
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Analysis of tryptophan metabolites and related compounds in human and murine tissue: development and validation of a quantitative and semi-quantitative method using high resolution mass spectrometry†
Sandy
Abujrais
 ab,
S. J. Kumari A.
Ubhayasekera
ab and
Jonas
Bergquist
*ab
ab,
S. J. Kumari A.
Ubhayasekera
ab and
Jonas
Bergquist
*ab
aAnalytical Chemistry and Neurochemistry, Department of Chemistry – BMC, Uppsala University, Box 599, 75124, Uppsala, Sweden. E-mail: jonas.bergquist@kemi.uu.se
bThe ME/CFS Collaborative Research Centre at Uppsala University, Sweden
First published on 29th January 2024
Abstract
This study explores the metabolic differences between human and murine plasma in addition to differences between murine subcutaneous and visceral white adipose tissue. A quantitative and semi-quantitative targeted method was developed and validated for this purpose. The quantitative method includes tryptophan and its metabolites in addition to tyrosine, phenylalanine, taurine, B vitamins, neopterin, cystathionine and hypoxanthine. While the semi-quantitative method includes; 3-indoleacetic acid, 5-hydroxyindoleacetic acid, acetylcholine, asymmetric dimethylarginine, citrulline and methionine. Sample preparation was based on protein precipitation, while quantification was conducted using ultrahigh-performance liquid chromatography coupled to a quadrupole Orbitrap tandem mass spectrometer with electrospray ionization in the parallel reaction monitoring (PRM) mode. The low limit of quantification for all metabolites ranged from 1 to 200 ng mL−1. Matrix effects and recoveries for stable isotope labelled internal standards were evaluated, with most having a coefficient of variation (CV) of less than 15%. Results showed that a majority of the analytes passed both the intra- and interday precision and accuracy criteria. The comparative analysis of human and murine plasma metabolites reveals species-specific variations within the tryptophan metabolic pathway. Notably, murine plasma generally exhibits elevated concentrations of most compounds in this pathway, with the exceptions of kynurenine and quinolinic acid. Moreover, the investigation uncovers noteworthy metabolic disparities between murine visceral and subcutaneous white adipose tissues, with the subcutaneous tissue demonstrating significantly higher concentrations of tryptophan, phenylalanine, tyrosine, and serotonin. The findings also show that even a semi-quantitative method can provide comparable results to quantitative methods from other studies and be effective for assessing metabolites in a complex sample. Overall, this study provides a robust platform to compare human and murine metabolism, providing a valuable insight to future investigations.
1 Introduction
Tryptophan (Trp) is an essential amino acid and has distinctive molecular properties. It is the only amino acid with two rings in its side-chain.1 Trp is degraded into three major pathways; the kynurenine, serotonin and indole as illustrated in the ESI Fig. S1.†2 In humans, kynurenine is primarily synthesized endogenously from tryptophan. The liver is primarily responsible for the production of kynurenine under physiological conditions through tryptophan 2,3-dioxygenase (TDO) enzyme, which is primarily expressed in the liver. TDO can also be present in other cell types like neurons.3–5 Additionally, a smaller portion of kynurenine is produced by indole amine dioxygenase (IDO) enzyme, which is present in various parts of the body.6 Increasing levels of inflammatory signals, such as interleukins, can activate the IDO enzyme, whereas stress hormones like cortisol can activate the TDO enzyme.7 Kynurenine levels are thus affected by our dietary intake of tryptophan, TDO and IDO activity, stress hormones and inflammatory signals. Kynurenine has two main pathways: i.e. to form kynurenic acid by the enzyme kynurenine aminotransferase (KAT) or to form 3-hydroxyanthranilic acid by kynureninase and or/kynurenine 3-monooxygenase (KMO).8 The kynurenine pathway plays a critical role in the development of neurological disorders, including Alzheimer's disease,9 Parkinson's disease10 and major depression,11 as it affects the nervous system and energy metabolism through its downstream metabolites. This makes the pathway a crucial target for treatment.2Tryptophan that escape hepatic degradation can traverse the blood–brain barrier and undergo hydroxylation, yielding 5-hydroxytryptophan which transforms into serotonin via decarboxylation catalyzed by aromatic L-amino acid decarboxylase.12 Serotonin is best known as a key neurotransmitter responsible for regulating mood and producing melatonin, a hormone that helps control sleep patterns.12 The indole pathway is a key part of tryptophan metabolism that involves the conversion of tryptophan into indole by gut bacteria. This produces substances like 3-hydroxy indole acetic acid, which has anti-inflammatory effects.13
The measurement of analytes beyond tryptophan metabolites is essential for gaining a comprehensive understanding of the complex biochemical pathways. For example, tyrosine and phenylalanine compete with tryptophan for transport across the blood–brain barrier. High levels of tyrosine and phenylalanine limit tryptophan's entry, decreasing its availability for serotonin synthesis. Measuring all three amino acid concentrations can thus help us assess metabolic competition.14 Also, adequate levels of B-vitamins, such as riboflavin (B2) and pyridoxal phosphate (PLP), are crucial for their role as essential coenzymes in the metabolic process of tryptophan. Similarly, assessing the levels of hypoxanthine and neopterin can provide insights into oxidative stress and activation of the kynurenine pathway, which have been linked to inflammation and various diseases such as cancer and autoimmune disorders.15
Metabolomic analysis utilizes mass spectrometry techniques, often combined with chromatography, to effectively reduce sample complexity and minimize ion suppression. This study employed liquid chromatography and quadrupole Orbitrap tandem MS in parallel reaction monitoring (PRM) mode. Our method enables the simultaneous monitoring of multiple precursor and product ion transitions with high resolution and mass accuracy, offering flexibility in selecting fragment ion transitions after data acquisition.16
This study offers a solution to the challenges encountered in analyzing multiple analytes in human and murine tissue samples. These challenges arise from variations in the sample matrix, polarity, ionization effects, and endogenous levels, which hinder the full understanding of tryptophan metabolism. The current techniques are limited in their ability to measure only a small number of metabolites. For instance, a study conducted by Silva et al. utilized three different detection methods to examine eight tryptophan-related compounds in human and murine plasma.17 This novel combination of analytes and unique application in the samples opens up new possibilities for a comprehensive understanding of tryptophan metabolism.
The aim of this research is to develop and validate a novel quantitative and semi-quantitative analytical method that utilizes liquid chromatography coupled to high resolution mass spectrometry (LC-HRMS) to accurately analyse more than 20 tryptophan metabolites and related compounds, as shown in the ESI Fig. S2,† in both human and murine tissue. The development of this method will facilitate future investigations into the relationship between these compounds and various physiological and pathological processes. Furthermore, it is of major importance to be able to compare results from animal models with clinical human samples, and thus facilitate further investigations of novel biomarkers and interventions. Our method is rapid with simple sample preparation, making it a valuable tool for investigating tryptophan metabolism across different species. However, some compounds did not meet the validation criteria and will require further adjustment.
2 Materials and methods
2.1 Chemicals
Purified deionized water with 18 MΩ cm conductivity was obtained using a Milli-Q water purification system (Millipore-Bedford, MA, USA). Whenever feasible, the analyte standards as shown in the ESI Table S1† were purchased from two different sources in order to allow independent preparation of calibration and quality control samples. Merck Life Science AB (Solna, Sweden) was the provider of the aforementioned standards except for quinolinic acid from Tocris Bioscience (Bristol, UK), dopamine hydrochloride and pantothenic acid calcium from the European Pharmacopoeia Reference Standard, serotonin and 5-hydroxytryptophan hydrochloride from Alfa Aesar, melatonin from Cayman Chemicals (Michigan, USA), and riboflavin from Supelco, while cyanocobalamin was sourced from Cerilliant. The vendor for stable isotope labeled internal standards as shown in the ESI Table S1† was Alsachim (Illkirch Graffenstaden, France), except for phenylalanine-[2H5], thiamine-[13C3], pyridoxine-[2H3], biotin-[2H2], folate-[13C5,15N] and cyanocobalamin-[13C7] were purchased from Merck. Cambridge isotopes was the supplier of cystathionine-[2H4], tyrosine-[2H7], and dopamine-[2H4], while Supelco provided nicotinamide-[13C6]. Neopterin-[13C5] was acquired from Toronto Research Chemicals (Ontario, Canada) and serotonin-[2H4] hydrochloride from Cayman Chemicals. Lastly, methanol (LC/MS grade) and acetonitrile (LC/MS grade) were purchased from Fisher Scientific (Göteborg, Sweden), along with formic acid (LC/MS grade), ammonia solution 25%, and dimethyl sulfoxide (DMSO) from Merck Life Science AB (Solna, Sweden).2.2 Calibration, quality controls and internal standard solutions
Standard stock solutions 1 mg mL−1 of picolinic acid, 3-hydroxyanthranilic acid, anthranilic acid, nicotinamide, phenylalanine, melatonin, serotonin, pantothenic acid, biotin, and cyanocobalamin were prepared in 100% methanol. Quinolinic acid, kynurenine, tryptophan, and 5-hydroxytryptophan were dissolved in a mixture of 50% acetonitrile and 50% water. 3-Hydroxykynurenine was prepared in a solution of 50% MeOH and 50% water with 0.1% formic acid. Xanthurenic acid, kynurenic acid, neopterin, and tyrosine were dissolved in 0.25 M NH4OH. Pyridoxal 5′-phosphate, hypoxanthine, taurine, and cystathionine were dissolved in Milli-Q water. Dopamine-hydrochloride, riboflavin, and folate were dissolved in DMSO. These standard stock solutions were prepared in duplicate and pooled into three groups (A, B and C) to create calibration and quality control solutions, as outlined in the ESI Table S2.† Stock solution A had the lowest concentration of the analyzed compounds, while stock B had an intermediate concentration; these two stocks were both diluted in methanol while stock C had the most concentrated level of analytes and was also diluted in methanol. Stock C was used for serial dilutions to prepare the calibration standards at eight different levels and quality control (QC) solutions at four levels; LLOQ, low, medium and high concentration levels for each targeted analyte as in the ESI Table S3.† Methanol was used for preparing all solutions. The final concentration for each analyte of the calibration and QC standards can be seen in the ESI Table S4.†2.3 Biological samples
2.4 Sample preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 10 min at 4 °C and the supernatant 425 μL was collected and concentrated using nitrogen stream supplied by TurboVap (Biotage, Uppsala, Sweden) at the flow rate of 1.2 mL min−1 at 25 °C. After this, the dry residues were reconstituted in 100 μL of 0.1% formic acid in water and vortexed for 15 s at 1600 rpm. At last, the samples were centrifuged for 10 min at 10000 × g at 4 °C to obtain the clear extract and the supernatant was transferred to amber LC vials for LC-HRMS analysis.
000 × g for 10 min at 4 °C and the supernatant 425 μL was collected and concentrated using nitrogen stream supplied by TurboVap (Biotage, Uppsala, Sweden) at the flow rate of 1.2 mL min−1 at 25 °C. After this, the dry residues were reconstituted in 100 μL of 0.1% formic acid in water and vortexed for 15 s at 1600 rpm. At last, the samples were centrifuged for 10 min at 10000 × g at 4 °C to obtain the clear extract and the supernatant was transferred to amber LC vials for LC-HRMS analysis.
2.5 LC-HRMS analysis
Analyses were conducted with a Waters Acquity UHPLC (Waters™) coupled to a high-resolution Q Exactive™ hybrid quadrupole-Orbitrap mass spectrometer (Thermo Scientific™). The LC column used was a Waters™ HSS T3 (1.8 μm: 2.1 × 100 mm) and the column oven temperature was set at 30 °C. The mobile phase consisted of Milli-Q water with 0.6% HCOOH (100:0.6%, v/v) (mobile phase A), and methanol with 0.6% HCOOH (100:0.6%, v/v) (mobile phase B) were delivered as a gradient at a flow rate of 300 μL min−1, with a sample injection of 5 μL. The gradient was as follows: 1–10% B (0.0–4.0 min), 10–90% B (4.0–7.0 min), hold at 90% B (7.0–8.0 min), 90–1% B (8.0–8.1 min) followed by equilibration of the column at 1% B (8.1–9.0 min). The heated electrospray ionization (HESI) probe parameters were as follows: sheath gas flow rate, 40 units; auxiliary gas unit flow rate, 15 units; sweep gas flow rate, 2 units; capillary temperature, 360 °C; auxiliary gas heater temperature, 400 °C; spray voltage, 4.00 kV; and S lens, RF level 60. Parallel reaction monitoring (PRM) with an inclusion list, as seen in the ESI Table S1,† was utilized for mass spectrometry detection with a 1 m/z isolation window in positive electrospray ionization mode (ESI+) and acquired at a resolving power of 17500 FWHM (full width half maximum) at m/z 200. The automatic gain control (AGC) was set at 2 × 105 and the injection time was set to 50 ms. Tune interface software (Tune v2.9, Thermo Scientific) was used for tuning and optimizing the analytes and Xcalibur® software (Version 4.1, Thermo Scientific) was used for data acquisition and processing by integrating selected product ion chromatographic peak area to calculate the metabolites concentration.
2.6 Method validation
The guidelines from the European Medicines Agency (EMA) on bioanalytical method validation were followed when carrying out the method validation. This guideline was released on July 21, 2011 and is titled “Guideline on bioanalytical method validation EMEA/CHMP/EWP/192217/2009”.2.7 Semi-quantification analysis
A semi-quantitative analysis of six analytes was performed in human plasma (n = 24) and murine plasma (n = 6) to estimate their concentrations using an internal standard of close retention time. The formulae, RF = peak area/concentration and C suspect concentration = peak area suspect compound/RF similar compound,19 were utilized to estimate the concentration of 3-indoleacetic acid using melatonin-[2H4] as IS, 5-hydroxyindoleacetic acid using kynurenine-[13C6]. Acetylcholine, asymmetric dimethylarginine and citrulline using thiamine-[13C3]. Lastly methionine using quinolinic acid-[13C4,15N]. The mzCloud database was used for mass transitions selections and appropriate collision energies for the monitored parent ions as shown in the ESI Table S6.†3 Results and discussion
Method development involved testing various solvents for protein precipitation, including methanol, methanol with water and 0.1% formic acid, and acetonitrile. Reconstitution solvents such as a mixture of methanol and water (1:9) and 0.1% formic acid in water were also compared. Our findings indicated that using methanol with 0.1% formic acid and reconstituting with 0.1% formic acid in water yielded slightly higher concentrations, although not significantly different from other solvent combinations. During our testing, we also encountered issues with double peaks for pyridoxine, pyridoxal phosphate, neopterin, hypoxanthine, and dopamine when using 0.1% formic acid in both mobile phases A and B. To address this problem, we increased the concentration of formic acid to 0.6% in both mobile phases A and B. This adjustment successfully resolved the issue and produced one peak in the chromatogram as shown in ESI Fig. S3.† Implementing high resolution in the measurement of complex samples, such as plasma and adipose tissue, is essential for accurate analysis and interpretation of results. The Orbitrap analyzer's high resolution allows for the separation of metabolite peaks in complex samples, reducing potential interferences and enhancing the accuracy of quantification. The LC-HRMS method developed in this study used retention times of precursor ions and at least one fragment ion to accurately measure metabolite levels as in the ESI Table S1.† The method was found to be effective in quickly analyzing 29 metabolites and estimate the concentration of six compounds in just 10 minutes. However, further adjustments are needed for some of the compounds as summarized in the ESI Table S7.†
3.1 Method validation
3.2 Application of the method to biological samples
Comparative analysis of murine and human plasma revealed that the concentrations of most of the compounds quantified were higher in murine plasma as shown in Fig. 1, except for kynurenine and quinolinic acid, with the median concentration of most analytes in murine plasma ranging from 4 ng mL−1 (biotin) to 16020 ng mL−1 (tryptophan). The comparison of tryptophan metabolites in mice and humans revealed an opposite trend; the levels of plasma serotonin in mice were found to be higher than those of kynurenine, whereas the opposite was observed in humans. This disparity can be attributed to the varying tryptophan metabolism and enzyme activities in the kynurenine pathway among different species, particularly in the central nervous system. For example, rats have lower IDO1 induction in response to immune stimulation compared to mice, while gerbils are more similar to humans in their increase of quinolinic acid levels during inflammation.24 These differences revealed the necessity to select the right species when studying the physiological and pathological effects of kynurenine pathway activity.
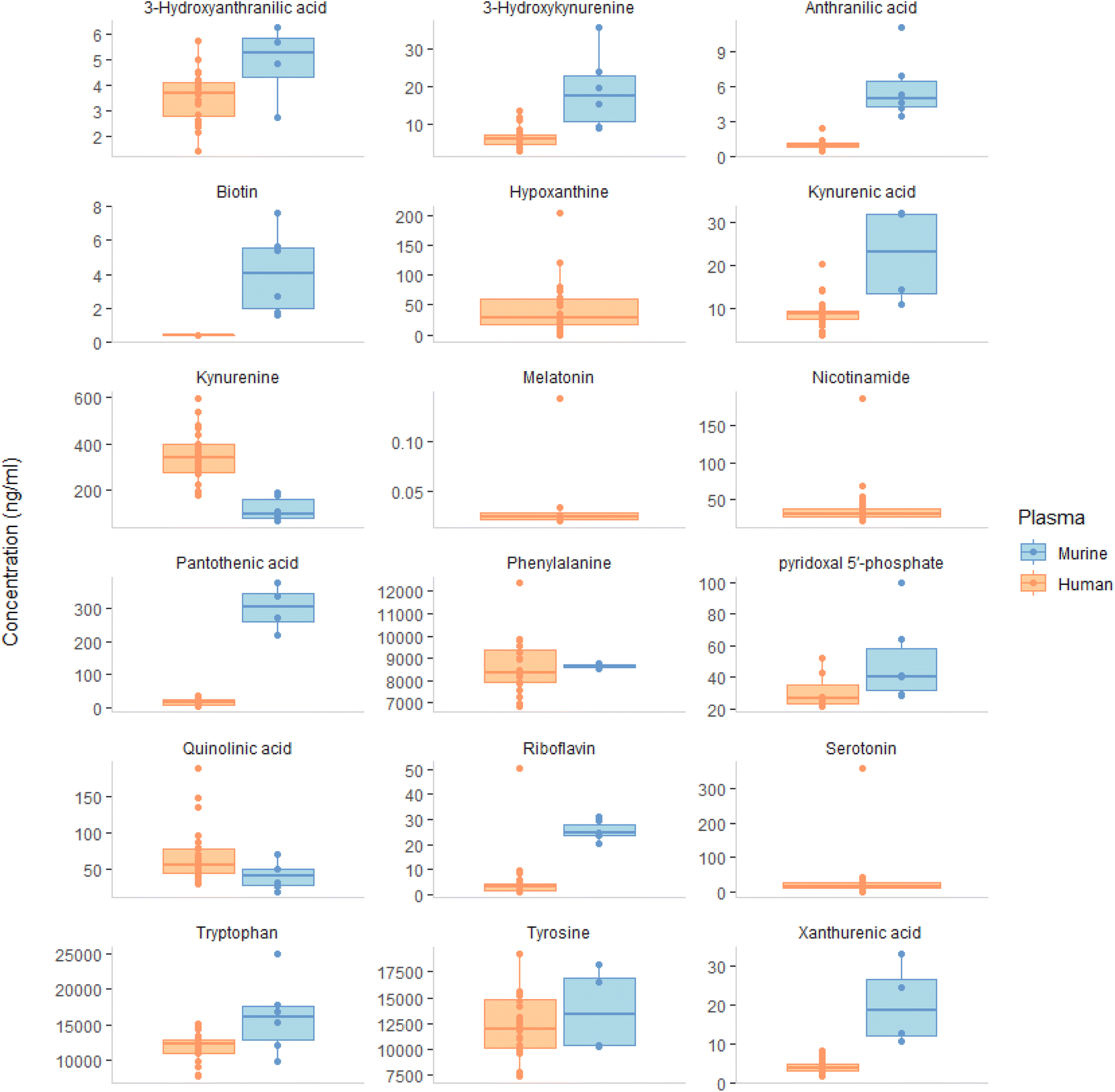 | ||
| Fig. 1 The median concentrations of the aromatic amino acids; tryptophan and its metabolites, tyrosine and phenylalanine. In addition to B vitamins (biotin, pantothenic acid, pyridoxal 5′-phosphate and riboflavin) and the oxidation marker hypoxanthine were determined in ng mL−1 in human (n = 24) and murine (n = 6) plasma from healthy controls. Comparing the concentration of analytes in murine and human plasma, the results showed that most compounds were higher in murine plasma. The only two exceptions were kynurenine and quinolinic acid. The analyte concentration in murine plasma ranged from 4 ng mL−1 (biotin) to 16020 ng mL−1 (tryptophan). Nicotinamide and serotonin had median values above ULOQ, while melatonin and hypoxanthine were not detected and thus omitted from the boxplot results. | ||
In our study, we measured the levels of (phenylalanine, pantothenic acid, tyrosine, 3-hydroxyanthranilic acid, kynurenic acid, and xanthurenic acid). The median concentrations of these analytes ranged from 5 to 8675 ng mL−1, with some values exceeding the upper limit of quantification (ULOQ) and thus being excluded from the calculation with the exception of 3-hydroxyanthranilic acid, which fell below LLOQ. Nicotinamide and serotonin had median values above ULOQ, while melatonin and hypoxanthine, were not detected and therefore excluded from the boxplot results. Our observed high levels of murine serotonin are consistent with previous research by Rubio et al.25
221, 11985, and 8361 ng mL−1), respectively. Our study found that biotin levels were much lower than another study with an average of 22.8 ng mL−1 for 2 out of 20 participants.26 Our riboflavin result of 3.01 ng mL−1 was similar to those reported from 4–9 ng mL−1.26–28 Moreover, our result for 3-hydroxy anthranilic acid concentration was 5.2 ng mL−1, which was consistent with a study conducted using Q-Exactive method in 214 healthy control subjects.29 The levels of tryptophan and tyrosine in our study were comparable to those found in other studies18,27,29–34 as displayed in ESI Fig. S8.† Tryptophan, tyrosine and phenylalanine, are used to produce neurotransmitters such as serotonin, dopamine, norepinephrine and GABA which contribute to physical and emotional wellbeing.34 Regarding the serotonin pathway, our method wasn't sensitive enough to measure melatonin and matrix effect might hinder the detection of 5 hydroxytryptophan in plasma. According to other studies, 5 hydroxytryptophan was found ranging from 0.9 to 10.9 ng mL−1 (ref. 29–34) and melatonin was found at 0.007 ng mL−1 with 102 participants.35 Our value for serotonin 17.1 ng mL−1 was within the 1.8–103 ng mL−1 range reported in other studies.34,36 Ultimately, it appears that using platelets are a superior marker of serotonin levels than plasma, as they reflect long-term serotonin activity and are not impacted by dietary intake. Siliconized glassware and plastic ware should be used to improve accuracy in serotonin measurement.37
A range of tryptophan metabolites reported from previous studies included anthranilic acid 0.5–13.4 ng mL−1,29,32 3 hydroxyanthranilic acid 3.2–10.7 ng mL−1,32,36 kynurenic acid 1.9–14 ng mL−1,32,36 3-hydroxykynurenine 1.9–47 ng mL−1,18,36 xanthurenic acid 1.2–68.1 ng mL−1,29,38 nicotinamide 21.1–147.3 ng mL−1,26,39 quinolinic acid 39–180 ng mL−1,36,38 kynurenine 43.7–1790 ng mL−1,34,36 and picolinic acid 3.2–49.4 ng mL−1.29,33 Using our method, it was observed that the isomers picolinic acid and niacin were unable to be differentiated through chromatography. Others have achieved to separate those compounds through the addition of 0.5% formic acid and 1 mM ammonium formate.36
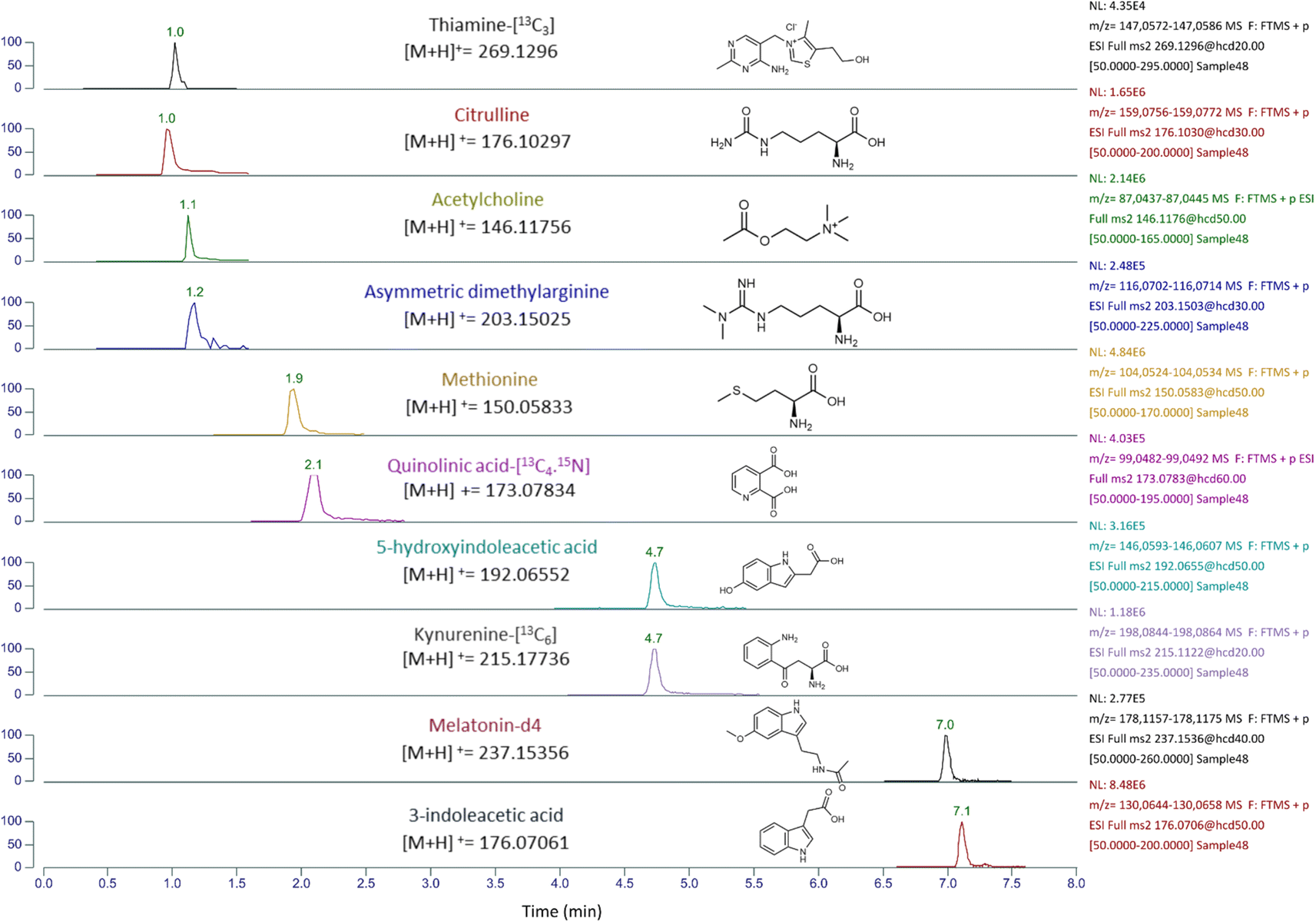 | ||
Fig. 2 An extracted ion chromatogram, of the semi quantification analysis, was generated for thiamine-[13C3] which was used as an internal standard (IS) to estimate the concentrations of citrulline, acetylcholine, and asymmetric dimethylarginine, which all elute at a similar retention time (RF similar compound). Additionally, quinolinic acid-[13C4,15N] was used as an IS for methionine, kynurenine-[13C6] as an IS for 5-hydroxyindoleacetic acid, and melatonin-[2H4] as an IS for 3-indoleacetic acid. The concentration estimation of these analytes was performed using the equations:  | ||
There are several potential explanations for why certain analytes may have failed to pass in the study. One possible reason could be the instability of these analytes to various environmental factors, such as light, heat, and acidity. For example, compounds like folic acid and cyanocobalamin may be prone to degradation in these conditions. Additionally, the combined effects of low ionization efficiency, matrix effects, and low concentration may have made it difficult to accurately detect and quantify other compounds listed in the ESI Table S7.† In order to address these issues, it is important to continue developing and exploring alternative LC gradient and sample preparation methods to mitigate the likelihood of analyte degradation.
4 Conclusions
In this study we present a fast and simple methodology for the investigation of tryptophan metabolism, applied in human plasma as well as murine plasma and adipose tissue. We want to highlight the importance of understanding the differences in tryptophan metabolism among species and tissues, as well as the need to consider species-specific differences when studying human pathological conditions as compared with animal models. Also, we provide valuable data for the need of future developments of standardized methodologies for accurate quantification of tryptophan metabolites and B vitamins. This study reveals that tryptophan metabolites vary widely across studies and can be influenced by different factors such as study population, sample preparation and types of analyzers used. Our high resolution mass spectrometric method for quantifying tryptophan metabolites and B vitamins in human and murine tissues offers a practical and effective option for use in clinical settings. In addition, our semi-quantitative technique allows for accurate identification of metabolites in a sample, with results similar to quantitative methods for some analytes.Author contributions
SA, KU, and JB designed the study. SA performed the experimental work. SA analysed the data. SA wrote the manuscript drafts. All authors revised and approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Open Medicine Foundation is acknowledged for the kind support of this study. We thank Bhagya Kolitha for his assistance in sample preparation, and also extend our thanks to Sooraj Baijnath and Juan R. Lopez-Edigo for their valuable input and review of our manuscript.References
- S. Khemaissa, S. Sagan and A. Walrant, Crystals, 2021, 11(9) DOI:10.3390/cryst11091032.
- S. Taleb, Front. Immunol., 2019, 10, 1–7 CrossRef PubMed.
- T. V. Lanz, S. K. Williams, A. Stojic, S. Iwantscheff, J. K. Sonner, C. Grabitz, S. Becker, L. I. Böhler, S. R. Mohapatra, F. Sahm, G. Küblbeck, T. Nakamura, H. Funakoshi, C. A. Opitz, W. Wick, R. Diem and M. Platten, Sci. Rep., 2017, 7, 1–13 CrossRef PubMed.
- M. Kanai, H. Funakoshi, H. Takahashi, T. Hayakawa, S. Mizuno, K. Matsumoto and T. Nakamura, Mol. Brain, 2009, 2, 1–16 CrossRef PubMed.
- R. Zulpaite, P. Miknevicius, B. Leber, K. Strupas, P. Stiegler and P. Schemmer, Int. J. Mol. Sci., 2021, 22, 1–32 CrossRef PubMed.
- A. A. B. Badawy, Int. J. Tryptophan Res., 2017, 10 DOI:10.1177/1178646917691938.
- S. Kim, B. J. Miller, M. E. Stefanek and A. H. Miller, Cancer, 2015, 121, 2129–2136 CrossRef CAS PubMed.
- J. Savitz, Mol. Psychiatry, 2020, 25, 131–147 CrossRef PubMed.
- M. Cespedes, K. R. Jacobs, P. Maruff, A. Rembach, C. J. Fowler, B. Trounson, K. K. Pertile, R. L. Rumble, S. R. Rainey-Smithe, C. C. Rowe, V. L. Villemagne, P. Bourgeat, C. K. Lim, P. Chatterjee, R. N. Martins, A. Ittner, C. L. Masters, J. D. Doecke, G. J. Guillemin and D. B. Lovejoy, Neurobiol. Dis., 2022, 171, 105783 CrossRef CAS PubMed.
- C. K. Lim, F. J. Fernández-Gomez, N. Braidy, C. Estrada, C. Costa, S. Costa, A. Bessede, E. Fernandez-Villalba, A. Zinger, M. T. Herrero and G. J. Guillemin, Prog. Neurobiol., 2017, 155, 76–95 CrossRef CAS PubMed.
- W. Marx, A. J. McGuinness, T. Rocks, A. Ruusunen, J. Cleminson, A. J. Walker, S. Gomes-da-Costa, M. Lane, M. Sanches, A. P. Diaz, P. T. Tseng, P. Y. Lin, M. Berk, G. Clarke, A. O'Neil, F. Jacka, B. Stubbs, A. F. Carvalho, J. Quevedo, J. C. Soares and B. S. Fernandes, Mol. Psychiatry, 2021, 26, 4158–4178 CrossRef CAS PubMed.
- E. Höglund, Ø. Øverli and S. Winberg, Front. Endocrinol., 2019 DOI:10.3389/fendo.2019.00158.
- H. Ormstad, C. S. Simonsen, L. Broch, D. M. Maes, G. Anderson and E. G. Celius, Mult. Scler. Relat. Disord., 2020, 46, 102533 CrossRef PubMed.
- H. Barone, Y. T. Bliksrud, I. B. Elgen, P. D. Szigetvari, R. Kleppe, S. Ghorbani, E. V. Hansen and J. Haavik, Am. J. Med. Genet., Part B, 2020, 183, 95–105 CrossRef CAS PubMed.
- R. Fuertig, A. Ceci, S. M. Camus, E. Bezard, A. H. Luippold and B. Hengerer, Bioanalysis, 2016, 8, 1903–1917 CrossRef CAS PubMed.
- J. Zhou, H. Liu, Y. Liu, J. Liu, X. Zhao and Y. Yin, Anal. Chem., 2016, 88, 4478–4486 CrossRef CAS PubMed.
- P. Valente-Silva, I. Cervenka, D. M. S. Ferreira, J. C. Correia, S. Edman, O. Horwath, B. Heng, S. Chow, K. R. Jacobs, G. J. Guillemin, E. Blomstrand and J. L. Ruas, Metabolites, 2021, 11(8) DOI:10.3390/metabo11080508.
- L. Whiley, L. C. Nye, I. Grant, N. Andreas, K. E. Chappell, M. H. Sarafian, R. Misra, R. S. Plumb, M. R. Lewis, J. K. Nicholson, E. Holmes, J. R. Swann and I. D. Wilson, Anal. Chem., 2019, 91, 5207–5216 CrossRef CAS PubMed.
- L. Malm, E. Palm, A. Souihi, M. Plassmann, J. Liigand and A. Kruve, Molecules, 2021, 26, 3524 CrossRef CAS PubMed.
- J. Marcos, N. Renau, O. Valverde, G. Aznar-Laín, I. Gracia-Rubio, M. Gonzalez-Sepulveda, L. A. Pérez-Jurado, R. Ventura, J. Segura and O. J. Pozo, J. Chromatogr. A, 2016, 1434, 91–101 CrossRef CAS PubMed.
- W. Hu, G. Yan, Q. Ding, J. Cai, Z. Zhang, Z. Zhao, H. Lei and Y. Z. Zhu, Biomed. Pharmacother., 2022, 150, 112957 CrossRef CAS PubMed.
- Q. Luong, J. Huang and K. Y. Lee, Biology, 2019, 8, 1–14 CrossRef PubMed.
- Y. Okamatsu-Ogura, M. Kuroda, R. Tsutsumi, A. Tsubota, M. Saito, K. Kimura and H. Sakaue, Metabolism, 2020, 113, 154396 CrossRef CAS PubMed.
- Y. Murakami and K. Saito, Int. J. Tryptophan Res., 2013, 6, 47–54 CAS.
- V. Y. Rubio, J. G. Cagmat, G. P. Wang, R. A. Yost and T. J. Garrett, Anal. Chem., 2020, 92, 2550–2557 CrossRef CAS PubMed.
- K. Meisser Redeuil, K. Longet, S. Bénet, C. Munari and E. Campos-Giménez, J. Chromatogr. A, 2015, 1422, 89–98 CrossRef CAS PubMed.
- O. Midttun, S. Hustad and P. M. Ueland, Rapid Commun. Mass Spectrom., 2009, 23, 1371–1379 CrossRef CAS PubMed.
- P. Chen, Y. Tang, Q. He, L. Liu, Z. Zhou, Y. Song, N. Zhang, B. Wang, H. Zhou, H. Shi and J. Jiang, J. Pharm. Biomed. Anal., 2022, 219, 114944 CrossRef CAS PubMed.
- R. Colle, P. Masson, C. Verstuyft, B. Fève, E. Werner, C. Boursier-Neyret, B. Walther, D. J. David, B. Boniface, B. Falissard, P. Chanson, E. Corruble and L. Becquemont, Psychiatry Clin. Neurosci., 2020, 74(2), 112–117 CrossRef CAS PubMed.
- B. A. Acids, A. Anesi, K. Berding, G. Clarke, C. Stanton, J. F. Cryan, N. Caplice, R. P. Ross, A. Doolan, U. Vrhovsek and F. Mattivi, J. Proteome Res., 2022, 21(5), 1262–1275 CrossRef PubMed.
- K. M. Ryan, K. A. Allers, D. M. McLoughlin and A. Harkin, Brain, Behav., Immun., 2020, 83, 153–162 CrossRef CAS PubMed.
- K. H. Chang, M. L. Cheng, H. Y. Tang, C. Y. Huang, Y. R. Wu and C. M. Chen, Mol. Neurobiol., 2018, 55, 6319–6328 CrossRef CAS PubMed.
- S. Adams, C. Teo, K. L. McDonald, A. Zinger, S. Bustamante, C. K. Lim, G. Sundaram, N. Braidy, B. J. Brew and G. J. Guillemin, PLoS One, 2014, 9, 1–28 Search PubMed.
- S. C. James, K. Fraser, J. Cooney, C. S. Günther, W. Young, R. B. Gearry, P. E. Heenan, T. Trower, J. I. Keenan, N. J. Talley, W. C. McNabb and N. C. Roy, Metabolites, 2023, 13 DOI:10.3390/metabo13020313.
- P. J. Eugster, M. Dunand, B. Grund, A. Ivanyuk, N. Fogarasi Szabo, C. Bardinet, K. Abid, T. Buclin, E. Grouzmann and H. Chtioui, Clin. Chim. Acta, 2022, 535, 19–26 CrossRef CAS PubMed.
- A. Anesi, K. Berding, G. Clarke, C. Stanton, J. F. Cryan, N. Caplice, R. P. Ross, A. Doolan, U. Vrhovsek and F. Mattivi, J. Proteome Res., 2022, 21(5), 1262–1275 CrossRef CAS PubMed.
- T. Audhya, J. B. Adams and L. Johansen, Biochim. Biophys. Acta, Gen. Subj., 2012, 1820, 1496–1501 CrossRef CAS PubMed.
- A. Desmons, L. Humbert, T. Eguether, P. Krasniqi, D. Rainteau, T. Mahdi, N. Kapel and A. Lamazière, J. Chromatogr. A, 2022, 1685, 463602 CrossRef CAS PubMed.
- G. R. Ibrahim, I. Shah, S. Gariballa, J. Yasin, J. Barker and S. S. Ashraf, Molecules, 2020, 25 DOI:10.3390/molecules25173932.
- J. Martens-Lobenhoffer and S. M. Bode-Böger, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2003, 798, 231–239 CrossRef CAS PubMed.
- K. Kawashima, T. Fujii, Y. Moriwaki and H. Misawa, Life Sci., 2012, 91, 1027–1032 CrossRef CAS PubMed.
- O. Galilea San Blas, F. Moreno Sanz, P. Herrero Espílez, B. Prieto García, F. V. Álvarez Menéndez, J. M. Marchante-Gayón and J. I. García Alonso, J. Anal. At. Spectrom., 2016, 31, 1885–1894 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ay01959d |
| This journal is © The Royal Society of Chemistry 2024 |