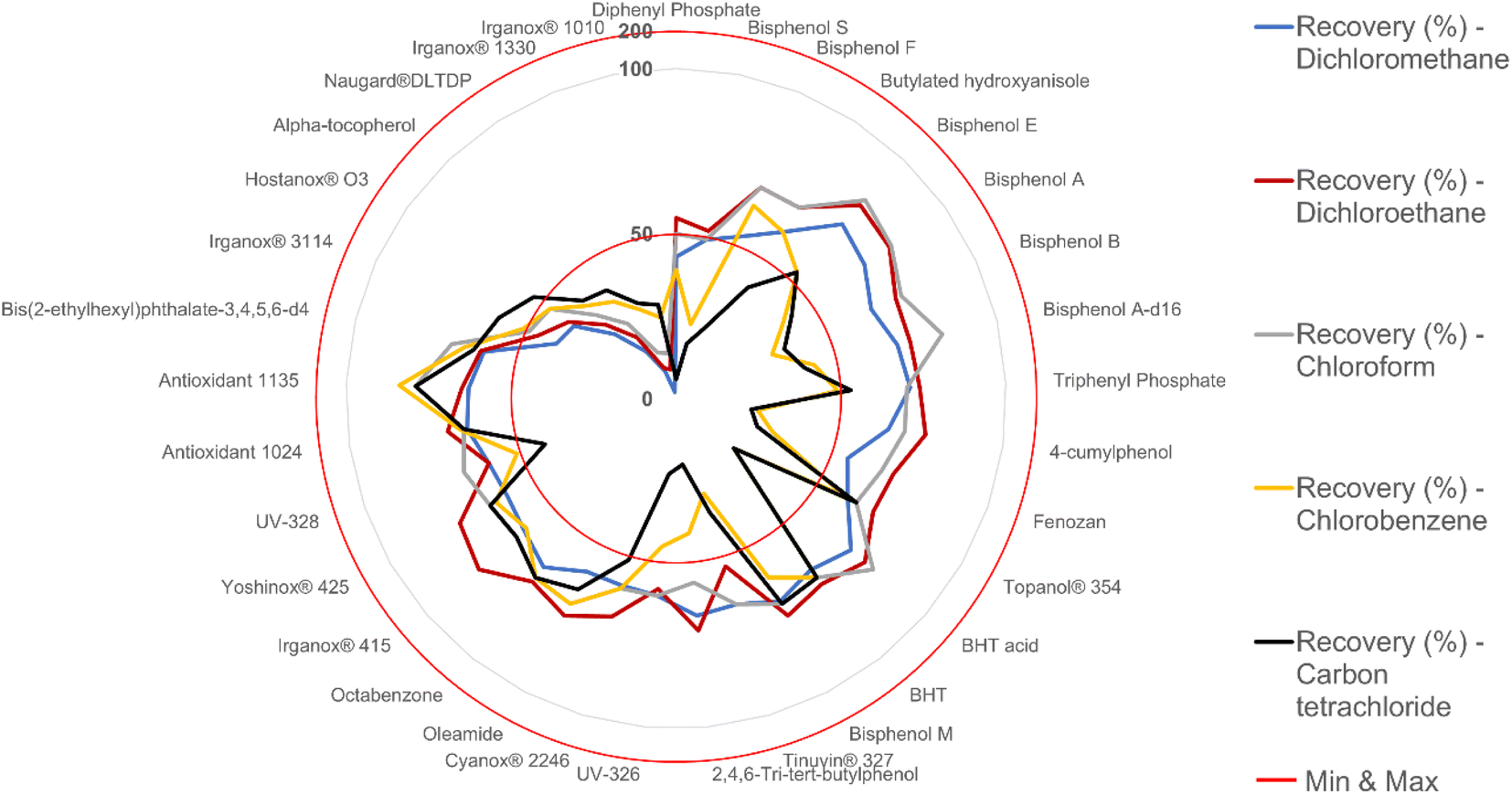 | ||
| Fig. 2 Different extraction solvents at a fixed volume of 0.35 mL tested for their performance in terms of extraction recovery (%) via DLLME for all 33 compounds (fixed volume of 2 mL of dispersion solvent acetone). | ||
Physical assistance
To improve the performance of DLLME, different physical assistance methods, i.e., vortexing and ultrasonication as well as a combination of both for different time durations (3, 5 and 10 minutes) were investigated. The results showed that using a ultrasonication bath for 5 min increased the recovery rates of the compounds of interest by approximately 10%. Ultrasonication breaks up larger microdroplets and disperses them further into smaller microdroplets, encouraging, in the process, better formation of microemulsions (homogeneous milky solution).Furthermore, vortex action alone and/or in combination with ultrasonication did not show any improvement. It is important to note that vortex action was executed before the sonication step. The vortex action did not seem to be fast enough to break apart droplets into microdroplets, which left them to aggregate and cause phase separation, hindering the transfer of analytes and negatively affecting the extraction recovery. Moreover, vortexing represents an additional step that compromises the analytical throughput of sample preparation. The results are presented in the histogram found in Fig. 3, showing ultrasonication enabling further extractions of compounds.
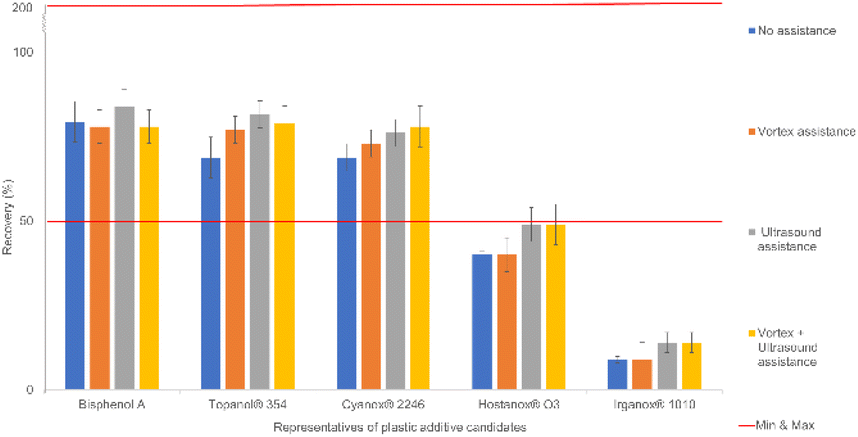 | ||
| Fig. 3 Different physical assistance methods for a duration of 5 minutes with 0.35 mL of extraction solvents tested for their performance in terms of extraction recovery (%) via DLLME for all 33 compounds (fixed volume of 2 mL of dispersion solvent acetone and 0.35 mL of extraction solvent 1,2-dichloroethane). | ||
Finally, the centrifugation parameters were also evaluated. A series of centrifugal forces as well as the duration of rotation were tested (3, 5 and 10 minutes at 2000, 2500, 3000 and 3500 rpm). It was observed that a maximum speed for a duration of 5 minutes was optimal.
Multiple solvent extractions
The concept of multiple extraction assays was tested with the latest optimal parameters of the DLLME established in the previous sections. To the best of our knowledge, this has never been attempted before. The idea of multiple solvent extraction came from LLE applications. Indeed, DLLME always underwent a single extraction step because the results would normally be optimal.29–34 According to the previous results, the DLLME extraction recovery did not seem optimal enough for a wide range of compounds. To increase the extraction recovery yield of the poorly extracted compounds, a multiple extraction step was set up to extend the range of extractable compounds. This approach consists of dispersing only the same volume of extraction solvent into a postextracted sample, which already contains acetone. The addition of a new volume of extraction solvent to the sample recreates the microemulsion, preparing it for another extraction process. The experiment was repeated again for a sequence of five maximum extractions to find the optimal extraction step and the best possible extraction recovery for analytes. Fifteen compounds were used for this experiment, i.e., butylated hydroxyanisole, bisphenol E, bisphenol A, bisphenol B, bisphenol A-d16, triphenyl phosphate, 4-cumylphenol, fenozan, Topanol® 354, BHT acid, BHT, bisphenol M, Tinuvin® 327, 2,4,6-tri-tert-butylphenol and UV-326. The results show that multiple extraction points increased the extraction recovery and reduced their relative standard deviations. It was also noted that for all fifteen compounds, at three serial extractions, an asymptotic curve was reached, and as this curve stabilized, so did the RSD. It was also verified that a negligible volume of acetone was consumed with each extraction solvent injection step since acetone is miscible with 1,2-dichloroethane. Only 0.03 mL of acetone was consumed after each extraction, and a total of 0.09 mL was consumed, which was considered negligible (<5% acetone lost). In conclusion, triple extraction was observed to be the best compromise in terms of recovery rates and analytical throughput. Some compounds demonstrated that two consecutive extractions may have been sufficient due to the negligible differences between the second and third extractions. However, the majority showed that a third extraction would greatly benefit the yield of poorly extracted compounds as well as compounds with high affinity with the DLLME organic solvents.To summarize, the final optimal parameters for the DLLME were 2 mL of acetone as the dispersion solvent and 0.35 mL of 1,2-dichloroethane as the extraction solvent for a 10 mL sample volume. After applying the solvents, a 5 min ultrasonication along with a 5-minute centrifugation at 3500 rpm were needed. The sediment phase at the tip of the tube was extracted with a glass syringe and transferred into a glass vial. This step was repeated 2 more times to obtain the optimal recovery rate for all 33 analyte candidates. As a summary, Fig. 4 describes the overall results of UA-DLLME in extraction recovery on candidate additives in water for injection. In the ESI, Fig. S1–S3† show the schematics of how the whole procedure is performed as well as chromatograms of all 33 compounds in both positive and negative modes. Moreover, Fig. S4 in the ESI† describes the results obtained from multiple extractions and the appearance of the asymptotic curve for all 5 representative sets of compounds, and vice versa, the decrease in the RSD in the form of a reverse asymptotic curve.
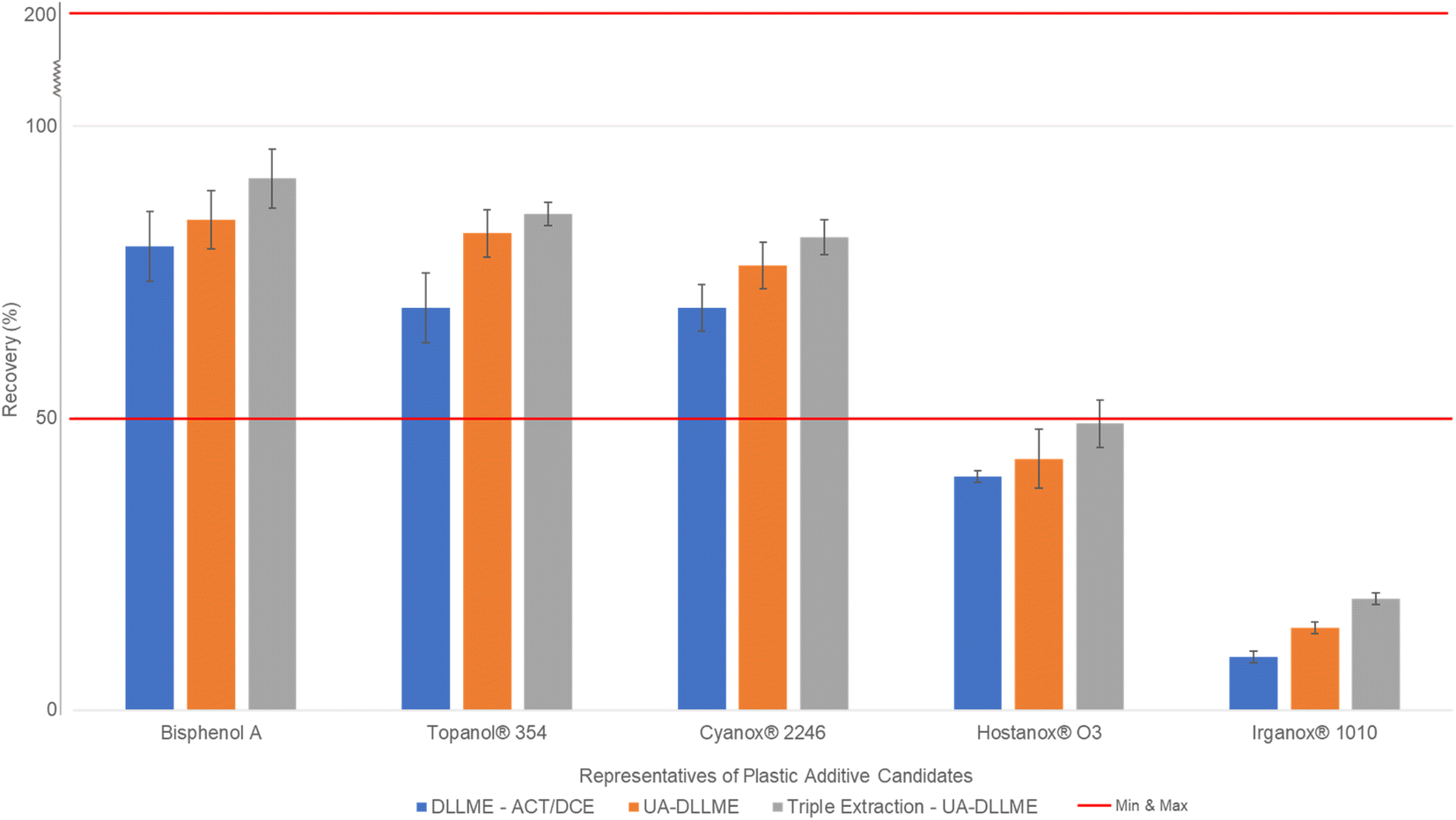 | ||
| Fig. 4 Histogram describing the overall results of UA-DLLME in terms of multiple development stages by using the candidate additives in water for injection. | ||
Matrix effect
In a hospital pharmacy setting, prefilled drug products are often made with different excipients to suit drug stability. An experiment was conducted on different common drug formulations, i.e., normal saline, 5 and 10% glucose solution, and buffer solution at pH 2.5, and pH 9.5. With the same test set of 33 compounds, different pharmaceutical matrices seemed to induce minor changes in the extraction recovery, with less than a 10% difference obtained. Multiple matrix solutions were compared to WFI. It was observed that solutions containing salts and other solutes did change the outcome of the recovery yield, whether it was positive or negative. The two aqueous solution matrices that gave the lowest extraction yield were 10% glucose and phosphate buffer at pH 9.5. For the first one, it was due to an increase in apparent viscosity, preventing as a result, a proper dispersion of droplets to form an optimal microemulsion. For the other matrix solutions, the decrease in extraction recovery was possibly due to the change in the molecular state since the majority of plastic additives are weak acids, possessing a phenolic-like structure and having a pKa of approximately 10–11. Therefore, at pH 9.5, these compounds would possess a proportion of molecules in their charged state. Hence, at this pH, 33% of bisphenol A possesses a negative charge on one of its phenol groups, transforming it into one of its ionized forms. For the other aqueous solutions, normal saline and acid buffer at pH 2.5 gave better extraction recovery (1.77–2.35 times higher compared to to WFI). The presence of the salt improved the extraction efficiency by reducing the solubility of the majority of the compounds, hence leading to the transfer of analytes in the microdroplets due to a much better distribution coefficient. This can be attributed to a salting out phenomenon, modifying the solubility of the analyte in the aqueous sample and promoting a better transfer of analytes to the organic phase.For the majority, the differences between water for injection and other matrices could be found to be less than 10%, and for the minority, some could be found between 10 and 20%. The matrices that happen to be above the 10% difference are 10% glucose and buffer solution at pH 9.5. Fig. 5 shows the different matrices influencing the extraction recoveries.
 | ||
| Fig. 5 Histogram describing the influence of different matrices on the extraction recovery (%) via UA-DLLME. | ||
Application of DLLME sample preparation in the semiquantitation of plastic additives in prefilled drug products for leachable studies
The aim of the sample preparation was to enable the isolation and enrichment of leachable compounds from hospital pharmacy-compounded prefilled drug products, stored for a defined period of time, corresponding to their maximum shelf lives. These analytes can be identified thanks to the developed database (DELTA).10 This sample preparation should be compatible with all compounds from the database. Therefore, they were tested for their extraction recovery (%), as observed in Fig. S5 and Table S2 in the ESI.† Several leachable compounds are not compatible with the developed sample preparation because some compounds are polar, and some are very lipophilic in nature. Upon testing the sample preparation on the list of compounds, it was observed that some compounds bearing logP values below 0 were not extracted at all, which is to be expected since they did not show any hydrophobic affinity with the halogenated extraction solvent. Moreover, this was also the case for compounds with very high logP values, starting from 11 and above, which could be due to the lack of lipophilic affinity with the DLLME combined extraction solvents. Extraction solvents such as 1,2-dichloroethane could only extract a range of lipophilic molecules but not highly apolar compounds. If tetracarbon chloride or chlorobenzene were to be selected as extraction solvents, the yield of compounds such as Irganox® 1010 or Naugard® DLTDP would have been favourably higher than that using the current solvent. However, other less lipophilic compounds, such as bisphenol A or diphenyl phosphate, would have had a poor extraction yield. Therefore, 5% of compounds outside the logP interval range of 0 and 11 possess low extraction recovery. However, this does not mean that 5% of the database serves no purpose. The excluded compounds could be included for extraction studies when using polar solvents such as acidic and basic solutions to extract polar compounds (logP < 0) and using very lipophilic solvents such as hexane and toluene to extract very lipophilic compounds (logP > 11). It is important to note that the semiquantitative approach brings about only a rough estimation of the concentration of targeted compounds. This means that the extraction recovery does not require absolute precision, and hence the wide extraction recovery range.
Moreover, it is important to note that this sample preparation will aid the operator in the sensitive detection of leachable compounds thanks to the fifty-fold enrichment factor. Therefore, this will render the instrumental LOD, LOQ and linear range of all listed compounds fifty times more sensitive.10
The overall experience of the sample preparation was fairly positive. It was developed for the preconcentration of identified additives as well as for their semiquantitation in hospital pharmacy-prepared prefilled aqueous drug products. This sample preparation would be most preferably applied for simple matrix prefilled drug products, since it provides low intervariability of extraction recovery of additives.
Conclusions
In this work, an original DLLME sample preparation protocol was established to perform leachable studies on hospital pharmacy-prepared prefilled drug products, such as long-term storage prefilled syringes, vials and IV bags, for the screening and semiquantitation of leachable compounds. Different parameters of the DLLME were investigated and compared in terms of performance for leachate recovery rates. Three dispersion solvents, five extraction solvents, vortexing, ultrasonication and multiple extraction procedures were investigated. Thirty-three plastic additive candidates were used to screen for optimal parameters in terms of extraction recovery. Extracted compound variability was assessed with the help of univariate data analysis via the relative standard deviation of the area of each additive. As a result, the selected optimal parameters were 2 mL acetone (dispersion solvent) and 0.35 mL 1,2-dichloroethane (extraction solvent). After this, the samples underwent a 5 min ultrasonication followed by a 5 min centrifugation (3500 rpm), and the sedimented phase was extracted. Finally, the extraction procedure was repeated three times in total. Moreover, recovery rates of the majority of the candidates were between 67 and 92%. This sample preparation appears to be eco-friendly due to the small consumption of halogenated solvent volume and allowed a preconcentration factor of 50. Various aqueous matrices were used to simulate the contents of different prefilled packages, such as 0.9% sodium chloride, 5% glucose, 10% glucose, and buffer solutions (pH 2.5 and 9.5), and an evaluation of the matrix effect was performed to establish which drug matrix solution was the most appropriate. As a result, less than 10% variation for normal saline solution and 5% for glucose is the most common matrix in prefilled drug products. However, a higher variability was observed for heavily salted and high glucose content matrices. The latter could affect the dispersion of the extraction solvent, which overall could affect the extraction recovery of analytes in a negative way. Finally, the developed DLLME procedure appears to be well suited to leachable experiments of hospital-prepared prefilled drug products.Author contributions
William Bello: conceptualization, investigation, methodology, formal analysis, data curation, visualization, writing – original draft, writing – reviewing and editing. Julian Pezzatti: conceptualization, methodology, supervision, writing – reviewing and editing. Serge Rudaz: conceptualization, methodology, writing – reviewing and editing, supervision, project administration. Farshid Sadeghipour: conceptualization, methodology, writing – reviewing and editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Makwana, B. Basu, Y. Makasana and A. Dharamsi, Prefilled syringes: An innovation in parenteral packaging, Int. J. Pharm. Invest., 2011, 1(4), 200–206, DOI:10.4103/2230-973X.93004.
- G. Sacha, J. A. Rogers and R. L. Miller, Pre-filled syringes: a review of the history, manufacturing and challenges, Pharm. Dev. Technol., 2015, 20(1), 1–11, DOI:10.3109/10837450.2014.982825.
- P. Bonnabry, Information Technologies for the Prevention of Medication Errors, Chimia, 2005, 59(6), 359 CrossRef CAS . Available from: https://www.chimia.ch/chimia/article/view/2005_359.
- S. Vrignaud, Resolution CM/Res(2016)2 and Centralised Intra Venous Additive Services (CIVAS): Challenges and Opportunities, Pharm. Technol. Hosp. Pharm., 2017, 2(3), 137–142, DOI:10.1515/pthp-2017-0023.
- P. Malik, M. Rangel and T. VonBriesen, Why the Utilization of Ready-to-Administer Syringes During High-Stress Situations Is More Important Than Ever, J. Infus. Nurs., 2022, 45(1), 27–36, DOI:10.1097/NAN.0000000000000451.
- L. Czuba, Application of Plastics in Medical Devices and Equipment, Handbook of Polymer Applications in Medicine and Medical Devices, 2014, pp. 9–19, DOI:10.1016/B978-0-323-22805-3.00002-5.
- E. J. North and R. U. Halden, Plastics and environmental health: the road ahead, Rev. Environ. Health, 2013, 28(1), 1–8, DOI:10.1515/reveh-2012-0030.
- L. Fishbein, Additives in synthetic polymers: an overview, Prog. Clin. Biol. Res., 1984, 141, 19–42 CAS.
- D. Paskiet, D. Jenke, D. Ball, C. Houston, D. L. Norwood and I. Markovic, The Product Quality Research Institute (PQRI) Leachables and Extractables Working Group Initiatives for Parenteral and Ophthalmic Drug Product (PODP), PDA J. Pharm. Sci. Technol., 2013, 67(5), 430–447, DOI:10.5731/pdajpst.2013.00936.
- W. Bello, J. Pezzatti, M. Berger-Gryllaki, S. Rudaz and F. Sadeghipour, Development of a generic approach for monitoring leachable compounds in hospital pharmacy-prepared prefilled plastic packaging by ultrahigh-performance liquid chromatography coupled to high-resolution mass spectrometry with postcolumn infusion, J. Pharm. Biomed. Anal., 2023, 236, 115640, DOI:10.1016/j.jpba.2023.115640.
- Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme. 2023-2027 Strategic Plan, PIC/S, Geneva, 2022, [cited 2023 Oct] available from: https://picscheme.org/docview/4768 Search PubMed.
- EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, https://health.ec.europa.eu/medicinal-products/eudralex/eudralex-volume-4_en, accessed 09 June 2023.
- Current Good Manufacturing Practice (CGMP) Regulations, https://www.fda.gov/drugs/pharmaceutical-quality-resources/current-good-manufacturing-practice-cgmp-regulations, accessed 17 June 2023.
- U.S. Food and Drug Administration, Guidance for Industry: Container Closure Systems for Packaging Human Drugs and Biologics, Center for Biologics Evaluation and Research, U.S. Department of Health and Human Services: Rockville, MD, 1999 Search PubMed.
- United States Pharmacopeia, General Chapter, 〈1663〉 Assessment of Extractables Associated with Pharmaceutical Packaging/Delivery Systems, USP-NF, United States Pharmacopeia, Rockville, MD Search PubMed.
- United States Pharmacopeia, General Chapter, 〈1664〉 Assessment of Drug Product Leachables Associated with Pharmaceutical Packaging/Delivery Systems, USP-NF, United States Pharmacopeia, Rockville, MD, 2022 Search PubMed.
- ISO, ISO 10993–18:2020 Biological Evaluation of Medical Devices - Part 18: Chemical Characterization of Medical Device Materials within a Risk Management Process, International Organization for Standardization (ISO), Geneva, Switzerland, 2020 Search PubMed.
- ISO, ISO 10993-13:2010 Biological Evaluation of Medical Devices Part 13: Identification and Quantification of Degradation Products from Polymeric Medical Devices, International Organization for Standardization (ISO), Geneva, Switzerland, 2010 Search PubMed.
- ISO, ISO 10993-17:2002 Biological Evaluation of Medical Devices Part 17: Establishment of Allowable Limits for Leachable Substances, International Organization for Standardization (ISO), Geneva, Switzerland, 2002 Search PubMed.
- Leachable/extractable issues in a pharmacy setting, https://hospitalpharmacyeurope.com/news/editors-pick/leachable-extractable-issues-in-a-pharmacy-setting/, accessed 11 October 2023.
- M. Tehranirokh, M. Van den Bronk, P. Smith, Z. Dai, K. Ragunathan, A. Muscalu, S. Mills, M. C. Breadmore and R. A. Shellie, Automated liquid-liquid extraction of organic compounds from aqueous samples using a multifunction autosampler syringe, J. Chromatogr. A, 2021, 1642, 462032, DOI:10.1016/j.chroma.2021.462032.
- S. Berijani, Y. Assadi, M. Anbia, M. R. Milani Hosseini and E. Aghaee, Dispersive liquid-liquid microextraction combined with gas chromatography-flame photometric detection. Very simple, rapid and sensitive method for the determination of organophosphorus pesticides in water, J. Chromatogr. A, 2006, 1123(1), 1–9, DOI:10.1016/j.chroma.2006.05.010.
- M. Rezaee, Y. Yamini and M. Faraji, Evolution of dispersive liquid-liquid microextraction method, J. Chromatogr. A, 2010, 1217(16), 2342–2357, DOI:10.1016/j.chroma.2009.11.088.
- Q. Andrew, C. Wayne and C. Damian, Dispersive Liquid-Liquid Microextraction in the Analysis of Milk and Dairy Products: A Review, J. Chem., 2016, 4040165, DOI:10.1155/2016/4040165.
- M. Rutkowska, K. Dubalska, P. Konieczka and J. Namieśnik, Microextraction techniques used in the procedures for determining organomercury and organotin compounds in environmental samples, Molecules, 2014, 19(6), 7581–7609, DOI:10.3390/molecules19067581.
- I. Notardonato, S. Passarella, A. Iannone, C. D. Fiore, M. V. Russo, C. Protano, M. Vitali and P. Avino, Comparison of Two Extraction Procedures, SPE and DLLME, for Determining Plasticizer Residues in Hot Drinks at Vending Machines, Processes, 2021, 9, 1588, DOI:10.3390/pr9091588.
- H. Yan and H. Wang, Recent development and applications of dispersive liquid-liquid microextraction, J. Chromatogr. A, 2013, 1295, 1–15, DOI:10.1016/j.chroma.2013.04.053.
- C. Pouech, F. Lafay, L. Wiest, R. Baudot, D. Léonard and C. Cren-Olivé, Monitoring the extraction of additives and additive degradation products from polymer packaging into solutions by multi-residue method including solid phase extraction and ultra-high performance liquid chromatography-tandem mass spectrometry analysis, Anal. Bioanal. Chem., 2014, 406(5), 1493–1507, DOI:10.1007/s00216-013-7551-4.
- H. Denghel and T. Göen, Dispersive liquid-liquid microextraction (DLLME) and external real matrix calibration for the determination of the UV absorber 2-(2H-benzotriazol-2-yl)-4,6-di-tert-pentylphenol (UV 328) and its metabolites in human blood, Talanta, 2021, 223(Pt 1), 121699, DOI:10.1016/j.talanta.2020.121699.
- A. Alves, G. Vanermen, A. Covaci and S. Voorspoels, Ultrasound assisted extraction combined with dispersive liquid-liquid microextraction (US-DLLME)-a fast new approach to measure phthalate metabolites in nails, Anal. Bioanal. Chem., 2016, 408(22), 6169–6180, DOI:10.1007/s00216-016-9727-1.
- I. Notardonato, S. Passarella, G. Ianiri, C. Di Fiore, M. V. Russo and P. Avino, Analytical Scheme for Simultaneous Determination of Phthalates and Bisphenol A in Honey Samples Based on Dispersive Liquid-Liquid Microextraction Followed by GC-IT/MS. Effect of the Thermal Stress on PAE/BP-A Levels, Methods Protoc., 2020, 3(1), 23, DOI:10.3390/mps3010023.
- C. K. Zacharis, I. Rotsias, P. G. Zachariadis and A. Zotos, Dispersive liquid-liquid microextraction for the determination of organochlorine pesticides residues in honey by gas chromatography-electron capture and ion trap mass spectrometric detection, Food Chem., 2012, 134(3), 1665–1672, DOI:10.1016/j.foodchem.2012.03.073.
- P. Fernández, M. Regenjo, A. M. Bermejo, A. M. Fernández, R. A. Lorenzo and A. M. Carro, Analysis of drugs of abuse in human plasma by dispersive liquid-liquid microextraction and high-performance liquid chromatography, J. Appl. Toxicol., 2015, 35(4), 418–425, DOI:10.1002/jat.3035.
- F. R. Mansour and M. A. Khairy, Pharmaceutical and biomedical applications of dispersive liquid-liquid microextraction, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1061–1062, 382–391, DOI:10.1016/j.jchromb.2017.07.055.
- ICH Harmonised Tripartite Guideline, Validation of Analytical Procedures Q2(R2), International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use, Geneva, Switzerland, 2022 Search PubMed.
- P. Christiaens, J. M. Beusen, P. Verlinde, J. Baeten and D. Jenke, Identifying and Mitigating Errors in Screening for Organic Extractables and Leachables: Part 2-Errors of Inexact Identification and Inaccurate Quantitation, PDA J. Pharm. Sci. Technol., 2020, 74(1), 108–133, DOI:10.5731/pdajpst.2018.009779.
- D. Jenke, Standardization of Chromatographic Screening Methods for Organic Extractables and Leachables by Managing Outcomes, PDA J. Pharm. Sci. Technol., 2023, 77(4), 329–338, DOI:10.5731/pdajpst.2021.012671.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ay02234j |
| This journal is © The Royal Society of Chemistry 2024 |