
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of a dispersive liquid–liquid microextraction method for the determination of plastic additives in seawater
María José
González-Castro
,
Jaime
Uribe-Ares
,
Soledad
Muniategui-Lorenzo
and
Elisa
Beceiro-González
 *
*
Departamento de Química, Facultade de Ciencias, Universidade da Coruña, Grupo Química Analítica Aplicada (QANAP), Instituto Universitario de Medio Ambiente (IUMA), Campus de A Coruña, 15071 A Coruña, Spain. E-mail: elisa@udc.es; Fax: +34981167065; Tel: +34981167000
First published on 26th February 2024
Abstract
A method using dispersive liquid–liquid microextraction (DLLME) prior to high performance liquid chromatography-diode array detection (HPLC-DAD) was developed to determine seven additives from the plastics industry (butylated hydroxytoluene, diisodecyl phthalate, irgafos 168, lawsone, quercetin, triclosan and vitamin E) in seawater samples. These compounds can reach seawater due to direct discharge from wastewater treatment plants and leaching from plastics and microplastics. The extraction was performed using 25 mL of seawater, 500 μL of 1-octanol (extraction solvent) and a stirring step instead of dispersive solvent. Additive concentrations were determined by LC-DAD on a C18 column with a mobile phase of acetonitrile and phosphoric acid aqueous solution (pH 3.5) by gradient elution. The analytical recoveries ranged from 82 to 93% for all compounds, except for lawsone (60%). Repeatability and intermediate precision were adequate with RSD < calculated values following the Horwitz equation at the concentration levels evaluated (0.06 and 0.24 mg L−1). All additives exhibited linear matrix calibration curves (R2 > 0.99). Detection limits ranged from 0.009 to 0.028 mg L−1 and quantification limits ranged from 0.027 to 0.084 mg L−1. Finally, the application of the method to real samples verified the method as accurate and applicable to seawater.
Introduction
Plastics have become a major environmental problem because of their increasing presence in marine ecosystems. These debris are a source of hazardous chemical compounds with negative effects on marine organisms. Thus, in addition to the harmful effects of the plastic particles ingested by organisms (zooplankton, bivalves, crustaceans, etc.), the associated chemical compounds can be transferred to them with toxic effects.1 To understand their impact, it is necessary to study the associated compounds which represent a risk to a healthy and productive ecosystem. However, research on chemicals associated with plastics has been limited.Chemical compounds associated with plastics include both additives added during manufacturing (plasticisers, antioxidants, stabilizers, flame retardants…) and organic compounds (polycyclic aromatic hydrocarbons, polychlorinated biphenyls, pesticides…) adsorbed on their surface from the surrounding environment.2 About this, it should be noted that additives have been studied considerably less than organic compounds adsorbed on the surface. However, environmental studies focusing on plastics should include additives as they can easily leach from the polymer matrix which implies a risk to marine organisms.3,4
Among the seven additives studied in this work, there are two natural antioxidants (quercetin and vitamin E) and a natural colouring agent with antimicrobial properties (lawsone). The latter one can be obtained from the leaves of the henna plant4 and quercetin is found naturally in some foods (onions, grapes and tea).5 The other four compounds are industrially produced chemicals. Butylated hydroxytoluene (BHT) is employed as an antioxidant in foodstuffs. Exposure to this compound can cause damage mainly due to its degradation products, especially in algae and fish.6 Irgafos 168 is also an antioxidant used in the manufacture of polymers for food preservation. Diisodecyl phthalate (DIDP) is a plasticiser belonging to the group of phthalates whose toxicity has been extensively investigated in recent years.7 Finally, triclosan is an antibacterial agent employed in household and personal care products. This additive accumulates in algae and its toxicity to aquatic organisms needs to be studied.8 The adverse effects on marine organisms of some of these additives are already known, but the lack of information on others makes it impossible to know what effects they may have. Therefore, it is necessary to develop analytical methods for the determination of these compounds in the marine environment.
High-performance liquid chromatography (HPLC) with ultraviolet (UV) detection or diode array detection (DAD) is the most commonly employed technique in the analysis of these additives.9–19 The reversed-phase mode using octadecyl columns10–14,20–23 and mixtures of acetonitrile/water or methanol/water as mobile phase11,12,14,16,17,22 is by far the most commonly used chromatographic conditions currently for the analysis of these additives.
Regarding the extraction procedures employed for the determination of additives in water samples, solid-phase extraction (SPE) is the most widely used method.15,20,24–26 However, this technique is being replaced by other fast techniques that minimize the waste of organic solvents such as liquid–liquid microextraction techniques. Thus, ultrasound-assisted salt-induced liquid–liquid microextraction followed by HPLC-DAD analysis has been used for the determination of triclosan in swimming pool, lake, and wastewater.16 Caldas et al. used dispersive liquid–liquid microextraction (DLLME) combined with liquid chromatography-tandem mass spectrometry (LC-MS/MS) to determine 58 organic compounds, including triclosan, in water samples. The DLLME procedure was carried out with 1-octanol as extraction solvent and acetone as dispersive solvent.27 Finally, DLLME has also been used in the extraction of some of the analytes under study from food samples. Asadollahi et al. used this microextraction technique for the analysis of quercetin in food samples.28 The sample pH was adjusted to ∼3 and methanol and 1-undecanol were employed as the dispersive and extraction solvent respectively. Moreover, a study for the analysis of vitamin E in infant formula by DLLME combined with HPLC-UV has also been developed.29 The advantages of DLLME are simplicity of operation, short extraction time, low cost and high enrichment factors.30 In addition, surface water matrices have little effect on DLLME and for seawater samples, the salt in the sample leads to increased ionic strength enhancing the extraction efficiency.
The present work has two objectives: (i) to develop a simple reversed-phase with ionic suppression for routine determination of the seven additives, and (ii) development and validation of a method based on DLLME for the extraction of the seven additives from seawater samples. It should be noted that the simultaneous chromatographic determination of these chemicals, as well as the use of DLLME for their extraction from seawater has not been reported before in the literature.
Experimental
Standards and reagents
Regarding additive standards, DIDP and vitamin E individual standards were supplied by AIMPLAS (an aliquot of the standards used in the fabrication of polymers). Lawsone, triclosan, BHT and irgafos 168 individual standards were supplied by Sigma Aldrich (Steinheim, Germany). Quercetin dihydrate was from Alfa Aesar (Thermo Fisher (Kandel) GmbH, Germany). The individual standard solutions of 2000 mg L−1 were prepared in different solvents: methanol (quercetin), acetonitrile (lawsone, triclosan, BHT and vitamin E) and ethyl acetate (DIDP and irgafos) by exact weighing of high-purity substances. To optimise the HPLC-DAD method, a mixture of all the compounds was prepared in acetonitrile containing 20 mg L−1 of each individual additive. To optimise the extraction method, since acetonitrile may compete with 1-octanol during extraction, a mixture of all the compounds containing 200 mg L−1 of each individual additive was prepared employing 1-octanol as solvent. Working solutions were daily prepared by appropriate dilution of 20 or 200 mg L−1 standard solutions. All solutions were stored at 4 °C in the dark. The additives studied are shown in Fig. 1.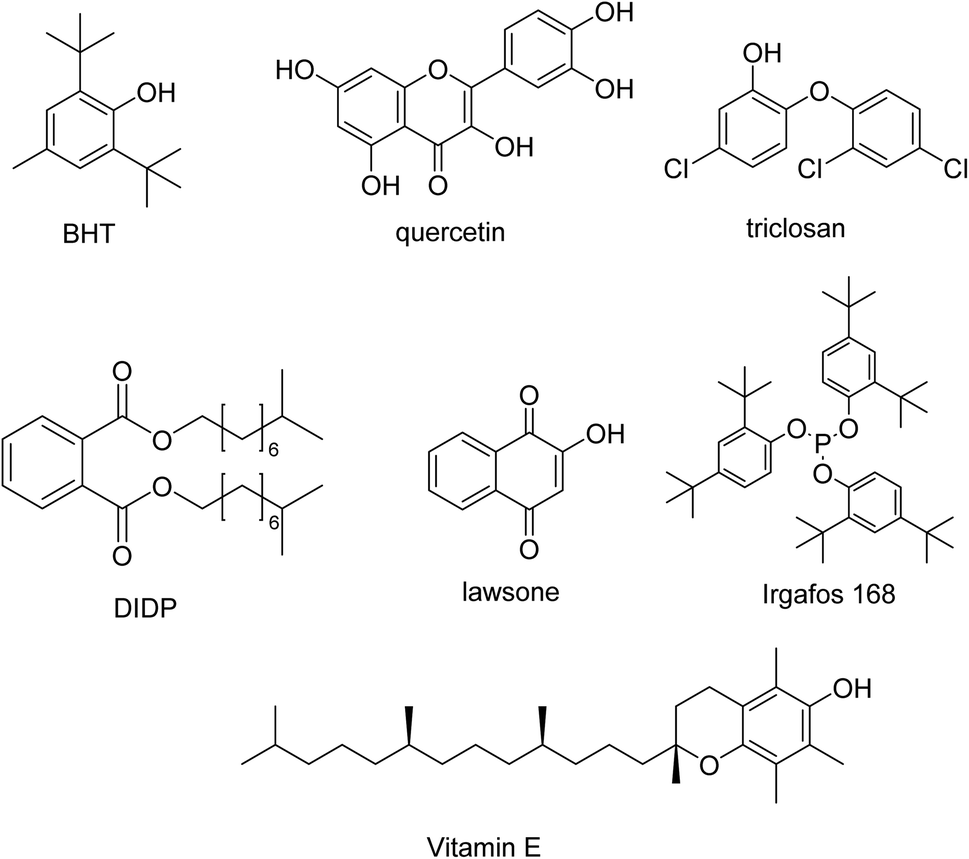 | ||
| Fig. 1 Structures of the additives. | ||
A synthetic saltwater solution of 35 g L−1 was prepared using sea salts from Sigma-Aldrich (chloride 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–20000 mg L−1; sodium 10700–11000 mg L−1; sulfate 2660 mg L−1; potassium 300–400 mg L−1; calcium 400 mg L−1; carbonate 140–200 mg L−1; boron 5.6 mg L−1; magnesium 1320 mg L−1; strontium 8.8 mg L−1 and insoluble matter ≤0.05%) (Steinheim, Germany). 1-Octanol Chromasolv® (grade HPLC 99%) was purchased from Sigma-Aldrich (Steinheim, Germany), acetonitrile HiPerSolv Chromanorm® (≥99.9%) was from VWR Chemicals (Leicester, UK) and phosphoric acid was from Panreac (Barcelona, Spain). Ultrapure water was obtained using a Millipore Milli-Q system (Millipore, Bedford, MA, USA). To prevent potential contamination, no plastic wares or equipment were used during sampling, extraction and analysis.
000–20000 mg L−1; sodium 10700–11000 mg L−1; sulfate 2660 mg L−1; potassium 300–400 mg L−1; calcium 400 mg L−1; carbonate 140–200 mg L−1; boron 5.6 mg L−1; magnesium 1320 mg L−1; strontium 8.8 mg L−1 and insoluble matter ≤0.05%) (Steinheim, Germany). 1-Octanol Chromasolv® (grade HPLC 99%) was purchased from Sigma-Aldrich (Steinheim, Germany), acetonitrile HiPerSolv Chromanorm® (≥99.9%) was from VWR Chemicals (Leicester, UK) and phosphoric acid was from Panreac (Barcelona, Spain). Ultrapure water was obtained using a Millipore Milli-Q system (Millipore, Bedford, MA, USA). To prevent potential contamination, no plastic wares or equipment were used during sampling, extraction and analysis.
Apparatus
The chromatographic system consisted of a 2695 pump with a 996 Diode Array Detector from Waters (Milford, MA, USA) and the Empower Pro software. The column was a stainless-steel column (150 mm × 4.6 mm ID, particle size 5 μm) packed with Hypersil GOLD C18 chemical bonded phase from Thermo Scientific (Austin, TX, USA).Chromatographic analysis
The separation of the seven additives is based on reverse phase partitioning with ionic suppression which allows the separation of ionic compounds by suppressing their ionisation state through pH control. For this purpose, a mixture of acetonitrile and an aqueous solution of phosphoric acid at pH 3.5 was employed as the mobile phase, using the following gradient elution: acetonitrile initial percentage of 20%, increased linearly to 30% in 3 min; increased to 100% in 3 min (9 min), after which the percentage was returned to the initial conditions in 4 min. A constant temperature (40 °C) and mobile phase flow rate (1 mL min−1) were used and 20 μL of sample was injected.The signal was monitored in the 190–600 nm range, to confirm its identification and also to check the homogeneity of the spectral peak. For quantification purposes 200 nm was selected in order to achieve maximum sensitivity.
Extraction procedure
500 μL of 1-octanol was added to 25 mL of seawater samples and the mixture was shaken using a Vibrax-VXR agitation plate from IKA (Staufen, Germany) during 20 min at 1200 rpm. The phases were separated by centrifugation (Eppendorf 5804, Madrid, Spain) at 3500 rpm for 5 min. Then, 200 μL of the 1-octanol drop was collected and the volume was adjusted to 600 μL with acetonitrile before chromatographic analysis. The schematic DLLME procedure is presented in Fig. 2. | ||
| Fig. 2 Scheme of the DLLME procedure. | ||
Results and discussion
HPLC-DAD optimization
Since lawsone and quercetin are weak acids, they are strongly dissociated in a neutral aqueous solution so they have an electrical charge which inhibits the interaction with the non-polar C18 stationary phase. For reverse phase chromatography, it is important that ionic species exist in a protonated form, the acidification of the mobile phase is necessary in order to obtain an acid analyte in non-ionized form and the separation takes place by partitioning. Therefore, it is recommended to work at a pH around 1.5–2 units lower than the pKa of the most acidic species to ensure that solutes are protonated and the reproducibility from one injection to another. Furthermore, it is important to consider that several studies devoted to this topic have shown that the pKa values of acids increase when increasing proportions of organic solvent content, which can be attributed to a decrease of the dielectric constant of the medium; for instance, in the case of using acetonitrile as organic solvent in the mobile phase, several authors have reported that an increase of 10% in the acetonitrile content led to an increase of 0.3log unit in the pKa value.31,32
On the other hand, silica-based columns usually contain free silanol groups on their surface, which are ionized at neutral pH and can participate on the analyte retention by electrostatic interactions because of their negative charge.33 Therefore, the adjustment of pH of the mobile phase also governs the state of the ionization of the silanol surface. Protonation of silanol groups occurs at pH values less than 4.5, thus working at lower pHs minimizes electrostatic interactions and helps to reduce peak tail problems.
Considering the points mentioned above, the pKa of lawsone and quercetin (4.31 and 6.31 respectively) and the range of pH of the aqueous mobile phase (2.5–3.7) employed in the literature for chromatographic analysis of lawsone, in this work the pH of the aqueous phase was adjusted to 3.5.18,21,34
The hydrophobicity of the seven additives is too large so a gradient elution will be necessary. The most employed acids for adjusting the pH of the mobile phase for the determination of these additives are acetic acid,19,21 formic acid18,23 and phosphoric acid.13,35 In this work phosphoric acid was selected since organic acids exhibit absorbance at 200 nm, and consequently they cannot be used in gradient elution.
Initially, because articles have not been found in which these seven additives were analysed simultaneously, isocratic runs were performed using individual solutions of the additives (10 mg L−1) at different ratios of acetonitrile and a solution of phosphoric acid at pH 3.5 as a mobile phase. Then several gradient conditions were carefully assayed which correspond closely to their isocratic counterparts. The best conditions for achieving an adequate chromatographic resolution of the seven additives were obtained by employing the gradient previously described in the Experimental section, starting the mobile phase gradient with 20% acetonitrile in order to avoid overlapping between the peaks of lawsone and quercetin. As can be seen in Fig. 3, adequate resolution was reached for all the additives in less than 20 min.
 | ||
| Fig. 3 HPLC chromatogram obtained under optimized conditions for a standard solution of additives (10 mg L−1). Target compounds are numbered as follows: (1) lawsone; (2) quercetin; (3) triclosan; (4) BHT; (5) DIDP; (6) vitamin E; (7) irgafos 168. | ||
Upon the chromatographic method being optimized, in order to check the precision of the chromatographic system, two standard solutions containing 0.5 and 6 mg L−1 of each additive were employed. For intra-day precision, ten consecutive measurements of each concentration level were analyzed; in the case of inter-day precision, three replicates of each concentration level were injected per day on five consecutive days. The obtained precision was good, with RSD values lower than 5% in both intra- and inter-day precision assays. In order to verify the linearity of the detector, eight concentration levels of each additive (0.25, 0.50, 1, 2, 4, 6, 8 and 10 mg L−1) were prepared and injected in triplicate. Coefficients of determination (R2) were higher than 0.999 for six of the seven additives, being a bit lower in the case of lawsone (0.998).
DLLME optimization
The DLLME method is based on a previous extraction procedure developed by the authors for the determination of triazines in seawater employing 1-octanol as the extraction solvent and a shaking step instead of a dispersive solvent.36 To carry out the optimization, 25 mL of synthetic saltwater solution spiked with 0.3 mg L−1 of each additive was used and three replicates were employed in each assay. Furthermore, procedural blanks were also analysed.The effect of the extraction solvent volume was studied employing 100, 300, 400 and 500 μL volumes of 1-octanol. The single drop formed by adding the organic solvent to the water sample was dispersed by using an agitation step (10 min, 1200 rpm). Then, the mixture was centrifuged (5 min at 3500 rpm) to separate the phases. For tested volumes below 500 μL, the formed drop was not easily distinguishable from the aqueous phase. This could be due to the solubility of 1-octanol in water, which is also reflected in the small volume of organic phase obtained (<300 μL) after the extraction when 500 μL were used. Consequently, 500 μL of 1-octanol was chosen as optimum extraction solvent volume.
Since the sample pH can affect the extraction efficiency of ionisable compounds, a study of sample pH was carried out. For this purpose, saltwater with unchanged pH and saltwater with pH adjusted to 3 using phosphoric acid were subjected to the extraction procedure under the above-mentioned shaking and centrifugation conditions. Then, 200 μL of the 1-octanol drop was collected and diluted with acetonitrile (1:3). To calculate the recoveries, standards of the additives with the final concentration of the extracts and the same ratio of 1-octanol/acetonitrile were used. The results obtained (see Table 1) showed that lawsone and quercetin were not extracted at saltwater pH because ionic suppression of these compounds is not achieved. For the other additives studied, recovery increased at acid pH, except for DIDP. Therefore, pH 3 was selected to carry out the extraction method.
| Compound | Recovery (%) | |
|---|---|---|
| pH = 3.5 | Saltwater pH | |
| Lawsone | 56.1 | — |
| Quercetin | 86.9 | — |
| Triclosan | 92.4 | 85.6 |
| BHT | 89.8 | 83.2 |
| DIDP | 83.5 | 89.7 |
| Vitamin E | 84.4 | 82.6 |
| Irgafos 168 | 88.1 | 84.2 |
The effect of agitation time was examined by testing three different times (5, 10 and 20 min) keeping constant the other experimental conditions. As can be seen in Fig. 4, the extraction efficiency slightly increased up to 20 min for most additives. Therefore, 20 min was chosen as extraction time. As an example, Fig. 5 shows the chromatogram corresponding to unspiked and spiked synthetic saltwater analysed with the developed procedure.
 | ||
| Fig. 4 Effect of agitation time on the extraction efficiency in DLLME (n = 3). | ||
 | ||
| Fig. 5 Chromatograms obtained after DLLME: (a) synthetic saltwater sample, (b) spiked synthetic saltwater sample. Target compounds are numbered as follows: (1) lawsone; (2) quercetin; (3) triclosan; (4) BHT; (5) DIDP; (6) vitamin E; (7) irgafos 168. | ||
Method validation
The developed DLLME method was validated by estimation of the linearity, limit of detection (LOD) and limit of quantification (LOQ), accuracy and precision. All quantitative results were calculated using 25 mL of synthetic saltwater spiked with a standard additive mixture. The 1-octanol phase obtained after DLLME was diluted with acetonitrile before LC injection as described in the extraction procedure. The validation data are shown in Table 2.| Compound | LOD (mg L−1) | LOQ (mg L−1) | Determination coefficient (R2) | Intra-day precision RSDa (%) | Inter-day precision RSDa (%) | Recoverya (%) | |||
|---|---|---|---|---|---|---|---|---|---|
| 0.06 mg L−1 | 0.24 mg L−1 | 0.06 mg L−1 | 0.24 mg L−1 | 0.06 mg L−1 | 0.24 mg L−1 | ||||
| a n = 5 for intraday precision and recovery; n = 3 for inter-day precision. | |||||||||
| Lawsone | 0.028 | 0.084 | 0.9929 | 7.98 | 5.92 | 11.45 | 8.94 | 62.1 | 59.6 |
| Quercetin | 0.027 | 0.082 | 0.9931 | 9.11 | 6.21 | 11.31 | 8.14 | 82.1 | 85.1 |
| Triclosan | 0.010 | 0.031 | 0.9990 | 5.10 | 3.42 | 7.80 | 7.42 | 87.4 | 92.6 |
| BHT | 0.009 | 0.027 | 0.9992 | 5.67 | 3.76 | 7.17 | 6.08 | 85.4 | 91.3 |
| DIDP | 0.020 | 0.059 | 0.9965 | 3.92 | 3.05 | 5.54 | 4.57 | 85.3 | 87.0 |
| Vitamin E | 0.025 | 0.077 | 0.9960 | 7.70 | 4.39 | 7.26 | 7.27 | 85.8 | 82.0 |
| Irgafos 168 | 0.013 | 0.041 | 0.9983 | 3.39 | 3.46 | 6.52 | 7.85 | 85.7 | 93.1 |
Linearity was investigated by triplicate analysis of standard synthetic saltwater solutions. Five-point calibration curves were constructed at the concentration range between 0.03 and 0.24 mg L−1. As can be seen in Table 2, determination coefficients (R2) higher than 0.9900 were achieved for all compounds.
The LODs were determined as 3 × Sy/x/b and the LOQs as 10 × Sy/x/b, where Sy/x is the residual standard deviation and b is the slope of the calibration curves. The detection and quantification limits obtained using only 25 mL of synthetic saltwater (between 0.009–0.028 and 0.027–0.084 mg L−1 respectively) can be considered adequate (see Table 2). Furthermore, in case more sensitivity is required, the method is easy to adapt for further analysis by LC-MS.
Precision and accuracy were investigated at two levels of concentration (0.06 and 0.24 mg L−1). The precision of the analytical method, expressed as relative standard deviation (RSD), was studied as intra-day and inter-day precision. To evaluate intra-day precision, RSDs were calculated using five replicates measured at the same day. Inter-day precision was investigated measuring three replicates on three consecutive days. The obtained values lower than 10 and 11.5% for intra-day and inter-day precision respectively (see Table 2) indicated that the developed method was reproducible.
The Horwitz equation was also employed as an additional criterion for checking the precision of the method. The equation is RSD (%) = 21−0.5logC, with C being the mass fraction expressed as a power of 10. For intra-day precision, the obtained values for RSD (%) would be lower than 1/2 (21−0.5logC), whereas in the case of inter-day precision, the values for RSD (%) would be lower than 2/3 (21−0.5logC). The values of RSDs obtained with the proposed methodology were highly satisfactory because, for the seven additives, the results achieved of RSD (%) for intra-day and inter-day precision were below the limits calculated by the Horwitz equation (intra-day 12.21 and 9.92% and inter-day 16.28 and 13.22% for 0.06 and 0.24 mg L−1 respectively).
The accuracy of the method was evaluated as analytical recovery. Five replicate analyses of 25 mL of synthetic saltwater solution spiked with the standard mixture of additives at each fortification level were employed. The method showed very good recoveries for all additives in the range of 82.1–87.4% for the low level and 82.0–93.1% for the high level, except for lawsone with a recovery value of around 60%, an accepted value in residue analysis (Table 2).
Analysis of real samples
The absence of organic compounds, humic acids and other materials in synthetic saltwater solution does not allow the evaluation of interferences due to matrix components present in real samples which may give rise to false positive or negative results. Therefore, it is necessary to validate the accuracy and suitability of the method for real samples.Seawater samples were collected using amber glass bottles from 3 different sites in the city of A Coruña during May of 2023: sample 1 from Santa Cristina beach located in the estuary formed by the mouth of Mero River, sample 2 from Riazor beach located in an open sea area and sample 3 from the harbour area. The samples were stored at 4 °C until further analysis.
The presence of the additives in the samples was studied by analyzing procedural blanks and peaks were not observed at the retention times of the compounds. Therefore, it was concluded that the additives were not present or present at concentrations below the LOD values (see Table 2).
Recovery experiments were carried out using sample 1 as a test sample. For this purpose, the sample was spiked at two concentration levels (0.06 and 0.24 mg L−1) and three replicates were assayed for both levels. The percent recovery results for additives are presented in Table 3. As can be seen, high recoveries were obtained for all additives at both concentration levels, except for lawsone. For this additive, the recovery reached (around 60%) was similar to that obtained when synthetic saltwater solution was used. Moreover, the RSD values were below 5% (see Table 3), which validates the high repeatability of the developed method.
| Compound | Recovery ± RSD (%) | |
|---|---|---|
| 0.06 mg L−1 | 0.24 mg L−1 | |
| Lawsone | 54.7 ± 1.2 | 64.3 ± 3.9 |
| Quercetin | 82.7 ± 3.6 | 100.7 ± 2.8 |
| Triclosan | 83.9 ± 3.3 | 95.9 ± 3.6 |
| BHT | 84.6 ± 2.4 | 94.0 ± 2.9 |
| DIDP | 84.5 ± 3.6 | 93.6 ± 4.0 |
| Vitamin E | 81.9 ± 3.4 | 92.9 ± 3.8 |
| Irgafos 168 | 84.7 ± 3.4 | 98.0 ± 3.7 |
Conclusions
A fast and simple method based on DLLME combined with HPLC-DAD for the analysis of seven additives, with different physicochemical properties, in terms of polarity and water solubility, from seawater samples has been developed and validated. Regarding the chromatographic method, both the type of elution and the pH of the mobile phase are decisive to obtain a satisfactory separation of the seven additives. The best results are obtained using gradient elution and acetonitrile/phosphoric acid aqueous solution at pH 3.5 as the mobile phase.The procedure employed for the extraction of the additives from seawater is mainly influenced by the sample pH. Under optimal conditions, the compounds are extracted from sample showing recoveries higher than 80% for all compounds, except for lawsone (60%).
Finally, it is important to note that there are few studies of these compounds in seawater and the presented method allows the separation and determination of these seven additives in seawater. Moreover, the method meets the requirements of Green Analytical Chemistry, as the score (PPs) calculated with the Analytical Eco-Scale metric was 86. This score is considered excellent for green analysis.37
Author contributions
M. J. González-Castro: supervision, conceptualization, formal analysis, validation, review & editing. J. Uribe-Ares: investigation, formal analysis, validation. S. Muniategui-Lorenzo: funding acquisition, resources. E. Beceiro-González: conceptualization, supervision, formal analysis, writing – original draft, review & editing. All authors approved the final manuscript.Conflicts of interest
There are not conflicts to declare.Acknowledgements
The authors acknowledge the financial support of the “Xunta de Galicia” (Project GRC ED431C 2021/56 potentially cofunded by the European Regional Development Fund) and the Spanish Ministry of Science and Innovation (Project PID2019-108857RB-C31).References
- S. L. Wright, R. C. Thompson and T. S. Galloway, The physical impacts of microplastics on marine organisms: a review, Environ. Pollut., 2013, 178, 483–492 CrossRef CAS PubMed.
- O. H. Fred-Ahmadu, G. Bhagwat, I. Oluyoye, N. U. Benson, O. O. Ayejuyo and T. Palanisami, Interaction of chemical contaminants with microplastics: Principles and perspectives, Sci. Total Environ., 2020, 706, 135978 CrossRef CAS PubMed.
- L. Hermabessiere, A. Dehaut, I. Paul-Pont, C. Lacroix, R. Jezequel, P. Soudant and G. Duflos, Occurrence and effects of plastic additives on marine environments and organisms: A review, Chemosphere, 2017, 182, 781–793 CrossRef CAS PubMed.
- R. Beiras, E. Verdejo, P. Campoy-Lopez and L. Vidal-Linan, Aquatic toxicity of chemically defined microplastics can be explained by functional additives, J. Hazard. Mater., 2021, 406, 124338 CrossRef CAS PubMed.
- M. D. Samper, E. Fages, O. Fenollar, T. Boronat and R. Balart, The potential of flavonoids as natural antioxidants and UV light stabilizers for polypropylene, J. Appl. Polym. Sci., 2013, 129, 1707–1716 CrossRef CAS.
- R. Liu and S. A. Mabury, Synthetic phenolic antioxidants: A review of environmental occurrence, fate, human exposure, and toxicity, Environ. Sci. Technol., 2020, 54, 11706–11719 CrossRef CAS PubMed.
- Y. S. Lee, J. E. Lim, S. Lee and H. B. Moon, Phthalates and non-phthalate plasticizers in sediment from Korean coastal waters: Occurrence, spatial distribution, and ecological risks, Mar. Pollut. Bull., 2020, 154, 111119 CrossRef CAS PubMed.
- A. B. Dann and A. Hontela, Triclosan: environmental exposure, toxicity and mechanisms of action, J. Appl. Toxicol., 2011, 31, 285–311 CrossRef CAS PubMed.
- E. Yıldız and H. Çabuk, Determination of the Synthetic Antioxidants Butylated Hydroxyanisole (BHA) and Butylated Hydroxytoluene (BHT) by Matrix Acidity-Induced Switchable Hydrophilicity Solvent-Based Homogeneous Liquid-Liquid Microextraction (MAI-SHS-HLLME) and High-Performance Liquid Chromatography with Ultraviolet Detection (HPLC-UV), Anal. Lett., 2022, 55, 480–494 CrossRef.
- M. Akkbik, Z. B. Assim and F. B. Ahmad, Optimization and Validation of RP-HPLC-UV/Vis Method for Determination Phenolic Compounds in Several Personal Care Products, Int. J. Anal. Chem., 2011, 2011(9), 858153 Search PubMed.
- H. Baloul, N. Belhaneche-Bensemra, A. Rodríguez-Bernaldo de Quirós and R. Sendón, Analysis and quantitative estimation of phenolic antioxidants in polypropylene packaging for fat products, J. Polym. Eng., 2018, 38, 899–904 CrossRef CAS.
- H. M. González, I. Almirall, J. Alpízar, R. Montes de Oca and V. Cerdà V, Determination of Vitamin E in Spirulina Platensis Extracts and Photoprotective Creams by Multi-Syringe Chromatography (MSC) and High-Performance Liquid Chromatography (HPLC), Anal. Lett., 2020, 53, 2949–2959 CrossRef.
- O. A. A. Ahmed, H. M. El-Bassossy, H. M. El-Sayed and S. S. A. El-Hay, RP-HPLC Determination of Quercetin in a Novel D-α-Tocopherol Polyethylene Glycol 1000 Succinate Based SNEDDS Formulation: Pharmacokinetics in Rat Plasma, Molecules, 2021, 26, 1435 CrossRef CAS PubMed.
- G. A. de Lima, J. Machado Santos, A. P. S. Paim and A. F. Lavorante, Bioaccessibility study and simultaneous quantification of endocrine disruptors (bisphenol A and phthalates) in utensils and toys for infants using HPLC–UV, Chem. Pap., 2022, 76, 189–202 CrossRef CAS.
- A. Alshishani, M. Saaid, C. Basheer and B. Saad, High performance liquid chromatographic determination of triclosan, triclocarban and methyl-triclosan in wastewater using mini-bar micro-solid phase extraction, Microchem. J., 2019, 147, 339–348 CrossRef CAS.
- M. N. H. Rozaini, B. Saad, N. Yahaya, J. W. Lim, M. N. M. Aris and M. R. Ramachandran, Determination of Three Endocrine Disruptors in Water Samples by Ultrasound-Assisted Salt-Induced Liquid-Liquid Microextraction (UA-SI-LLME) and High-Performance Liquid Chromatography–Diode Array Detection (HPLC-DAD), Anal. Lett., 2022, 55, 132–145 CrossRef CAS.
- P. Legrand, A. Desdion, G. Boccadifuoco, A. D. Wojcicki, A. Worsley, V. Boudy and S. G. Dufay, Development of an HPLC/UV method for the evaluation of extractables and leachables in plastic: Application to a plastic-packaged calcium gluconate glucoheptonate solution, J. Pharm. Biomed. Anal., 2018, 155, 298–305 CrossRef CAS PubMed.
- A. Restivo, I. Degano, E. Ribechini and M. P. Colombini, Development and optimisation of an HPLC-DAD-ESI-Q-TOF method for the determination of phenolic acids and derivatives, PLoS One, 2014, 9, e88762 CrossRef PubMed.
- V. S. Chaudhari, R. M. Borkar, U. S. Murty and S. Banerjee, Analytical method development and validation of reverse-phase high-performance liquid chromatography (RP-HPLC) method for simultaneous quantifications of quercetin and piperine in dual-drug loaded nanostructured lipid carriers, J. Pharm. Biomed. Anal., 2020, 186, 113325 CrossRef CAS PubMed.
- L. M. Madikizela, S. F. Muthwa and L. Chimuka, Determination of triclosan and ketoprofen in river water and wastewater by solid phase extraction and high-performance liquid chromatography, S. Afr. J. Chem., 2014, 67, 143–150 Search PubMed.
- G. P. Jyotshna, D. K. Singh, S. Luqman and K. Shanker, Validated method for quality assessment of henna (Lawsonia inermis L.) leaves after postharvest blanching and its cosmetic application, Ind. Crops Prod., 2017, 95, 33–42 CrossRef.
- J. Hernández-Fernández, H. Cano and A. F. Reyes, Valoration of the Synthetic Antioxidant Tris-(Diterbutyl-Phenol)-Phosphite (Irgafos P-168) from Industrial Wastewater and Application in Polypropylene Matrices to Minimize Its Thermal Degradation, Molecules, 2023, 28, 3163 CrossRef PubMed.
- I. de Araújo Rodrigues, S. M. Gomes, I. P. Garrido Fernandes and A. M. Oliveira-Brett, Phenolic composition and total antioxidant capacity by electrochemical, spectrophotometric and HPLC-EC evaluation in portuguese red and white wines, Electroanalysis, 2019, 31, 936–945 CrossRef.
- E. Fries and W. Püttmann, Analysis of the antioxidant butylated hydroxytoluene (BHT) in water by means of solid phase extraction combined with GC/MS, Water Res., 2002, 36, 2319–2327 CrossRef CAS PubMed.
- A. Paluselli, Y. Aminot, F. Galgani, S. Net and R. Sempere, Occurrence of phthalate acid esters (PAEs) in the northwestern Mediterranean Sea and the Rhone River, Prog. Oceanogr., 2018, 163, 221–231 CrossRef.
- H. Arkaban, M. Mirzaei and M. Behzadi, Magnetic solid-phase extraction of lawsone using polyphenol-coated magnetic nanoparticles: synthesis, characterization and examination, Chromatographia, 2021, 84, 455–462 CrossRef CAS.
- S. S. Caldas, C. Rombaldi, J. L. Oliveira Arias, L. C. Marube and E. G. Primel, Multi-residue method for determination of 58 pesticides, pharmaceuticals and personal care products in water using solvent demulsification dispersive liquid–liquid microextraction combined with liquid chromatography-tandem mass spectrometry, Talanta, 2016, 146, 676–688 CrossRef CAS PubMed.
- T. Asadollahi, S. Dadfarnia, A. M. H. Shabani and M. Amirkavei, Separation/preconcentration and determination of quercetin in food samples by dispersive liquid–liquid microextraction based on solidification of floating organic drop-flow injection spectrophotometry, J. Food Sci. Technol., 2015, 52, 1103–1109 CrossRef CAS PubMed.
- F. Sadrykia, A. Shayanfar, H. Valizadeh and M. Nemati, A Fast and Simple Method for Determination of Vitamin E in Infant Formula by Dispersive Liquid-Liquid Microextraction Combined with HPLC-UV, Food Anal. Methods, 2019, 12, 23–31 CrossRef.
- M. Rezaee, Y. Assadi, M.-R. M. Hosseini, E. Aghaee, F. Ahmadi and S. Berijani, Determination of organic compounds in water using dispersive liquid-liquid microextraction, J. Chromatogr. A, 2006, 1116, 1–9 CrossRef CAS PubMed.
- D. S. Bell and A. D. Jones, Solute attributes and molecular interactions contributing to “U-shape” retention on a fluorinated high-performance liquid chromatography stationary phase, J. Chromatogr. A, 2005, 1073, 99–109 CrossRef CAS PubMed.
- J. Hammer, J. J.-H. Haftka, P. Scherpeniss, J. L. M. Hermens and P. de Voogt, Investigating hydrophilic and electrostatic properties of surfactants using retention on two mixed-mode liquid chromatographic columns, J. Chromatogr. A, 2018, 1571, 185–192 CrossRef CAS PubMed.
- J. Hammer, J. J.-H. Haftka, P. Scherpeniss, J. L. M. Hermens and P. de Voogt, Fragment-based approach to calculate hydrophobicity of anionic and nonionic surfactants derived from chromatographic retention on a C18 stationary phase, Environ. Toxicol. Chem., 2017, 36, 329–336 CrossRef CAS PubMed.
- G. Muruganathan, B. Mandala and T. K. Ravi, Separation, identification and quantification of lawsone and metabolites by chromatographic methods, World J. Pharm. Res., 2014, 3, 726–734 CAS.
- R. Gevrenova, Determination of natural colorants in plant extracts by high-performance liquid chromatography, J. Serb. Chem. Soc., 2010, 75, 903–915 CrossRef CAS.
- N. Rodríguez-González, E. Beceiro-González, M. J. González-Castro and S. Muniategui-Lorenzo, An environmentally friendly method for the determination of triazine herbicides in estuarine seawater samples by dispersive liquid–liquid microextraction, Environ. Sci. Pollut. Res., 2015, 22, 618–626 CrossRef.
- M. Sajid and J. Plotka-Wasylka, Green analytical chemistry metrics: A review, Talanta, 2022, 238, 123046 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |