
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Field method for preservation of total mercury in waters, including those associated with artisanal scale gold mining†
David C. P.
King
 ab,
Michael J.
Watts
b,
Elliott M.
Hamilton
*b,
Robert J. G.
Mortimer
c,
Mike
Coffey
a,
Odipo
Osano
d,
Maureene Auma
Ondayo
d and
Marcello
Di Bonito
ae
ab,
Michael J.
Watts
b,
Elliott M.
Hamilton
*b,
Robert J. G.
Mortimer
c,
Mike
Coffey
a,
Odipo
Osano
d,
Maureene Auma
Ondayo
d and
Marcello
Di Bonito
ae
aNottingham Trent University, Nottingham, UK
bInorganic Geochemistry Facility, Centre for Environmental Geochemistry, British Geological Survey, Nottingham, UK
cYork St John University, York, UK
dUniversity of Eldoret, Eldoret, Kenya
eUnversità di Bologna, Bologna, Italy
First published on 16th April 2024
Abstract
Analysis of mercury (Hg) in natural water samples has routinely been impractical in many environments, for example, artisanal and small-scale gold mines (ASGM), where difficult conditions make monitoring of harmful elements and chemicals used in the processes highly challenging. Current sampling methods require the use of hazardous or expensive materials, and so difficulties in sample collection and transport are elevated. To solve this problem, a solid-phase extraction-based method was developed for the sampling and preservation of dissolved Hg in natural water samples, particularly those found around ASGM sites. Recoveries of 85% ± 10% total Hg were obtained during 4 weeks of storage in refrigerated (4 °C, dark) and unrefrigerated (16 °C, dark) conditions, and from a representative river water spiked to 1 μg L−1 Hg2+, 94% ± 1% Hg recovery was obtained. Solid-phase extraction loading flow rates were tested at 2, 5, and 10 mL min−1 with no breakthrough of Hg, and sorbent stability showed no breakthrough of Hg up to 2 weeks after functionalisation. The method was deployed across five artisanal gold mines in Kakamega gold belt, Kenya, to assess Hg concentrations in mine shaft water, ore washing ponds, and river and stream water, including drinking water sources. In all waters, Hg concentrations were below the WHO guideline limit value of 6 μg L−1, but drinking water sources contained trace concentrations of up to 0.35 μg L−1 total Hg, which may result in negative health effects from long-term exposure. The SPE method developed and deployed here is a robust sampling method that can therefore be applied in future Hg monitoring, toxicology, and environmental work to provide improved data that is representative of total dissolved Hg in water samples.
1. Introduction
1.1 Mercury (Hg) as a pollutant
Mercury (Hg) is a highly toxic element that is ubiquitous in the environment, arising from both natural and anthropogenic processes.1 Due to its ability to bioaccumulate in animals and biomagnify up the food chain,2 this element can be persistent and problematic for wildlife, where exposure to low concentrations of just 0.03 μg L−1 Hg in the aquatic environment can result in considerable accumulation up the various trophic levels and top predators in the environment.3 Consumption of contaminated fish, a key protein source, may subsequently result in severe negative human health effects in the neurological, renal, cardiovascular, immunological, and reproductive systems.4,5 Unpolluted concentrations for environmental water sources, which may affect human exposure routes, are usually 0.01 μg L−1 Hg but can be as high as 0.1 μg L−1 Hg.4 However, guideline values for the protection of aquatic wildlife from chronic Hg exposure are 0.77 μg L−1 Hg as defined by the US EPA,6 while drinking water guidelines are defined at 6 μg L−1 Hg by the WHO.7To prevent anthropogenic pollution of Hg, the 2013 Minamata Convention limits the use of Hg in all sectors of society, but in most middle- and lower-income countries the metal is relied upon for many industries. The most prevalent use of Hg is in artisanal and small-scale gold mining (ASGM),8,9 where Hg is used to amalgamate gold in mined and pulverised ores and is subsequently released by burning to obtain gold. Through the amalgamation and burning processes, surrounding soil and atmosphere is polluted with Hg.8,9 This practice occurs globally and has been routinely investigated over the past 20 years.10–13 Across over 40 African countries, approximately 10 million people work in ASGM sites14 and are potentially exposed to large quantities of potentially harmful elements, including Hg. In a review of ASGM across Africa14 concentrations of Hg in waters (rivers, drinking water sources, and ore washing ponds) ranged from 0.01 to 8040 μg L−1 Hg, with two-thirds of studies (18 out of 27) reporting Hg water concentrations above the 0.77 μg L−1 Hg chronic exposure limit. This underscores the need for monitoring and possible intervention, to limit environmental harm. The most extreme concentrations, up to 8040 μg L−1 Hg, were reported in Kenya;15 however sample collection and preservation methods are not specified and so may cast doubt on the reported values. In addition, this study reported data from an acid digestion of the sample, which may release Hg bound to particulate matter which would otherwise have been filtered off in standard procedures and thereby leading to a report of a Hg concentration above what local populations would be exposed to through drinking water sources.
Since 2003, it is estimated that up to 100 tonnes of Hg have been imported to Kenya for ASGM activities,9 but the ultimate fate of this metal is unknown due to difficulties in sampling and analysis. Workers, communities, and environments near ASGM sites in Migori and Kakamega gold belts, Kenya, are reported to be adversely exposed to Hg,12,15,16 with soils and sediments reported to contain up to 150 mg kg−1 Hg,16 residential soils containing 1.07 mg kg−1 Hg and fish pond sediments containing 2.4 mg kg−1 Hg,12 all above WHO, USEPA, and European Union standards for Hg in environmental media. Concentrations of Hg in water sources at these sites are not reported and therefore the true extent of Hg exposure of the local populations is not determined due to difficulties with sampling and preservation of the metal in the field.
1.2 Sampling methods and their challenges
Instability of Hg dissolved in water samples is a well-known challenge when monitoring the metal; sorption to container walls and loss through volatilisation are the main mechanisms of loss from a water sample.17,18 Conventional sampling processes for dissolved Hg often have attributes of risk and difficulty, exacerbated by the need for use in challenging environments such as ASGM sites. It is recommended to send samples overnight to a laboratory for preservation,19 however, when sampling waters at ASGM sites, samples often must be shipped internationally, which increases the time between sampling and analysis and thus potentially results in considerable Hg losses. Acidification of samples with hydrochloric acid stabilises the metal but can result in spillage and acid burn accidents if not carefully handled, and increased restrictions on international transportation of acids limits the shipping of samples.20 There is also a potential for contamination during handling and transportation. Collection of >500 mL sample volume can mitigate loss of Hg by wall sorption due to a decreased volume-to-surface area ratio and allows for preconcentration methods except, where multiple samples must be collected, as transportation and storage may become impractical due to the large total sample volumes. This also increases the cost of collection due to bottle material costs. Glass (amber glass or borosilicate glass quality 3.3) and PTFE containers are recommended to minimize sorption of Hg to the container walls, but glass is fragile while PTFE is expensive ($100 per 500 mL PTFE bottle).21 If unpreserved or inadequately preserved, considerable losses of Hg are noted within few days of sampling and 90% of total Hg can be lost after just 1 week.17,18,22 In-field monitoring of Hg in water sources is particularly challenging, due to analysis requiring sensitive detectors, which currently do not achieve adequate detection limits,23 thus analysis in a laboratory using robust and sensitive instrumentation, such as CV-AAS and ICP-MS, is currently necessary.19 Some emerging technologies demonstrate appropriate sensitivity but are not currently widespread or commercially available.24–26Solid-phase extraction offers a method to stabilise dissolved Hg in water for a substantial amount of time.2 Through immobilisation to a solid-phase, the risk of analyte loss is minimised and representative data can be collected, particularly in challenging environments where there is difficulty in transporting materials and samples. Previous work for the preservation of other metals (arsenic, As and vanadium, V) has shown the usefulness of SPE techniques in sampling and stabilisation of metals in water samples.27–29 Significant losses of V data are reported over less than 1 week of storage, which can limit sample collection for many studies. An optimised SPE-based sampling method was developed, improving sample storage to up to 2 weeks with recoveries of 95–101% V(IV) and V(V) from river water samples.29 For As concentrations, there is a known instability of the metal species in a 1 mg L−1 Fe water matrix, often found in rivers and lakes.30 To avoid this issue, an SPE-based sampling method was developed by Watts et al. (2008) and O'Reilly et al. (2010) to preserve As(III), As(V), monomethylarsonic acid, and dimethylarsinic acid.27,31 This method uses anionic and cationic SPE cartridges to allow in-field separation and preservation of As for up to 4 weeks with 100% recovery of each species. Parameters were optimised based upon field conditions. For example, the flow rate for sample loading was determined by the physical ability of the operator to push water through the cartridges. A 4 weeks storage time was chosen as a typical time from field to laboratory analysis, accounting for a field sampling campaign and potential international travel. Previous research into the SPE method for Hg sampling and preservation methods have shown >90% recovery of total Hg from river and lake water samples over a 1 week period,29,30 but extending this preservative timeframe is essential for an effective field method that can be used in all environments.
1.3 Considerations for a field-method
For SPE to be used as a field-based sampling method, it is crucial to consider practical characteristics that affect the immobilisation of dissolved Hg and stability of the metal once retained (Fig. 1). Initially, the cartridges must be stable from the point of functionalisation to sampling in the field. Degradation of the functionalised phase may result in a decrease in immobilised Hg during the loading process and therefore underrepresent the dissolved Hg concentration in the environmental samples. This is a characteristic not often explored in the literature, as typical speciation and preconcentration methods apply the cartridge immediately after functionalisation,23 thus not requiring a storage time for the solid phase. However, to ensure representative data collection, the functionalised solid-phase must be stable during transfer from laboratory to sampling location, approximately 2 weeks between functionalisation and application in the field. Flow rates for loading samples onto the cartridges must be appropriate for field-use, to ensure feasible sampling times and thus overall cost-effectiveness. If flow rates are too slow, the time required to load a sample onto a cartridge will not be appropriate for field studies. This is generally not explored in the literature, as SPE methods are often developed for on-line laboratory speciation or preconcentration, and so flow rate is controlled by optimised chromatographic flow rates. The quantity of functionalising agent immobilised on the solid-phase is also important, as quantities below the optimum will fail to retain sufficient Hg concentrations, while quantities above the optimum may impede elution processes and result in partial elution of Hg. Once immobilised, Hg must be retained to the phase with adequate time for transportation to a laboratory. This may range from hours to weeks depending on the sampling environment and location, so Hg must be eluted with good recovery after at least a 4 weeks period. Similar characteristics were used in previous literature related to SPE as a sampling technique,27–29 which is representative of necessary parameters for a functional sampling and preservation method for dissolved Hg. The development of an SPE field method for sampling and preserving Hg from water samples will allow for the determination and monitoring of Hg in waters that affect some of the most vulnerable populations. | ||
| Fig. 1 Main considerations of an SPE-based field sampling method, including quantity of functionalising agent applied, stability of the functionalised cartridge prior to use, flow rate of sample loading, and stability of Hg on the cartridge after immobilisation. | ||
1.4 Aim and objectives
There is an identified gap in the literature to safely and reliably sample total dissolved Hg from water matrices in challenging environments.23 Therefore, the aim of this study was to develop a robust method for the sampling and accurate measurement of Hg in water matrices based on SPE techniques. To achieve this aim, the objectives were to: (1) develop a practical functionalised SPE method to retain and preserve Hg from water matrices and for elution on return of samples to a controlled laboratory environment; (2) optimise parameters of sorption and desorption of Hg to the stationary phase for subsequent measurement; (3) demonstrate robustness of method in water matrices.2. Method
Based on literature methods,32,33 a dithizone-functionalised C18 cartridge was assessed for the extraction and recovery of Hg from 30 mL of water, eluted using a 2-mercaptoethanol solution.The effect of flow rate of the sample through the cartridge was assessed, as well as the stability of the functionalised cartridge prior to use in the field, approximately 2 weeks from functionalisation to use. The stability of Hg immobilised on the cartridge was assessed over a 4 weeks period. This is an expected time from sampling to analysis for projects conducted in challenging environments such as ASGM sites, including time needed for international shipping of samples, and ensures preservation over shorter time periods, i.e., hours or days.
The workflow of functionalisation and field-use of SPE cartridges are illustrated in Fig. 2.
 | ||
| Fig. 2 The functionalisation of SPE cartridges using 50 μg dithizone, loading of water samples in the field at 10 mL min−1 to immobilise Hg, and elution of Hg using 30 mL of a 1% v/v 2-mercaptoethanol solution. | ||
The functionalisation of the cartridges was conducted following methods reported by Yin et al. (2010).32,33 Briefly, C18 cartridges (Bond Elut Jr, Agilent, UK) were flushed with 5 mL of methanol (SigmaAldrich, UK) to wet the resin bed, followed by 5 mL of sodium formate–formic acid (0.5 mol L−1, pH 4; SigmaAldrich, UK) to condition the resin bed. A dithizone solution (1 mL, 0.5 mg L−1 dithizone; AlfaAesar, UK) was immobilised on the cartridge as the functional compound in the phase. The resin bed was then conditioned by passing through a 3 mL sodium formate–formic acid solution (0.5 mol L−1, pH 4; SigmaAldrich, UK) and 3 mL of sodium formate–formic acid solution (0.05 mol L−1, pH 4; SigmaAldrich, UK).
Samples were analysed by ICP-MS analysis. Other analytical techniques are possible, but the sensitivity and robustness of measurement provided by ICP-MS analysis was deemed appropriate for this study. Parameters and conditions for ICP-MS analysis are given in ESI Table 2.† A method detection limit was calculated as 3σ from measurements of a blank solution (n = 10) (ESI Table 3†). Other water chemistry of real samples was analysed by ion chromatography, pH and alkalinity analysis, and conductivity probe at the British Geological Survey, Nottingham, UK (ESI Table 4†).
2.1 Quality control
Due to the instability of dissolved Hg in water samples, certified reference materials for Hg in environmental waters are not readily available. Commercially available certified reference materials are often preserved using BrCl solutions, (ORMS-5, NRCC, Canada)34 and therefore may not be truly representative of Hg concentrations in unpreserved river waters, as required by this study. During analysis, a 0.25 μg L−1 Hg2+ quality control sample was periodically analysed (every 15 samples) to ensure accuracy and precision between experiments. To ensure the quality and performance of the cartridge would not be impacted by major anion and cation chemistry commonly found at ASGM sites (ESI Table 1†),12 a synthetic hard water was used as a matrix in experimental work. The matrix was made following a method described by Smith et al. (2002).35 To represent total dissolved Hg, it was spiked to 1 μg L−1 inorganic Hg2+ (1000 μg L−1 Hg2+ standard, Romil, UK), which is the most common form of Hg in water. The use of a standard hard water matrix across all experimental work demonstrates the robustness of the method under expected matrix conditions and the effects of common major ionic constituents.2.2 Quantity of dithizone immobilised onto the cartridge
An important factor for ensuring the sorption and stability of Hg on the solid phase is the quantity of dithizone immobilised on the cartridges. To assess this, dithizone was loaded onto C18 cartridges in quantities of 25, 50, 100, and 200 μg dithizone. A 1 μg L−1 Hg2+ synthetic freshwater solution (30 mL) was passed through the functionalised cartridges. The effluent was collected and acidified to 1% v/v HNO3 (Romil, UK) and 0.5% v/v HCl (Romil, UK). A 2-mercaptoethanol eluent solution (1% v/v, 30 mL) was then passed through the cartridges and collected for analysis.2.3 Sample flow rate and sorbent stability
It is necessary to establish an optimum sample flow rate for immobilisation of Hg that is achievable in field conditions. To ensure adequate flow rates could be achieved, a 1 μg L−1 Hg2+ synthetic freshwater solution (30 mL) was passed through the functionalised cartridges at a flow rate of 2, 5, or 10 mL min−1 using a vacuum box. Prior to use, cartridges were functionalised with a 50 μg dose of dithizone (1 mL, 0.05 mg L−1 dithizone solution). The cartridge effluents were collected and acidified to 1% v/v HNO3 (Romil, UK) and 0.5% v/v HCl (Romil, UK) for analysis.The stability of the functionalised cartridge was investigated over 2 weeks. Cartridges were functionalised and stored for 0, 7 and 14 days (16 °C, in the dark and 4 °C, in the dark), to simulate potential storage conditions for transfer from the laboratory to the field. A 1 μg L−1 Hg2+ solution (30 mL) was loaded onto functionalised cartridges at a flow rate of 10 mL min−1, and the effluent was collected and acidified to 1% v/v HNO3 (Romil, UK) + 0.5% v/v HCl (Romil, UK).
2.4 Recovery of immobilised Hg over time
The stability of Hg on the solid-phase must be established to determine the efficacy of SPE as an in-field sampling method. A 1 μg L−1 Hg2+ spiked synthetic freshwater matrix (30 mL) was passed through functionalised C18 cartridges. These cartridges were stored in the dark at either 4 °C or 16 °C, temperatures of cooled and non-cooled storage respectively, for up to 4 weeks, a typical timescale between sampling in challenging environments and analysis in a laboratory. Retained Hg was eluted following the previously described method at 0, 1, 2, 3, and 4 weeks after extraction of Hg.2.5 Sampling sites
 | ||
| Fig. 3 Map of (b) Kakamega and Vihiga counties, (a) Kenya, the ASGM sites sampled: (c) Viyalo, (d) Bushiangala, (e) Malinya, (f) Rosterman, and (g) Lunyerere mine sites, and sampling locations. | ||
3. Results and discussion
3.1 Dithizone load quantity
Concentrations of Hg measured in the effluent were below the 0.008 μg L−1 detection limit across 50–200 μg dithizone, indicating total immobilisation of Hg to the resin bed. At 25 μg dithizone, breakthrough of the metal at 0.01 μg L−1 Hg2+ was observed. This is a potential issue for polluted water sources (>1 μg L−1 Hg), such as those found by Ogola (2001) and Ngure et al. (2017) where Hg concentrations were found up to 8 and 19 μg L−1 Hg respectively and thus are likely to exceed the capacity of the functionalised cartridge at 25 μg dithizone.15,37 Complex matrices which contain increased quantities of particulate matter and mine-related elements, may also compete for active sites on the solid-phase, and therefore may decrease the sorption of Hg thus requiring a dithizone quantity greater than 25 μg.Elution from all assessed dithizone quantities showed Hg recoveries above 85%, with the most optimal recovery of 92% at 50 μg dithizone, indicating suitable recovery for representative data collection (Fig. 4). Using a cartridge functionalised with a lower quantity of dithizone, Wang et al. (2022),33 12.5 μg dithizone, reported high recoveries (95% ± 8% Hg), however, this study focused on trace unpolluted concentrations of Hg, <0.2 μg L−1 Hg. The lower quantity of dithizone may not appropriately immobilise higher concentrations of Hg such as those reported by Ngure et al. (2014) in the Migori gold belt, Kenya, 0.36–52 μg L−1 Hg.38 The comparable recoveries achieved by this work, 92% (±3%) Hg, demonstrate the suitability of a 50 μg dithizone quantity for the extraction and recovery of Hg, and can fully immobilise Hg from polluted water sources up to 1 μg L−1. A 50 μg dithizone load was used for all other experiments.
 | ||
| Fig. 4 Recovery and standard deviation of Hg2+ from SPE cartridges with various quantities of immobilised dithizone (25, 50, 100 and 200 μg) (n = 5), indicating a load of 25 μg dithizone or greater is suitable for immobilisation of Hg2+ from water samples with 50 μg showing the most optimal performance. | ||
3.2 Sample flow rate and sorbent stability
Two key factors of a viable field sampling SPE method are the maximum flow rate of sample loading and the stability of the cartridge prior to use. A moderate sample flow rate is necessary to ensure sampling does not require unreasonable lengths of time to conduct when in the field. At loading speeds of 2, 5, and 10 mL min−1, all effluent samples had Hg concentrations below the detection limit (0.008 μg L−1 Hg2+), showing total sorption of Hg to the solid-phase. This is approximately the speed that can be achieved when manually pushing solution through a filter and cartridge using a Luer-lock syringe, and so typically, this is what may be achieved by operators in the field with minimal equipment requirements.The stability of the cartridges prior to use was also investigated. Cartridges were stored for up to 14 days after functionalisation, and an extraction was conducted. Across the 14 days period, Hg in the effluent samples was consistently below the detection limit of 0.008 μg L−1 Hg. This means that a 30 mL sample of 1 μg L−1 Hg completely absorbed into the stationary phase after being stored in the dark for up to two weeks at a temperature of 4 or 16 °C. Prepared cartridges can therefore be stored prior to use with no breakthrough of Hg, even at significant concentrations of Hg pollution.
3.3 Four-week preservation
Stability of the sorbed Hg was investigated over 0–4 weeks in the dark in either refrigerated (4 °C) or unrefrigerated (16 °C) conditions. As illustrated in Fig. 5, recovery of Hg2+ was consistently high at both 4 °C and 16 °C in the dark across 4 weeks. At either 4 °C or 16 °C in the dark, average recoveries of Hg were 88% ± 8% and 85% ± 10%, respectively, showing good reproducibility and demonstrating adequate Hg preservation for typical transportation and storage times from field to laboratory. The lowest recorded recovery was 68% after 3 weeks of storage at 16 °C in the dark, which is still considered an acceptable recovery for SPE techniques.39 Blanco et al. (2000) used a diethyldithiocarbamate functionalised microcolumn for dissolved Hg preservation but noted a significant decrease in recovery from 102% to just 40% after just 1 week of storage.40 A similar dithizone-based microcolumn was reported to stabilise dissolved Hg from waters over 10 days of storage, which may not be an adequate timeframe in many environments.33 The recorded improvement using the dithizone functionalised cartridges allows for representative data to be collected and analysed with a reasonable time limit from field to laboratory with minimal losses over time. A comparison of the performance of the developed cartridge and literature methods is shown in Table 1.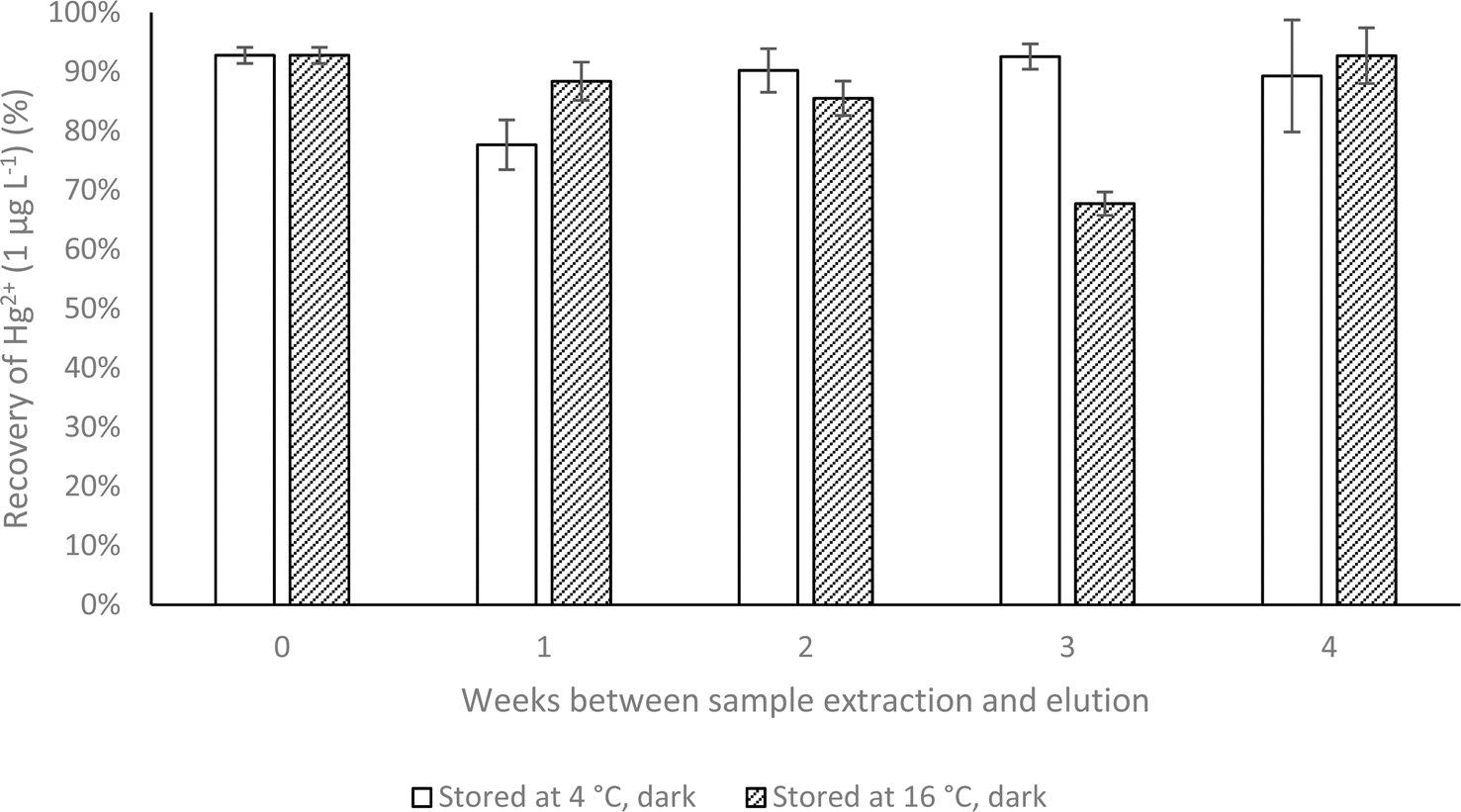 | ||
| Fig. 5 Recovery (%) and standard deviation (n = 5) of Hg (30 mL, 1 μg L−1 Hg2+ solution) immobilised on a functionalised cartridge over 4 weeks storage and eluted with a 2-mercaptoethanol solution (1% v/v, 30 mL). | ||
| Study | Functional agent | Hg spike concentration assessed (μg L−1 Hg2+) | Sample volume (mL) | Eluent volume (mL) | Recovery (%) | Effective storage time | Replicates |
|---|---|---|---|---|---|---|---|
| This work | Dithizone (50 μg) | 1 | 30 | 30 | 92 ± 3 | 4 weeks | 5 |
| Blanco et al. (2000)40 | Diethyldithiocarbamate | 0.05–0.15 | 50 | 0.5 | 70 ± 3 | 1 week | 3 |
| Yin et al. (2010)32 | Dithizone (200 μg) | 0.2 | 100 | 3 | 83 ± 4 | 4 days | 3 |
| Wang et al. (2022)33 | Dithizone (12.5 μg) | 0.005 | 1.5 | N/A (online elution) | 95 ± 8 | 1 week | 7 |
3.4 Natural water samples water
Mercury was not detected in the effluent samples, indicating total sorption of Hg to the solid-phase from the river water matrix. Recoveries from the spiked water samples were >90%, which is comparable to literature recoveries of typical preservation methods such as acidification of the samples (Table 2).18,22
| References | Sample | Hg concentration (μg L−1) | Sample volume (mL) | Elution volume (mL) | Spike (μg L−1) | Recovery (%) | Relative standard deviation |
|---|---|---|---|---|---|---|---|
| This work | River water (River Trent) | <0.008 | 30 | 30 | 1.004 Hg | 94 | 1% |
| Blanco et al. (2000)40 | River water | Not reported | 50 | 0.5 | 0.05–0.15 Hg2+ | 90 | 6.7% |
| Yin et al. (2010)32 | River water | 0.025 | 100 | 3 | — | 86 | Not reported |
| Sample | Water type | Location | Hg concentration (μg L−1) | Weekly exposure (μg (Hg) per kg (body weight) per week) |
|---|---|---|---|---|
| M1 | Mine shaft water | Lunyerere | 0.179 | 0.063 |
| M2 | Mine shaft water | Malinya | 0.255 | 0.089 |
| M3 | Mine shaft water | Bushiangala | 0.348 | 0.122 |
| M4 | Mine shaft water | Bushiangala | 0.041 | 0.014 |
| M5 | Mine shaft water | Rosterman | 0.045 | 0.016 |
| R1 | River water (near mine shaft) | Lunyerere | 0.166 | 0.058 |
| R2 | River water (near mine shaft) | Lunyerere | 0.148 | 0.052 |
| R3 | River water (drinking water) | Malinya | 0.023 | 0.008 |
| R4 | River water (mineshaft water released to river) | Bushiangala | 0.084 | 0.029 |
| R5 | River water (upstream of new alluvial mining operation) | Rosterman | 0.02 | 0.007 |
| R6 | River water (downstream of new alluvial mining operation) | Rosterman | 0.032 | 0.011 |
| R7 | River water (new alluvial mining operation) | Rosterman | 0.058 | 0.02 |
| R8 | River water (from a separate mining village) | Rosterman | 0.141 | 0.049 |
| R9 | River water (downstream of ASGM site) | Rosterman | 0.028 | 0.01 |
| S1 | Spring water (drinking water) | Lunyerere | 0.132 | 0.046 |
| S2 | Spring water (drinking water) | Viyalo | 0.043 | 0.015 |
| S3 | Spring water (drinking water) | Bushiangala | 0.036 | 0.013 |
| S4 | Spring water (drinking water) | Rosterman | 0.055 | 0.019 |
| S5 | Spring water (upstream of ore washing ponds) | Rosterman | 0.01 | 0.004 |
| S6 | Spring water (downstream of ore washing ponds) | Rosterman | 0.043 | 0.015 |
| O1 | Ore washing pond | Lunyerere | 0.151 | 0.053 |
| O2 | Ore washing pond | Viyalo | 0.079 | 0.028 |
| O3 | Ore washing pond | Malinya | 0.03 | 0.011 |
| O4 | Ore washing pond | Malinya | 0.046 | 0.016 |
| O5 | Ore washing pond | Bushiangala | 0.162 | 0.057 |
| O6 | Ore washing pond | Bushiangala | 0.102 | 0.036 |
| O7 | Hg amalgamation pan for ore washing | Rosterman | 0.277 | 0.097 |
| O8 | Ore washing pond | Rosterman | 0.162 | 0.057 |
| O9 | Ore washing pond | Rosterman | 0.032 | 0.011 |
| (WHO guideline value)7 | 6.000 | |||
| (US EPA no effect guideline – aquatic wildlife)6 | 0.770 |
The measured Hg concentrations are lower than previously reported values from ASGM sites in Kenya by Ogola et al. (2001), 8040 μg L−1 Hg, however, methods of sampling, filtration, or preservation were not reported for waters, so the data should be considered with this in mind.15 The analysis method from this literature study also included a HNO3 digestion step, which may digest particulate matter-bound Hg from unfiltered samples and therefore increase the measured concentration of Hg. A study by Ngure et al. (2017) reported Hg concentrations of 0.06–19 μg L−1 Hg in drinking water in the Migori gold belt, Kenya, where the most elevated concentrations were found at sampling points closer to the mouth of the river system and indicating potential pollution from ASGM activities along the rivers length.37 The lower concentration samples, 0.06 and 0.92 μg L−1 Hg, are comparable to those found in this study. In both the literature study and this work, the sample locations are similarly positioned in the Kenyan gold belts with respect to potential nearby ASGM sites.
For a more accurate assessment of associated health risks, Hg speciation should be taken into consideration due to differing bioavailability and toxicity. Future work on SPE-based sampling methods should examine the potential for species sampling and preservation to better understand Hg speciation and transformations in the environment.
4. Conclusion
Due to poor stability of Hg in water samples, previous literature assessing Hg concentrations in waters in challenging environments may not accurately report representative values, likely as a result of inherent difficulties in the sampling, preservation, and subsequent analysis methods. This study successfully assessed an SPE-based method for the sampling and preservation of Hg in water samples, providing a robust method applicable in a wide variety of environments. Total dissolved Hg can be completely extracted from a natural water sample and preserved over a 4 weeks period with a >75% recovery, an improvement over previous literature preservation of just 1 week. While this was determined using ICP-MS analysis, the predominantly water-based matrix of the eluent is compatible for analysis with other techniques such as CV-AAS. The method was applied at Kenyan ASGM sites, finding Hg concentrations between 0.020 and 0.348 μg L−1 Hg, with water sources used as drinking waters found to not pose a significant health risk individually but do contribute up to 17% of the total weekly intake limit. The improved sampling method presented in this work will allow the collection of representative data at concentrations below the guidance limit for drinking water and thus improve future Hg monitoring schemes and toxicological studies, notably in vulnerable areas which currently cannot be assessed effectively. The assignment of such guidance limits is often constrained by the analytical chemistry detection limit capability and availability of equipment to reliably measure potentially toxic elements in environmental matrices at low concentrations that may better inform chronic exposure.Author contributions
David C. P. King: conceptualisation, writing – original draft, methodology. Michael J. Watts: conceptualisation, funding acquisition, writing – review & editing. Elliott M. Hamilton: writing – review & editing. Robert Mortimer: writing – review & editing. Mike Coffey: writing – review & editing. Odipo Osano: writing – review & editing. Maureene Auma Ondayo: writing – review & editing. Marcello Di Bonito: conceptualisation, funding acquisition, writing – review & editing.Conflicts of interest
The authors report no conflict of interest.Acknowledgements
Funding for D. C. P. K. was provided by Nottingham Trent University and The British Geological Survey University Funding Initiative (contract number: GA/21S/002). This work is published with the permission of the Executive Director, British Geological Survey.References
- J. F. Risher, Elemental mercury and inorganic mercury compounds: human health aspects, WHO, Geneva, Switzerland, 2003 Search PubMed.
- M. K. Kim and K. D. Zoh, Journal of Preventive Medicine and Public Health, 2012, 45, 335–343, DOI:10.3961/jpmph.2012.45.6.335.
- P. Morcillo, M. A. Esteban and A. Cuesta, AIMS Environ. Sci., 2017, 4(3), 386–402, DOI:10.3934/environsci.2017.3.386.
- Z. Svobodova, R. Lloyd, J. Machova and B. Vykusova, Water Quality and Fish Health, Food and Agriculture Organization of the United Nations, 1993 Search PubMed.
- United States DHHS, Toxicological Profile for Mercury, US DHHS, Georgia, USA, 2022 Search PubMed.
- United States EPA, 1995 Water Quality Criteria, US EPA, Washington, DC, USA, 1995 Search PubMed.
- World Health Organisation, Guidelines for drinking-water quality, WHO, Geneva, Switzerland, 4th edn, 2022 Search PubMed.
- A. K. Kristensen, J. F. Thomsen and S. Mikkelsen, Int. Arch. Occup. Environ. Health, 2014, 87, 579–590, DOI:10.1007/s00420-013-0902-9.
- L. J. Esdaile and J. M. Chalker, Chem.–Eur. J., 2018, 24, 6905–6916, DOI:10.1002/chem.201704840.
- T. Hentschel, F. Hruschka and M. Priester, Global Report on Artisanal & Small-Scale Mining, International Institute for Environmental Development, 2002 Search PubMed.
- P. Li, X. B. Feng, G. L. Qiu and Z. G. Li, J. Hazard. Mater., 2009, 168, 591–601, DOI:10.1016/j.jhazmat.2009.03.031.
- M. A. Ondayo, M. J. Watts, E. M. Hamilton, C. Mitchell, J. Mankelow and O. Osano, Environ. Geochem. Health, 2023, 45, 6543–6565, DOI:10.1007/s10653-023-01647-z.
- M. A. Ondayo, M. J. Watts, C. Mitchell, D. C. P. King and O. Osano, Exposure Health, 2023, 29 DOI:10.1007/s12403-023-00611-7.
- R. Grynberg, V. Kandaswamy and F. Singogo, Development Southern Africa, 2022, 39, 151–164, DOI:10.1080/0376835X.2020.1868288.
- J. S. Olgola, W. V. Mitullah and M. A. Omulo, Environ. Geochem. Health, 2001, 24, 141–158, DOI:10.1023/A:1014207832471.
- O. B. Odumo, A. O. Mustapha, J. P. Patel and H. K. Angeyo, Bull. Environ. Contam. Toxicol., 2011, 86, 484–489, DOI:10.1007/s00128-011-0242-y.
- L.-P. Yu and X.-P. Yan, TrAC, Trends Anal. Chem., 2003, 22, 245–253, DOI:10.1016/S0165-9936(03)00407-2.
- J. L. Parker and N. S. Bloom, Sci. Total Environ., 2005, 337, 253–263, DOI:10.1016/j.scitotenv.2004.07.006.
- United States EPA, Method 1669 sampling ambient water for trace metals at EPA water quality criteria levels, USEPA, Washington, DC, USA, 1996 Search PubMed.
- International Air Transport Association, Dangerous Goods Regulations, IATA, Motreal, Canada, 2022 Search PubMed.
- Camlab, PTFE bottle with screw cap 500 mL, 2023, https://www.camlab.co.uk/ptfe-bottlewith-screw-cap-500ml, accessed 26/10/2023 Search PubMed.
- H. Louie, C. Wong, Y. J. Huang and S. Fredrickson, Anal. Methods, 2012, 4, 522–529, 10.1039/C2AY05182F.
- D. C. P. King, M. J. Watts, E. M. Hamilton, R. Mortimer, D. P. A. Kilgour and M. Di Bonito, Environ. Sci.: Processes Impacts, 2023, 25, 351–363, 10.1039/D2EM00409G.
- X. Zhao, Y. Liu, B. Shu, Z. Guo, C. Song, X. Huang and Z. Jiao, Int. J. Environ. Anal. Chem., 2020, 1–10, DOI:10.1080/03067319.2020.1756274.
- S. N. Karuk Elmas, S. Dinckan, F. N. Arslan, D. Aydin, T. Savran and I. Yilmaz, J. Photochem. Photobiol., A, 2021, 421, 113521, DOI:10.1016/j.jphotochem.2021.113521.
- A. Lopreside, L. Montali, B. Wang, A. Tassoni, M. Ferri, M. M. Calabretta and E. Michelini, Biosens. Bioelectron., 2021, 194, 113569, DOI:10.1016/j.bios.2021.113569.
- M. J. Watts, M. Button, T. S. Brewer, G. R. T. Jenkin and C. F. Harrington, J. Environ. Monit., 2008, 10, 753–759, 10.1039/B800567B.
- M. J. Watts, J. O’Reilly, A. L. Marcilla, R. Shaw and N. I. Ward, Environ. Geochem. Health, 2010, 32, 479–490, DOI:10.1007/s10653-010-9321-y.
- W. A. Al Rawahi and N. I. Ward, Talanta, 2017, 165, 391–397, DOI:10.1016/j.talanta.2016.12.078.
- S. J. Stetson, M. L. Erickson, J. Brenner, E. C. Berquist, C. Kanagy, S. Whitcomb and C. Lawrence, Appl. Geochem., 2021, 125, 104814, DOI:10.1016/j.apgeochem.2020.104814.
- J. O'Reilly, M. J. Watts, R. Shaw, A. L. Marcilla and N. I. Ward, Environ. Geochem. Health, 2010, 32, 491–515, DOI:10.1007/s10653-010-9317-7.
- Y.-G. Yin, M. Chen, J.-F. Peng, J.-F. Liu and G.-B. Jiang, Talanta, 2010, 81, 1788–1792, DOI:10.1016/j.talanta.2010.03.039.
- Y. Wang, A. Zhu, Y. Fang, C. Fan, Y. Guo, Z. Tan, Y. Yin, Y. Cai and G. Jiang, J. Environ. Sci., 2022, 115, 403–410, DOI:10.1016/j.jes.2022.08.031.
- S. Willie, P. Grinberg, Z. Mester and L. Yang, ORMS-5, NRCC, 2011, DOI:10.4224/crm.2011.orms-5.
- J. Smith, W. Davidson and J. Hamilton-Taylor, Water Res., 2002, 36, 1286–1296, DOI:10.1016/S0043-1354(01)00341-4.
- DEFRA, Classifications Data for Trent River Operational Catchment: 2019 Cycle 3, 2019, London, UK, accessed 13/06/2023 Search PubMed.
- V. Ngure, F. Lelo and B. Obwanga, J. Nat. Sci., 2017, 7, 46–53 Search PubMed.
- V. Ngure, T. Davies, G. Kinuthia, N. Sitati, S. Shisia and E. Oyoo-Okoth, J. Geochem. Explor., 2014, 144, 511–516, DOI:10.1016/j.gexplo.2014.04.004.
- United States EPA, Method 3535A Solid-Phase Extraction (SPE), US EPA, Washington DC, USA, 2007 Search PubMed.
- R. M. Blanco, M. T. Villanueva, J. E. Sánchez Urıa and A. Sanz-Medel, Anal. Chim. Acta, 2000, 419, 137–144, DOI:10.1016/S0003-2670(00)01002-3.
- J. Margetínová, P. Houserová-Pelcová and V. Kubáň, Anal. Chim. Acta, 2008, 615, 115–123, DOI:10.1016/j.aca.2008.03.061.
- R. Kuras, B. Janasik, M. Stanislawska, L. Kozlowska and W. Wasowicz, Biol. Trace Elem. Res., 2017, 179, 23–31, DOI:10.1007/s12011-017-0939-9.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ay02216a |
| This journal is © The Royal Society of Chemistry 2024 |