
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Beyond helium: hydrogen as a carrier gas in multiresidue pesticide analysis in fruits and vegetables by GC-MS/MS†
Víctor
Cutillas
 ,
Guillermo
García-Gallego
,
María
Murcia-Morales
,
Carmen
Ferrer
and
Amadeo R.
Fernández-Alba
*
,
Guillermo
García-Gallego
,
María
Murcia-Morales
,
Carmen
Ferrer
and
Amadeo R.
Fernández-Alba
*
European Union Reference Laboratory for Pesticide Residues in Fruit & Vegetables, Agrifood Campus of International Excellence (ceiA3), University of Almeria, Ctra. Sacramento S/N, La Cañada de San Urbano, 04120, Almería, Spain. E-mail: amadeo@ual.es; Fax: +34 950 015 034; Tel: +34 950 015 034
First published on 26th February 2024
Abstract
In this comprehensive study, we evaluated the feasibility of using hydrogen instead of helium as a carrier gas in a GC-MS/MS system for pesticide residue analysis, spanning three matrices: pepper, tomato, and zucchini. Initial assessments focused on the ion source's chemical inertness, employing nitrobenzene as a benchmark to monitor the hydrogenation process. A method with a duration of less than 12 minutes was developed, achieving good chromatographic peak resolution attributable to the enhanced chromatographic performance of hydrogen as a carrier gas. The study emphasized the optimization of system parameters, testing various ion source temperatures, detector voltages, and injection volumes. Sensitivity assessments, based on the DG-SANTE criteria, indicated that the majority of compounds were identifiable at a concentration of 5 μg kg−1 (81% in tomato, 84% in pepper and 73% in zucchini). Detailed validation for reproducibility, matrix effects, and linearity across 150 pesticides unveiled generally favorable outcomes, with a notable majority of compounds displaying low matrix effects, satisfactory linearity ranges and good reproducibility with most compounds returning a relative standard deviation (RSD) below 10%. When applied to 15 real samples, the hydrogen-based system's performance was juxtaposed against a helium-based counterpart, revealing that results are very comparable between both systems. This comparative approach highlights hydrogen's potential as a reliable and efficient carrier gas in pesticide residue analysis for routine food control laboratories, overcoming difficulties resulting from the lack of helium supplies.
1. Introduction
Gas chromatography coupled with mass spectrometry (GC-MS) is a powerful analytical technique used for the identification and quantification of volatile and thermostable organic compounds, including pesticide residues. The choice of carrier gas used in GC-MS analysis plays a critical role in the separation of individual components of the sample. Hydrogen, helium and nitrogen provide the same optimal plate height at substantially different flow rates.1Helium is the most commonly used carrier gas in GC-MS analysis because it is inert, faster than nitrogen and due to its compatibility with most of detectors. Helium is an element used in a wide range of scientific applications such as nuclear magnetic resonance (NMR), particle accelerators, or gas chromatography. However, helium it is also known for not being a renewable source and its limited global production capacity. Helium cannot be manufactured as it main source in earth comes from the radioactive decay of uranium during millions of years.2 Only a reduced number of natural gas wells have the enough helium concentration to perform an efficient extraction.3 In fact, some shortages have taken place in recent years and this has a huge impact on the scientific fields using helium.4 The shortage is the result of a combination of factors, including the closure of some production plants and the growing demand from the manufacturing industries. These shortages have been solved as new gas wells are found. However, the price of helium has skyrocketed, increasing the overall price to conduct experiments at laboratories and industries.
The need to find helium alternatives made the use of nitrogen as gas carrier something of interest. There are not supply issues regarding nitrogen and it has not any flammable nature like hydrogen. Nitrogen has a narrow optimum range for linear velocity, so this is the gas which provides the lowest efficiency if you want to perform analysis in a reasonable time. In addition, there is a noticeable sensitivity reduction since the ionization yield is approximately nine times higher than of helium.5 However, it has demonstrated to be an alternative to helium for the analysis of petroleum biomarkers6 and pesticides using atmospheric pressure chemical ionization (APCI).7
Hydrogen has the higher diffusion coefficient of the carrier gases used in GC-MS and about half the viscosity of helium and nitrogen. These properties allow analysis to run faster than hydrogen optimal linear velocity with little penalty in resolution. In summary, using H2 the resolution and chromatographic performance are improved while the analysis time is reduced.8–10 The benefits of substitution of helium by hydrogen have been demonstrated by many researchers in the last years, for example for the analysis of steroids by GC-MS,11 the analysis of forensic samples,12 or the analysis of dioxins and furans.13 The improvements of resolution and efficiency of hydrogen can be observed with other detectors like Flame Ionization Detection (FID).14,15 In addition, the ready availability of H2 is a huge advantage as it can be produce onsite through electrolysis of water using a gas generator, making hydrogen a renewable, more sustainable, and greener source.
H2 requires special precautions because of its flammable nature,16,17 and this is the main reason is not used more often as carrier gas despite its benefits. Concentrations higher than 4 vol% in air are explosive, however, the actual electrolytic generators produce high purity H2 in a safer way with internal leak detections and automatic shutdown features. Nonetheless, current GC ovens have a capacity around 20 L and hence it requires a considerable time to reach the 4% using typical 1.5–2 mL min−1 flow rates used for multiresidue analysis of pesticides. Furthermore, nowadays GC usually are equipped with built-in H2 sensors, these detect the concentration of H2 inside the GC oven and when it reaches levels near 1–2% they stop the flow and automatically shut down the system. In short, the instruments developed in the last years have the capability to overcome the H2 safety issues without interfering with the analysis quality.
The main disadvantage using H2 is the sensitivity drop. H2 is less inert than Helium and reacts in the ion source. These chemical interactions can decrease the response of some analytes and there is a general increase of the background noise. The main reactivity produced by H2 is the hydrogenation of unsaturated compounds. Smith (1982) reported one of the first described hydrogenation and isomerization of alkene bonds in a fatty acid moiety using this type of carrier gas.18 The effect of the hydrogenation reaction has been found to be present even during the pyrolysis of polymer samples using H2.19 The difficulties using library search were negligible for most polymer samples, but caution must be taken as libraries usually are He based. In addition, due to its small molecular size and low viscosity, H2 can penetrate through certain materials like vacuum seals leading to a reduction of the vacuum integrity. The use of specialized materials for hydrogen as well as employing reduced carrier gas flow rates and narrow-bore columns is important to avoid any decrease in the instrument performance.20 Also, it's important to take into account that transforming an existing helium-based instrument to a hydrogen one requires increased vigilance, not just regarding safety but also because we are introducing a less inert gas into the system. Moving the existing helium multiresidue method to hydrogen, on the other hand, is quite straightforward since there are many method translation software available.
In this work, we evaluated a GC-MS/MS method with 150 pesticides using H2 as carrier gas. The optimization of the different parameters to overcome the sensitivity issues are discussed along with the evaluation of the identified compounds, matrix effects, linearity, and reproducibility in different matrices. With this study, it can be tested whether using H2 in a multiresidue method of pesticides is able to reach the threshold of maximum residue levels required by routine laboratories.
2. Material and methods
2.1 Reagents and materials
The standards of pesticides included in the multiresidue method of study were provided by LGC (Teddington, United Kingdom) and Dr Ehrenstorfer (Augsburg, Germany). The analytical standards were stored at −30 °C. The standard-mix solution was prepared using individual stock solutions. Individual stock solutions (800–1000 mg L−1) were prepared from each standard and stored in the dark at −40 °C in amber glass vials.Acetone and ethyl acetate were obtained from Fluka Analytical (Steinheim, Germany). The salts employed in the QuEChERS extraction (anhydrous magnesium sulphate, sodium chloride, sodium hydrogenocitrate sesquihydrate, and sodium citrate tribasic dihydrate) were supplied by Sigma-Aldrich, (Steinheim, Germany) except for PSA that was obtained from Supelco (Bellefonte, PA, USA). Ultra-gradient HPLC-grade acetonitrile was obtained from Honeywell (Charlotte, NC, USA).
2.2 Sample preparation
The three matrices (tomato, pepper, and zucchini) used in this study were sourced from a local market in Almería, Spain. These samples were analyzed to ensure that these matrices were free from detectable pesticide residues that could interfere with the study, a citrate-buffered QuEChERS extraction method with dispersive solid-phase extraction (dSPE)21 clean-up was employed. The resulting extracts, containing 1 g of matrix per mL, were analyzed. Matrix-matched vials were prepared by evaporating 50 μL of each blank extract under a gentle stream of nitrogen and reconstituting them with an equal volume of ethyl acetate containing the desired concentration of the pesticide mixture.2.3 Matrix effects evaluation
To evaluate the matrix effects, the slopes of the calibration curves derived from the extracts of each matrix were compared to those from calibration curves prepared in solvent. The latter serves as a reference for no suppression.
A positive matrix effect value indicates a stronger signal in the matrix compared to the solvent, signifying signal enhancement. Conversely, a negative value indicates signal suppression. A zero matrix effect occurs when the slopes of the calibration curves in both the solvent and the matrix are identical.
2.4 Gas chromatography coupled with mass spectrometry
The analysis were performed using a GCMS-TQ8050 NX (Shimadzu Corporation, Kyoto, Japan) equipped with an AOC 30i autosampler and a 2030 PTV injector. The data acquisition was developed by GCMS Real Time Analysis and the data were processed in Labsolution Insight software version 4.0. Samples were injected using a Shimadzu inlet liner siltek deactivated with wool. The injection volume was 1 μL. During the solvent evaporation stage (0.1 min), the injector temperature was maintained at 70 °C. Subsequently, it was increased to 280 °C at a rate of 350 °C min−1 and held for 5 min. A Shimadzu column with the following specifications was utilized: SH-I-5 MS, 20 m length, 0.18 mm inner diameter, and 0.15 μm film thickness. Five different glass liners were acquired from two different manufacturers. Topaz liners, both with and without wool, were acquired from Restek (Bellefonte, PA, USA). Another three liners with different deactivations (Siltek, IP, Base) were acquired from Shimadzu.The oven temperature program was as follows: 60 °C for 0.5 min, increased to 170 °C at 60 °C min−1 and finally up to 280 °C at 15 °C min−1. The total run time was 11.8 min with additional 3 min for cleaning. The solvent cut time was 2 min. The instrument worked at a constant linear velocity of 70 cm s−1. The system worked in MRM acquisiton mode, acquiring the transitions in a ±0.25 min window from the retention time of each analyte. Hydrogen (99.99999% purity) was generated using a Trace 250 GC Carrier gas generator (Peak Scientific Instruments, Inchinnan, Reino Unido), and Argon (99.999% purity) as the collision gas. The system operated using electron ionization (EI). Both the ion source and the interface were maintained at 280 °C. The detector was set at 1.6 kV.
2.5 Optimization of the MS/MS parameters
The Shimadzu Smart Pesticides Database version 2.0 was used for the development of the multiresidue method. Only the top three transitions per pesticide were chosen from the numerous transitions available based on their sensitivity ranking. To confirm the selected transitions, individual standard solutions of the pesticides were injected. The transition with the highest signal (quantifier) and the second most sensitive transition (qualifier) were selected for each pesticide.For accurate identification, the guidelines established by the SANTE Document No. 11312/2021 (ref. 22) were employed: two transitions must be detected with an ion ratio difference less than 30% and a retention time drift below ±0.1 min from the average of the calibration standards. Acquisition windows of ±0.25 min were established for each pesticide. The 150 pesticides of the multiresidue method and their MS parameters appear on the ESI Table.† The retention times are also described in the table.
3. Results and discussion
3.1 Ion source chemical inertness check
The ion source employed in this system is equipped with a specialized amorphous silicon coating that enhances inertness during the ionization process. Nevertheless, it is essential to assess the hydrogenation status prior to analysis. To accomplish this, a standard solution of nitrobenzene at a concentration of 1 mg L−1 in ethyl acetate was injected. The mass spectra for single ion monitoring (SIM) of nitrobenzene and its commonly observed fragments are presented in Fig. 1. The hydrogenation process of nitrobenzene results in the reduction of the nitro functional group (–NO2) to an amino group (–NH2). Consequently, the ratios of nitrobenzene (123 m/z) and aniline (93 m/z) can be compared to evaluate the inertness of the ion source. The nitrobenzene 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 aniline ratio is considered an optimal situation; any higher predominance of the 93 m/z means there is an excess of the hydrogenation reaction. The daily check is of great significance due to the system's capacity to enhance the hydrogenation process in situations where it has not been used for several days.
:1 aniline ratio is considered an optimal situation; any higher predominance of the 93 m/z means there is an excess of the hydrogenation reaction. The daily check is of great significance due to the system's capacity to enhance the hydrogenation process in situations where it has not been used for several days.
 | ||
| Fig. 1 Mass spectrum of a 1 mg kg−1 nitrobenzene vial injection. The left spectrum indicates hydrogenation in the ion source, with the predominant presence of the aniline mass (93 m/z). In contrast, the right spectrum represents the optimal condition, with the nitrobenzene mass (123 m/z) equaling or exceeding that of aniline. | ||
3.2 Optimization of system parameters
It's widely recognized that hydrogen offers slightly reduced sensitivity compared to helium. Consequently, precise optimization of instrument parameters is necessary to achieve maximum sensitivity. In this regard, three different temperatures (250 °C, 280 °C, and 300 °C) were tested for the ion source. The variation between them was found to be within 3%, so 280 °C was established as the definitive ion source temperature. The detector voltage plays a critical role in the process. Several voltages (1.1 kV, 1.3 kV, 1.5 kV, 1.6 kV, and 1.7 kV) were evaluated, alongside three injection volumes, as part of the experiment. Increasing the detector voltage beyond 1.3 kV and injecting 1.5–2 μL of the sample resulted in detector saturation at 100 μg kg−1 for approximately 30% of the pesticides in the multiresidue method. To avoid saturation while achieving maximum sensitivity, it was decided to reduce the injected sample volume to 1 μL and employ a detector voltage of 1.6 kV. It should be noted that excessively high voltage can lead to a decrease in the detector's lifespan. However, the manufacturer assures that MRM experiments can be conducted using detector voltages of up to 2 kV. In addition, 5 different glass liners were evaluated (experimental subsection 2.3). Those equipped with wool provided similar results; the different deactivation does not have a significant impact on peak shape or sensitivity. However, if the liner does not have wool, there is a significant decrease in the peak performance for a large number of pesticides.3.3 Sensitivity
To assess the system's sensitivity, the mixture of 150 compounds was fortified in three different matrices at varying concentrations (2, 5, 10, 20, 50, 100 and 200 μg kg−1). Sensitivity evaluation was conducted based on the number of identified compounds, adhering to the DG-SANTE criteria.22 The number of compounds identified in pepper, tomato, and zucchini is presented in Fig. 2. While the majority of compounds were identifiable at 10 μg kg−1, certain compounds, such as chlorfenapyr and phenothrin, weren't identified at this concentration in the studied matrices due to their limited sensitivity. In tomato and pepper, more than 80% of the compounds were identified at 5 μg kg−1, and the rate was 73% for zucchini. Additionally, around half of the method's compounds were detectable at 2 μg kg−1. The individual instrumental limits of quantification can be observed on the ESI Table.† This data emphasizes that the multiresidue method meets the sensitivity benchmarks of current analytical standards.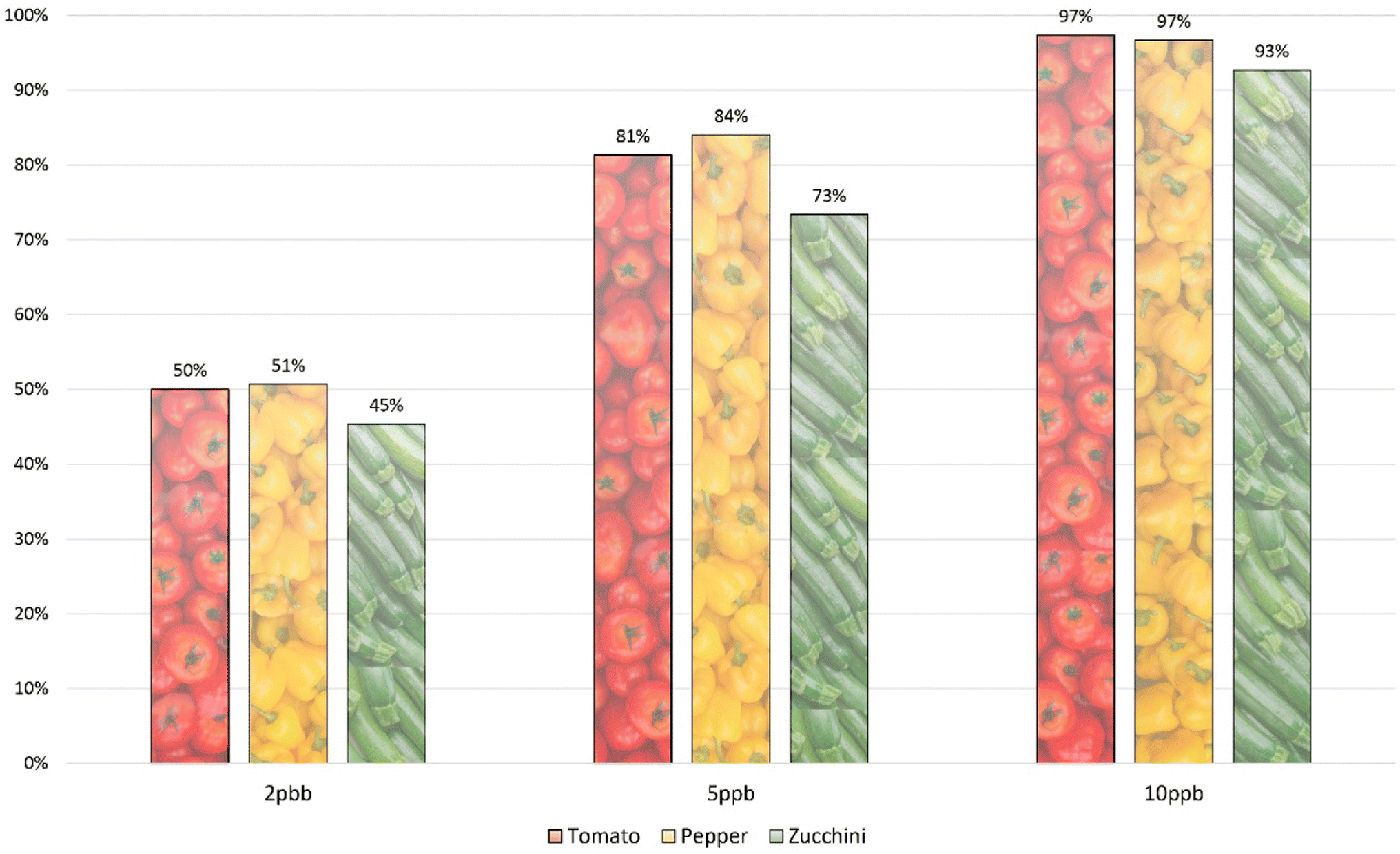 | ||
| Fig. 2 Percentage of identified compounds in the three matrices studied: tomato, pepper, and zucchini. | ||
3.4 Reproducibility, matrix effects and linearity
These validation parameters were evaluated for the 150 pesticides across the three matrices previously mentioned. Reproducibility was assessed by injecting each vial five times at a concentration of 10 μg kg−1 in each matrix over three consecutive days. We calculated the relative standard deviation (RSD). Most of the results were below 20%. Only 1% of compounds exceeded this threshold in tomato and pepper, and 3% in zucchini. These compounds that showed low area values due to sensitivity include phorate, propaphos, folpet, and sulprofos. More than half of the compounds exhibited RSDs below 10%: 63% for tomato, 59% for pepper, and 68% for zucchini. Regarding matrix effects, the tomato calibration curve was used as a reference. A modification of the signal between 0 and 20% is considered a low or non-existent matrix effect. However, alterations between 20 and 50% and those greater than 50% are deemed medium and strong matrix effects, respectively. In pepper, 86% of the compounds showed no matrix effects, 8% displayed medium matrix effects, and 4% indicated strong matrix effects. For zucchini, 72% showed no matrix effects, 19% medium effects, and 7% strong effects. Finally, linearity was examined in the range of 2–200 μg kg−1. In most instances, the response was linear throughout the entire range. For those pesticides which remained unidentified at 2, 5, or 10 μg kg−1, the linearity corresponds to a more narrow range (see ESI Table†).3.5 Real samples
To complete the study, 15 real samples were analyzed using this system and compared with the results obtained by a GC-MS/MS system using helium as a carrier gas. The parameters of the system available in the laboratory used for the comparison can be found elsewhere.23,24 The results of the nine sample that presented pesticides can be observed in Table 1. Quantification was conducted using two different matrix-matched calibration curves in tomato and orange matrices. Nine samples tested positive, and they were from various matrices. The outcomes between the two systems were comparable; however, the hydrogen GC-MS/MS typically quantified slightly lower than the helium system. In all instances, the differences were below 30%, typically around 20% or less, with the exception of pyriproxyfen in the orange sample, where the difference reached 36%. Additionally, the latest proficiency test sample from EURL-FV, which had a melon matrix, was analyzed, and the results fell within the acceptable Z-score range (chlorpyrifos: −1.1, chlorpyrifos-methyl: −0.8, diazinon: −0.7, fenazaquin: −0.5, fenitothrion: −0.4, flutriafol: 0.0, mepanipyrim: −0.3, profenofos: −0.6 and pyriproxyfen: −0.2). This data demonstrates that using hydrogen as a carrier gas does not adversely impact the sample quantification.| Conc. (μg kg−1) | GC-MS/MS system | Potato | Spinach | Lemon | Peach | Pear | Grape | Orange | Nectarine | Tomato |
|---|---|---|---|---|---|---|---|---|---|---|
| Boscalid | Hydrogen | 0.036 | ||||||||
| Helium | 0.046 | |||||||||
| Cypermethrin | Hydrogen | 0.008 | ||||||||
| Helium | 0.011 | |||||||||
| Cyprodinil | Hydrogen | 0.015 | 0.058 | 0.232 | 0.106 | |||||
| Helium | 0.017 | 0.07 | 0.289 | 0.137 | ||||||
| Flutolanil | Hydrogen | 0.024 | ||||||||
| Helium | 0.031 | |||||||||
| Lambda-cyhalothrin | Hydrogen | 0.109 | ||||||||
| Helium | 0.111 | |||||||||
| Pyrimethanil | Hydrogen | 0.009 | ||||||||
| Helium | 0.011 | |||||||||
| Pyriproxyfen | Hydrogen | 0.014 | 0.009 | |||||||
| Helium | 0.018 | 0.014 |
4. Conclusion
Our comprehensive evaluation of the GC-MS/MS system utilizing hydrogen as the carrier gas has underscored its potential in pesticide residue analysis. Utilizing the chromatographic enhancements provided by hydrogen, a rapid method was established, capable of analyzing 150 pesticides within a 12 minute timeframe. The ion source inertness check revealed the necessity of regular monitoring, particularly if the system has been dormant. Through system optimization, we determined the optimal parameters for injection volume, ion source temperatures, and detector voltage. The sensitivity analysis, based on the DG-SANTE criteria, found that most compounds were identifiable at 5 μg kg−1 across the matrices examined. The reproducibility, matrix effects, and linearity assessments reaffirm the system's proficiency in achieving reliable and consistent results. Comparison of real sample analyses between the hydrogen and helium GC-MS/MS systems indicated similar outcomes. This evaluation demonstrates that using hydrogen as a carrier gas remains a valid alternative without compromising the quantification quality.Author contributions
Víctor Cutillas (conceptualization, methodology, validation, investigation, writing – original draft, writing – review & editing, visualization), Guillermo García Gallego (methodology, validation), María Murcia-Morales (methodology, validation) Carmen Ferrer (conceptualization, writing – review & editing) Amadeo Rodríguez Fernández-Alba (conceptualization, writing – original draft, writing – review & editing, supervision, project administration, funding acquisition).Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors received funding support provided by the European Commission, DG SANTE (Grant decision SI2.870831). Views and opinions expressed are however those of the author(s) only and do not necessarily reflect those of the European Union or the European Health and Digital Executive Agency (HaDEA). Neither the European Union nor the granting authority can be held responsible for them. The authors would like to thank Uwe Oppermann for his continuous support of the work.References
- D. C. Harris, Quantitative Chemical Analysis, W. H. Freeman and Company, New York City, New York, USA, 9th edn, 2015 Search PubMed.
- P. Airey, T. Hinton, J. Twining, Chapter 1 – The Scientific Basis, in Radioactivity in the Environment, ed. J. R. Twining, Elsevier, vol.18, 2012, pp. 1–57, ISSN 1569-4860 Search PubMed.
- W. P. Halperin, Nat. Phys., 2014, 10, 467–470 Search PubMed.
- W. J. Nuttall, R. H. Clarke and B. A. Glowacki, Nature, 2012, 485, 573–575 CrossRef CAS PubMed.
- K. J. Margolin Eren, H. F. Prest and A. Amirav, J. Mass Spectrom., 2022, 57, e4830 CrossRef CAS PubMed.
- C. W. Taylor and S. A. Bowden, J. Chromatogr. A, 2023, 1697, 463989 CrossRef CAS PubMed.
- S. Saito-Shida, M. Nagata, S. Nemoto and H. Akiyama, Food Addit. Contam.: Part A, 2021, 38, 125–135 CrossRef CAS.
- C. A. Cramers, H.-G. Janssen, M. M. van Deursen and P. A. Leclercq, J. Chromatogr. A, 1999, 856, 315–329 CrossRef CAS.
- P. Korytár, H.-G. Janssen, E. Matisová and U. A. T. Brinkman, TrAC, Trends Anal. Chem., 2002, 21, 558–572 CrossRef.
- M. Galletta, M. Zoccali, N. Jones, L. Mondello and P. Q. Tranchida, Anal. Bioanal. Chem., 2022, 414, 6371–6378 CrossRef CAS PubMed.
- J. A. Muñoz-Guerra, P. Prado and S. V. García-Tenorio, J. Chromatogr. A, 2011, 1218, 7365–7370 CrossRef PubMed.
- C. N. Nnaji, K. C. Williams, J. M. Bishop and G. F. Verbeck, Sci. Justice, 2015, 55, 162–167 CrossRef PubMed.
- P. Benedetti, E. Guerriero, S. Mosca and M. Rotatori, J. Sep. Sci., 2017, 40, 3469–3478 CrossRef CAS PubMed.
- F. Bernardoni, H. M. Halsey, R. Hartman, T. Nowak and E. L. Regalado, J. Pharm. Biomed. Anal., 2019, 165, 366–373 CrossRef CAS PubMed.
- J. Buse, J. L. Robinson, R. Shyne, Q. Chi, D. Affleck, D. Duce and I. Seiden-Long, Clin. Biochem., 2019, 73, 98–104 CrossRef CAS.
- E. Yamada, N. Kitabayashi, A. K. Hayashi and N. Tsuboi, Int. J. Hydrogen Energy, 2011, 36, 2560–2566 CrossRef CAS.
- A. L. Sánchez and F. A. Williams, Prog. Energy Combust. Sci., 2014, 41, 1–55 CrossRef.
- N. B. Smith, J. Chromatogr. A, 1982, 249, 57–63 CrossRef CAS.
- A. Watanabe, C. Watanabe, R. R. Freeman, N. Teramae and H. Ohtani, Anal. Chem., 2016, 88, 5462–5468 CrossRef CAS PubMed.
- J. Hinshaw, LCGC North Am., 2011, 29, 36–43 CAS.
- M. Anastassiades, S. J. Lehotay, D. Štajnbaher and F. J. Schenck, J. AOAC Int., 2003, 86, 412–431 CrossRef CAS.
- SANTE 11312/2021 – Analytical Quality Control and Method Validation Procedures for Pesticide Residues Analysis in Food and Feed, European Commission, 2021, pp. 1–51 Search PubMed.
- M. Murcia Morales, M. J. Gómez Ramos, P. Parrilla Vázquez, F. J. Díaz Galiano, M. García Valverde, V. Gámiz López, J. Manuel Flores and A. R. Fernández-Alba, Sci. Total Environ., 2020, 710, 136288 CrossRef CAS PubMed.
- E. Hakme, A. Lozano, S. Uclés and A. R. Fernández-Alba, Anal. Bioanal. Chem., 2018, 410, 5491–5506 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ay02119j |
| This journal is © The Royal Society of Chemistry 2024 |