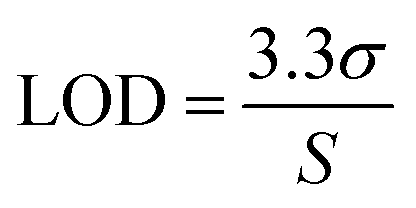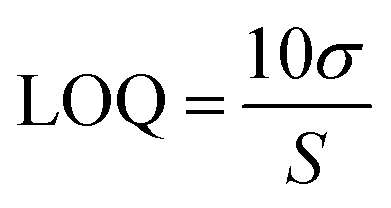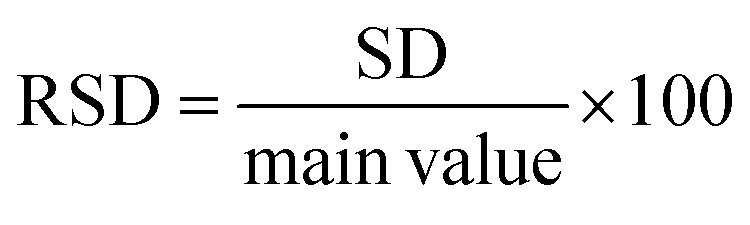DOI:
10.1039/D3AY01833D
(Paper)
Anal. Methods, 2024,
16, 979-989
An isocratic RP-HPLC-UV method for simultaneous quantification of tizanidine and lidocaine: application to in vitro release studies of a subcutaneous implant
Received
16th October 2023
, Accepted 11th December 2023
First published on 18th December 2023
Abstract
Implantable devices have been widely investigated to improve the treatment of multiple diseases. Even with low drug loadings, these devices can achieve effective delivery and increase patient compliance by minimizing potential side effects, consequently enhancing the quality of life of the patients. Moreover, multi-drug products are emerging in the pharmaceutical field, capable of treating more than one ailment concurrently. Therefore, a simple analytical method is essential for detecting and quantifying different analytes used in formulation development and evaluation. Here, we present, for the first time, an isocratic method for tizanidine hydrochloride (TZ) and lidocaine (LD) loaded into a subcutaneous implant, utilizing reversed-phase high-performance liquid chromatography (RP-HPLC) coupled with a UV detector. These implants have the potential to treat muscular spasticity while providing pain relief for several days after implantation. Chromatographic separation of the two drugs was accomplished using a C18 column, with a mobile phase consisting of 0.1% TFA in water and MeOH in a 58![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :42 ratio, flowing at 0.7 ml min−1. The method exhibited specificity and robustness, providing accurate and precise results. It displayed linearity within the range of 0.79 to 100 μg ml−1, with an R2 value of 1 for the simultaneous analysis of TZ and LD. The developed method demonstrated selectivity, offering limits of detection and quantification of 0.16 and 0.49 μg ml−1 for TZ, and 0.30 and 0.93 μg ml−1 for LD, respectively. Furthermore, the solution containing both TZ and LD proved stable under various storage conditions. While this study applied the method to assess an implant device, it has broader applicability for analysing and quantifying the in vitro drug release of TZ and LD from diverse dosage forms in preclinical settings.
:42 ratio, flowing at 0.7 ml min−1. The method exhibited specificity and robustness, providing accurate and precise results. It displayed linearity within the range of 0.79 to 100 μg ml−1, with an R2 value of 1 for the simultaneous analysis of TZ and LD. The developed method demonstrated selectivity, offering limits of detection and quantification of 0.16 and 0.49 μg ml−1 for TZ, and 0.30 and 0.93 μg ml−1 for LD, respectively. Furthermore, the solution containing both TZ and LD proved stable under various storage conditions. While this study applied the method to assess an implant device, it has broader applicability for analysing and quantifying the in vitro drug release of TZ and LD from diverse dosage forms in preclinical settings.
1 Introduction
Tizanidine (TZ) serves as a central alpha-2 adrenoreceptor agonist, commonly prescribed to manage spasticity in patients afflicted with multiple sclerosis or spinal cord injuries.1–3 Spasticity, a potentially debilitating condition, can lead to severe consequences in the absence of treatment, causing deformities and irreversible contractures.4 This condition disrupts daily activities and diminishes the patient's quality of life. The manifestation of symptoms varies based on the lesion within the central nervous system, determining the location of spasms. The lower back and legs are the most frequently affected body parts.5 Spasticity's severity spans from mild stiffness to painful and uncontrollable muscle spasms.6 TZ is also used to alleviate lower back pain and unpleasant muscular spasms induced by musculoskeletal issues. Additionally, the drug has a short half-life of 2.5 hours, necessitating multiple administrations that can lead to treatment fatigue and non-adherence. Exploring alternative methods, such as subcutaneous implants, is a potential alternative to address these limitations.
Implantable devices have garnered extensive interest over recent years as they are ideal candidates to treat chronic conditions requiring long-term treatment.7,8 These devices can achieve effective delivery, promoting patient compliance and minimizing potential side effects,9 thereby improving patients' quality of life. This aspect is particularly vital for chronic conditions, where treatment is often lifelong.10 Furthermore, drug delivery implant designs often employ biodegradable materials like poly(lactic acid), poly(lactic-co-glycolic acid) or poly(caprolactone) (PCL). These polymers have been extensively used for the development of drug eluting medical devices.11–15 Over time, they degrade into non-toxic by products, rendering them compatible with the human body.16 The use of biodegradable implants may obviate the need for implant removal, enhancing patient well-being while reducing healthcare costs.17 Implants are known to cause local side effects, such as pain at the insertion site, stemming from the procedure involved in placing the implant inside the body. To alleviate this, lidocaine (LD), a widely used local anaesthetic agent, can be used to reduce pain.
Previous literature reports have described the quantification of both drugs for various purposes. Methods for quantifying LD were developed in conjunction with preservatives18 and other drugs, serving as antiseptics,19 antiparasitic,20 antifungal,20 anti-inflammatory,21 analgesic,22 mucolytic,23 antiviral24,25 and corticosteroids.21 Additionally, other validated methods exist for determining the drug in polymeric matrices,26–28 plasma,29,30 and in new topical dosage forms.25,31–33 Methods for the quantification of TZ, on the other hand, were described for analysing the drug alone,34,35 concurrently with other drugs like muscle relaxants,36 analgesics,37–39 and anti-inflammatories,40–48 and from pharmaceutical formulations.2,38,42,44–51 Moreover, a method has been described for detecting TZ in skin deposition52 and plasma.35,41,49 While methods for detecting and quantifying both TZ and LD have been reported, none of these studies have presented an HPLC-UV method capable of simultaneously quantifying both. In the present work, for the first time, we developed a combined implant containing TZ for the treatment of muscular spasticity and LD to reduce pain at the implantation site. To evaluate the performance of this type of implant, a simple analytical method was required. No documented studies have reported an HPLC-UV method capable of simultaneously quantifying both TZ and LD. Hence, in this work, we present a straightforward quantification method for TZ and LD using HPLC-UV, which underwent validation following ICH guidelines.
2 Materials and methods
2.1. Materials
TZ powder (hydrochloride salt) was purchased from Tokyo Chemical Industry (Oxford, England, UK). LD powder (hydrochloride salt), dichloromethane (DCM), methanol (MeOH), trifluoracetic (TFA) and phosphate buffer saline tablets (PBS) pH 7.4 were procured from Sigma-Aldrich (Gillingham, Dorset, UK). PCL – CAPA™ 6505 (MW = 50000 Da, i.e., high molecular weight) was obtained from Ingevity (North Charleston, South Carolina, U.S.A).
2.2. Stock solution, working standards, quality control solutions of tizanidine and lidocaine
A 1 mg ml−1 stock solution of TZ and LD was prepared in PBS pH: 7.4. Eight standard solutions were prepared by further dilution of TZ–LD stock solution with PBS, which were used for the calibration curve (0.78, 1.56, 3.125, 6.25, 12.5, 25, 50 and 100 μg ml−1). Quality control (QC) solution at three concentrations was prepared from the same stock solution of 1 mg ml−1 of TZ–LD (2, 10, and 75 μg ml−1), which were utilised for accuracy and precision studies.
2.3. Chromatographic conditions
HPLC analysis was conducted using an Agilent Technologies 1200 Infinity compact LC Series, comprising an Agilent degasser, binary Pump, auto standard injector, and detector (Agilent Technologies UK Ltd, Stockport, UK). A Phenomenex® LC Column (5 μm particle size, C18 100 Å, 250 × 4.6 mm) from (Macclesfield, England, UK) was employed for chromatographic separation. The separation of analytes occurred at 30 °C, utilising a mobile phase composed of 0.1% TFA in water and MeOH in a 58:42 ratio. A flow rate of 0.7 ml min−1 and an injection volume of 50 μL were used. Detection of the analytes transpired at 227 nm absorbance after 5.7 minutes for TZ and 9 minutes for LD.
The UV absorption maximum of each drug was determined using a FLUOstar Omega Microplate Reader UV-vis spectrometer (BMG LABTECH, Ortenberg, Germany) to identify the wavelength necessary to validate the HPLC method. A 1 cm2 quartz cell filled with PBS (pH 7.4) was used as a blank for TZ and LD. Standard solutions containing each drug were scanned at room temperature between 220 and 350/400 nm to produce a spectrum for TZ and LD.
2.4. Method validation
The analytical method was validated according to ICH guidelines,53 covering aspects such as linearity, limit of detection (LOD), limit of quantification (LOQ), specificity, accuracy, precision, and robustness.
2.4.1. Specificity and selectivity.
The specificity and selectivity of method was evaluated by comparing chromatograms of the lowest and highest concentrations from the calibration curve (0.79 and 100 μg ml−1) containing both drugs (TZ and LD) with those of blank release media (PBS pH 7.4).
2.4.2. Linearity, LOD, and LOQ.
Standard solutions at different concentration were employed to check the linearity of the analytical method.54 Calibration curves were generated by analysing the peak area against drug concentration using least squares linear regression. The solution of containing TZ and LD was prepared in concentration of 0.79, 1.56, 3.125, 6.25, 12.5, 25, 50, 100 μg ml−1. Assuming a normal distribution, the limit of detection (LOD) and limit of quantification (LOQ) were calculated as the signal equal to three and ten times the noise level (signal-to-noise ratio), respectively53 (eqn (1) and (2), respectively).| | 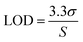 | (1) |
| | 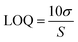 | (2) |
2.4.3. Accuracy and precision.
Accuracy refers to how closely the calculated value aligns with the accepted reference value. This is often referred to as the relative error of the nominal solution concentrations. To be deemed acceptable, all concentrations must exhibit accuracy within ±10%. Precision, on the other hand, evaluates the level of agreement between multiple measurements taken using various samples of the same homogenous drug solution under predetermined conditions. This assesses the presence of random error. Metrics like relative standard deviation (RSD) (eqn (3)), repeatability, and intermediate precision were computed based on the estimated concentrations.55 For concentrations under analysis, an RSD value lower than 10% is considered acceptable.52,56| |  | (3) |
2.4.4. Carry over.
The carry-over study was conducted by injecting a blank sample of PBS after the highest concentration of the calibration standard solution of TZ–LD (100 μg ml−1).57 Acceptable performance is determined when the area of TZ and LD observed in the blank samples is not greater than 20% of the area of a sample solution at the LOQ concentration. This analysis is based on the chromatogram of the blank solution.58
2.4.5. Dilution integrity.
The dilution integrity was assessed by spiking a sample of TZ–LD in PBS at a concentration higher than the maximum concentration from the calibration curve. Specifically, the samples were initially prepared at a concentration of 250 μg ml−1 and then diluted 5 and 10 times with PBS. Accuracy and precision were determined based on three replicates for each dilution.
2.4.6. Robustness.
Slight adjustments in the chromatographic parameters were examined to assess the robustness of the method, demonstrating the reliability of the suggested analytical technique under normal conditions. Variations in column oven temperature, flow rate, and mobile phase composition were considered as chromatographic factors. Changes in retention time and peak area for TZ and LD were recorded during these modifications.
2.5. Stability studies
To assess the degradation of TZ and LD in the release media (PBS pH 7.4), stability studies were conducted. A standard solution containing 2.5 μg ml−1 of TZ and LD was prepared in PBS pH 7.4 and stored under various conditions: room temperature, in an incubator (37 °C), in the refrigerator (2–8 °C), and in the freezer (−20 °C). The concentration of each drug was quantified using the HPLC method at intervals of 1, 2, 3, 7, 14, 21, and 28 days.59
2.6. Preparation of the implant device
Implants containing TZ and PCL, as well as LD and PCL, in a 1:1 mass ratio were produced using the Vacuum Compression Moulding (VCM) technique (MeltPrep®, Graz, Austria). In this procedure, films comprising the drug–polymer mixture were first prepared by dissolving the PCL in DCM (12% w/w). The mixture was then left to evaporate the organic solvent for a day in a glass Petri dish. After drying the films, separate implants were formed. Quantities needed to obtain tablets containing 45.81 ± 4.9 mg of TZ, and 23.21 ± 4.4 mg of LD from each film was placed in the heating chamber at 80 °C for 5 minutes, followed by cooling without heat for another 5 minutes. Subsequently, one mini tablet containing TZ and other containing LD were combined inside the vacuum compression chamber at 80 °C for 5 minutes, following the same process as described above. This procedure resulted in the fusion of both implants due to melting. The resulting implants took the form of cylindrical rods (2.9 × 15 mm) containing 45.81 ± 4.9 mg of TZ and 23.21 ± 4.4 mg of LD.
2.7. Application of the method to in vitro tizanidine/lidocaine release
An analytical method was employed to assess the release profiles of TZ and LD. The implant device, as previously described in Section 2.6, was evaluated through an in vitro release study. Four implant devices were placed in vials containing 20 ml of PBS (pH: 7.4) at 37 °C, with agitation at 400 rpm in a shaker incubator (Jeio Tech ISF-7100, Medline Scientific, Chalgrove Oxon, UK). Samples were collected at various time intervals over a period of 32 days. To ensure sink conditions, the media was replaced with fresh PBS at each time point, and the samples were quantified using HPLC. After each time point, 1 ml of sample was withdrawn and analysed using the HPLC. If necessary, samples were diluted with PBS (pH: 7.4) before analysis.
2.8. Statistical analysis
The mean, standard deviation, relative standard deviation (RSD), and relative error (RE) were calculated using Microsoft® Excel® 2016 (Microsoft Corporation, Redmond, USA). GraphPad Prism® version 6 (GraphPad Software, San Diego, California, USA) was employed for statistical analysis of all the data.
3 Results and discussion
3.1. Method validation
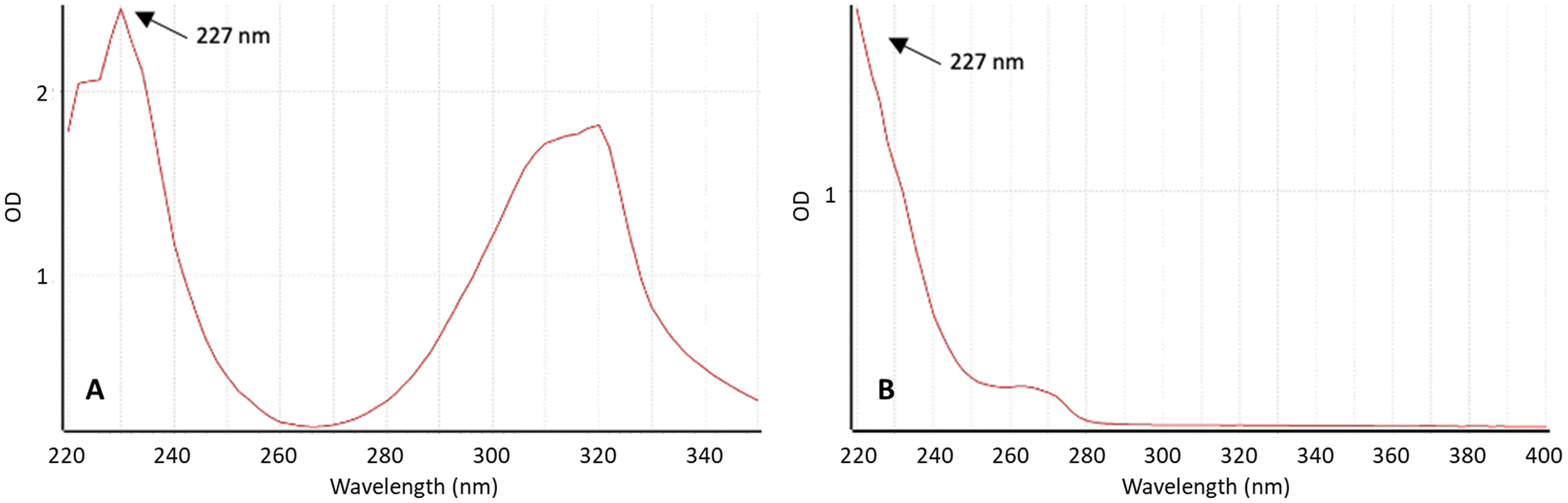
To quantify the drug released from the implantable devices, a reverse-phase high-performance liquid chromatography (HPLC) method was employed. The UV absorbance spectra of TZ and LD were examined using a UV-vis spectrophotometer (Fig. 1) to identify the wavelength where the signal is most intense and exhibits the desired specificity. The maximum absorbance of TZ in PBS pH 7.4 was 227 nm. Interestingly, LD presents high absorption at this wavelength too. This method can be simplified by using a single wavelength to quantify both drugs in HPLC. This is especially interesting for HPLC setups where only a single wavelength can be used at a time. Moreover, these findings align with data previously published in the literature.2,26,52
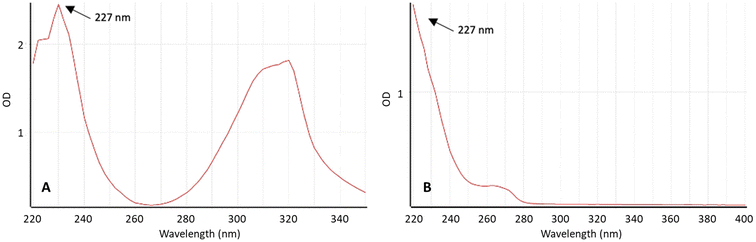 |
| | Fig. 1 Absorbance spectrum of (A) TZ and (B) LD at concentration 50 μg ml−1 in PBS (pH 7.4) measured using UV spectroscopy. | |
This HPLC method used methanol and water/TFA 0.1% as the mobile phase. It was decided to use methanol since TZ and LD are both soluble in it, and TFA 0.1% was added to the water phase to achieve a pH of 2.10. Knowing the pKa of the drugs allows for an appropriate selection of mobile phase pH.60 Because the pKa of TZ and LD are 7.48 and 7.75, respectively, a lower pH in the medium will lead to drug ionisation. This is important as this factor contributes to better separation and quantification.60
3.1.1. Specificity and selectivity.
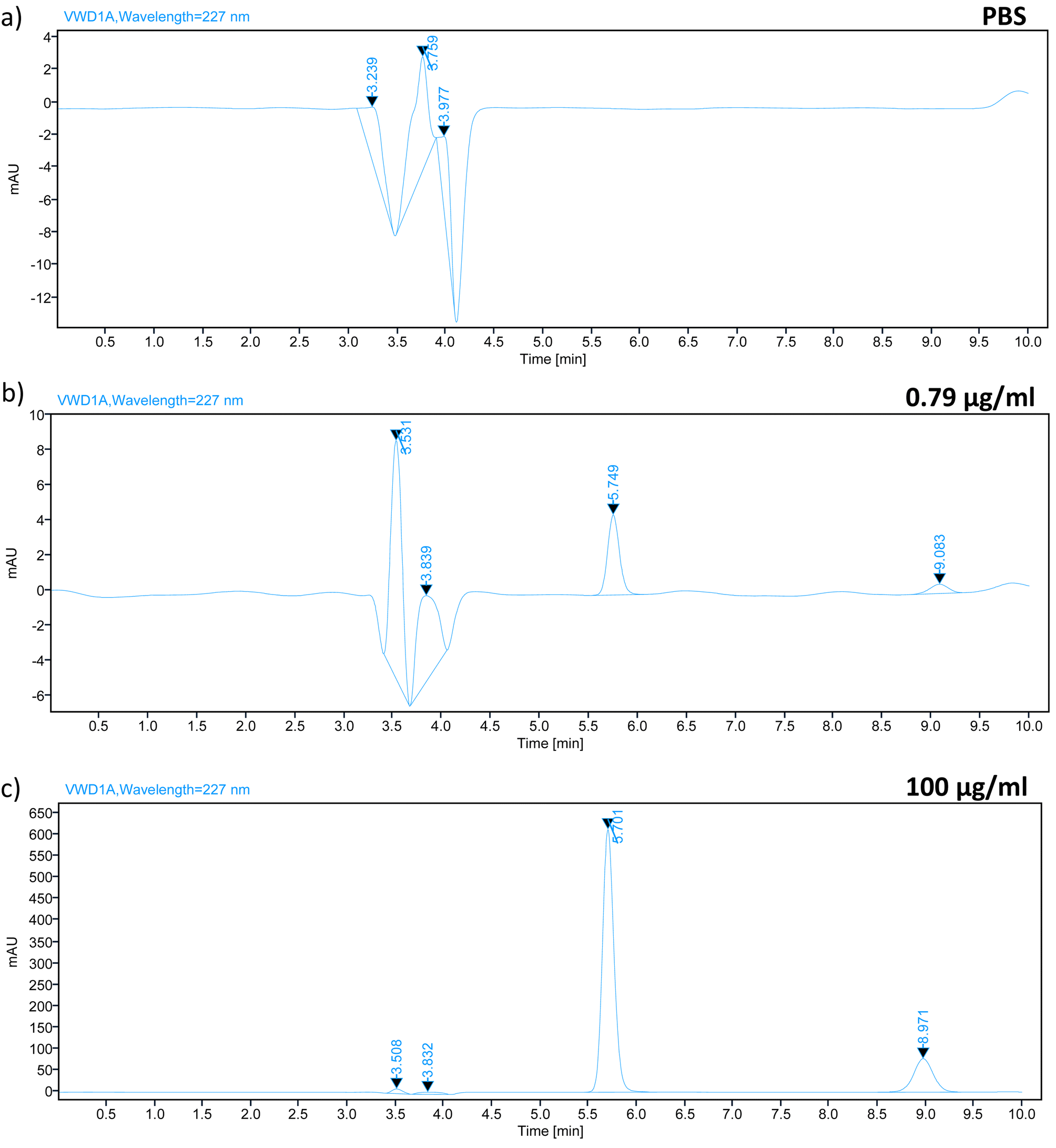
In order to identify compounds that could potentially interfere with the elution of analytes in the chromatogram, we conducted a study on specificity and selectivity. The specificity study of the HPLC method for TZ–LD was carried out using a wavelength of 227 nm. A blank sample of PBS pH 7.4 was analysed alongside a sample containing TZ–LD at concentrations of 0.79 and 100 μg ml−1 for comparison. The retention times for TZ and LD under the selected chromatographic conditions were 5.7 minutes and 8.9 minutes, respectively, within a total running time of 10 minutes. As depicted in Fig. 2, the presence of both drugs in the sample is well-defined and clearly separated without any interaction, indicating the method's specificity for the selected drugs. Conversely, the blank sample exhibited no peak at the same retention time as the drugs. In summary, these data confirm that the analytical method for quantifying TZ–LD demonstrates good specificity and selectivity.
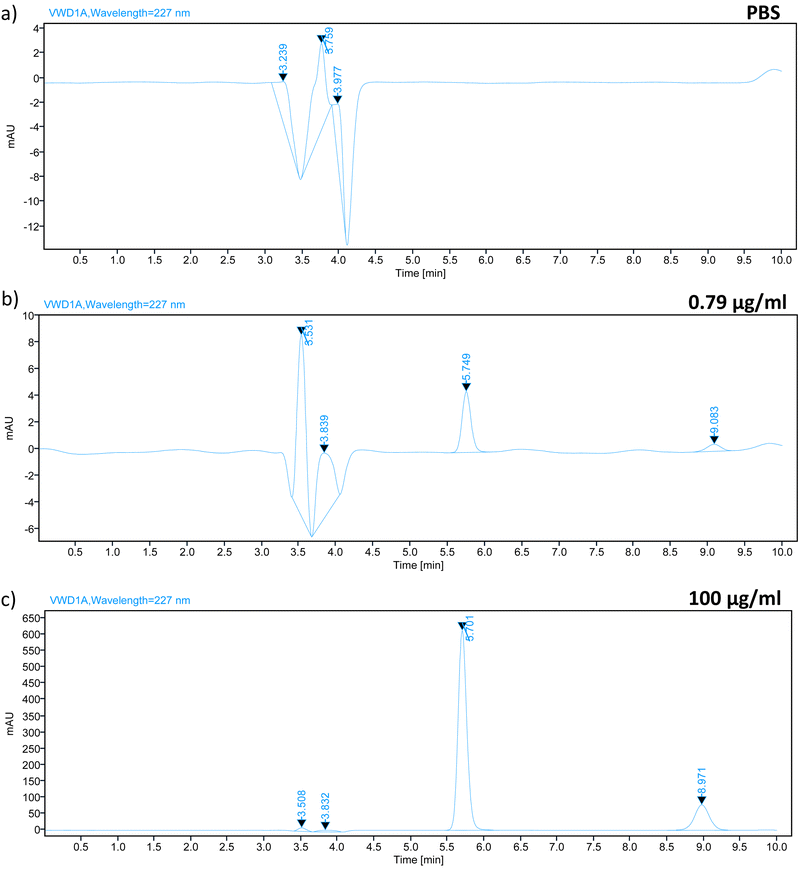 |
| | Fig. 2 Chromatograph spectrum of (a) PBS, (b) TZ–LD in a concentration of 0.79 μg ml−1, and (c) 100 μg ml−1, respectively where the X axis represents the time in minutes and the Y axis represents the area under the curve in mAU. | |
3.1.2. Linearity, LOD, and LOQ.
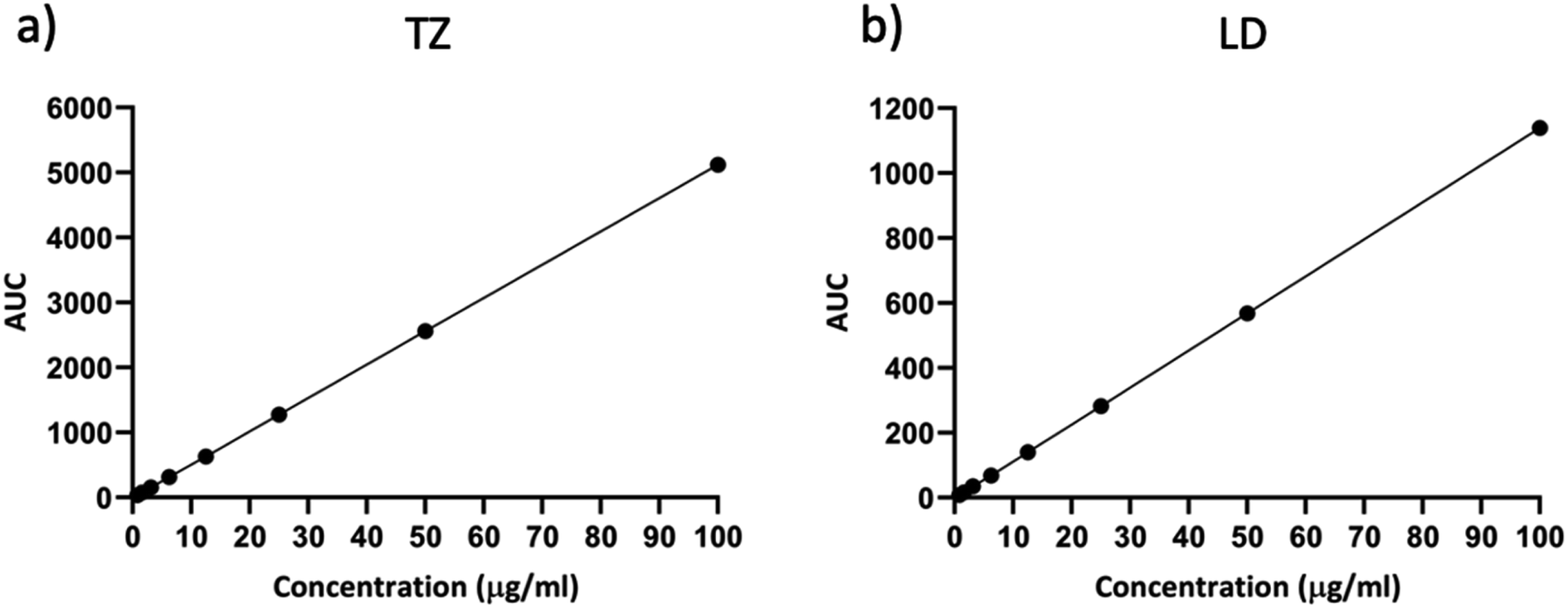
The linearity of the analytical method was assessed over a concentration range from 0.79 μg ml−1 to 100 μg ml−1. These samples were run in triplicate and analysed over three consecutive days. Fig. 3 illustrates the calibration curve for TZ and LD.
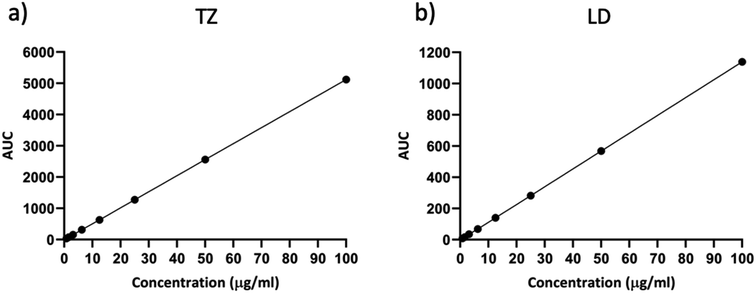 |
| | Fig. 3 Representative calibration curve of (a) TZ and (b) LD in PBS pH 7.4 (means ± SD, n = 9). | |
Table 1 presents the values for LOD, LOQ, slope, Y-intercept, and the coefficient of correlation derived from this method. The correlation coefficient R2 was 1 for both drugs, indicating a robust linear response across the concentration range evaluated. It is worth noting that the LOD and LOQ values in our study are relatively higher compared to previous publications.2,39,52 Previously published studies often dealt with lower concentration ranges,2,26 in nanograms52,61 or employed more sensitive HPLC techniques like Ultra High Performance Liquid Chromatography (UHPLC), mainly because of the necessity to quantify the drug in complex samples such as blood or human serum.39 The purpose behind validating this method was its intended application in in vitro release studies. As a result, the chosen range for linearity studies is within the microgram range, as there is no requirement for blood samples or human serum. This ensures the method's relevance for the intended research.
Table 1 Physicochemical properties of the calibration curve for quantification of TZ and LD in PBS pH 7.4
| Drug |
Range (μg ml−1) |
Slope |
y-Intercept |
R
2
|
LOD (μg ml−1) |
LOQ (μg ml−1) |
| TZ |
0.79–100 |
51.25 |
−3.09 |
1 |
0.16 |
0.48 |
| LD |
0.79–100 |
11.41 |
−1.59 |
1 |
0.30 |
0.92 |
3.1.3. Accuracy and precision.
To assess the accuracy of the method, QC solutions at three concentrations within the range of the calibration curve were measured (2, 10, and 75 μg ml−1). The samples were injected in triplicate over three consecutive days. By comparing the nominal concentrations with those obtained using the developed method, it is possible to calculate the percentage of recovery. The results are presented in Tables 2 and 3. Across the various concentrations chosen for this experiment, the mean recovery percentage falls within the acceptable range of 95–110%, as stipulated by the ICH guidelines.53
Table 2 Determination of accuracy TZ HPLC analysis method in PBS pH 7.4 (means ± SD, n = 3)
| Actual value (μg ml−1) |
Percentage recovery (%) |
Mean recovery ± SD (%) |
| 2 |
Day 1 |
100.94 |
100.79 ± 0.29 |
| Day 2 |
101.43 |
| Day 3 |
100 |
| 10 |
Day 1 |
99.12 |
99.17 ± 0.17 |
| Day 2 |
99.32 |
| Day 3 |
99.06 |
| 75 |
Day 1 |
100.54 |
100.11 ± 1.65 |
| Day 2 |
99.81 |
| Day 3 |
99.99 |
Table 3 Determination of accuracy LD HPLC analysis method in PBS pH 7.4 (means ± SD, n = 3)
| Actual value (μg ml−1) |
Percentage recovery (%) |
Mean recovery ± SD (%) |
| 2 |
Day 1 |
104.69 |
102.89 ± 0.74 |
| Day 2 |
103.31 |
| Day 3 |
100.68 |
| 10 |
Day 1 |
104.48 |
103.11 ± 0.65 |
| Day 2 |
104.85 |
| Day 3 |
99.99 |
| 75 |
Day 1 |
99.22 |
97.58 ± 0.32 |
| Day 2 |
98.5 |
| Day 3 |
95.01 |
Moreover, to ensure the reliability and repeatability of the method for analysing TZ and LD within the concentration range described earlier, precision was assessed. For this test, samples of three different concentrations were quantified in triplicate on a single day (inter-day) and across three consecutive days (intra-day). The results are presented in Tables 4 and 5. Accuracy and RSD values, indicative of precision, remained within acceptable limits, as the variability for all concentrations was below 4%.52,56
Table 4 Determination of precision TZ HPLC analysis method in PBS pH 7.4 (means ± SD, n = 3)
| Selected concentration (μg ml−1) |
Inter-day precision |
Intra-day precision |
| Mean concentration found ± SD (μg ml−1) |
RSD (%) |
Mean concentration found ± SD (μg ml−1) |
RSD (%) |
| 2 |
2.02 ± 0.02 |
0.92 |
2.01 ± 0.02 |
0.78 |
| 10 |
9.91 ± 0.05 |
0.48 |
9.94 ± 0.07 |
0.74 |
| 75 |
75.08 ± 1.57 |
2.09 |
74.69 ± 0.54 |
0.72 |
Table 5 Determination of precision LD analysis method in PBS pH 7.4 (means ± SD, n = 3)
| Selected concentration (μg ml−1) |
Inter-day precision |
Intra-day precision |
| Mean concentration found ± SD (μg ml−1) |
RSD (%) |
Mean concentration found ± SD (μg ml−1) |
RSD (%) |
| 2 |
2.06 ± 0.04 |
2.07 |
2.01 ± 0.05 |
2.82 |
| 10 |
10.31 ± 0.26 |
2.53 |
9.85 ± 0.18 |
1.88 |
| 75 |
73.18 ± 1.48 |
2.02 |
71.81 ± 0.42 |
0.59 |
The aim of this study was to develop an analytical method to establish a sensitive and accurate approach for the detection and quantification of TZ and LD simultaneously. HPLC was chosen as the analytical technique for quantifying TZ–LD due to its capability to handle complex molecules with diverse polarity and molecular mass. The HPLC method for TZ–LD was successfully developed and validated following the ICH guidelines, including testing for specificity, linearity, range LOD and LOQ, accuracy, and precision. The results obtained demonstrate the accuracy and validation of the method, making it suitable for quantifying the drug in in vitro studies.
3.1.4. Carry-over.
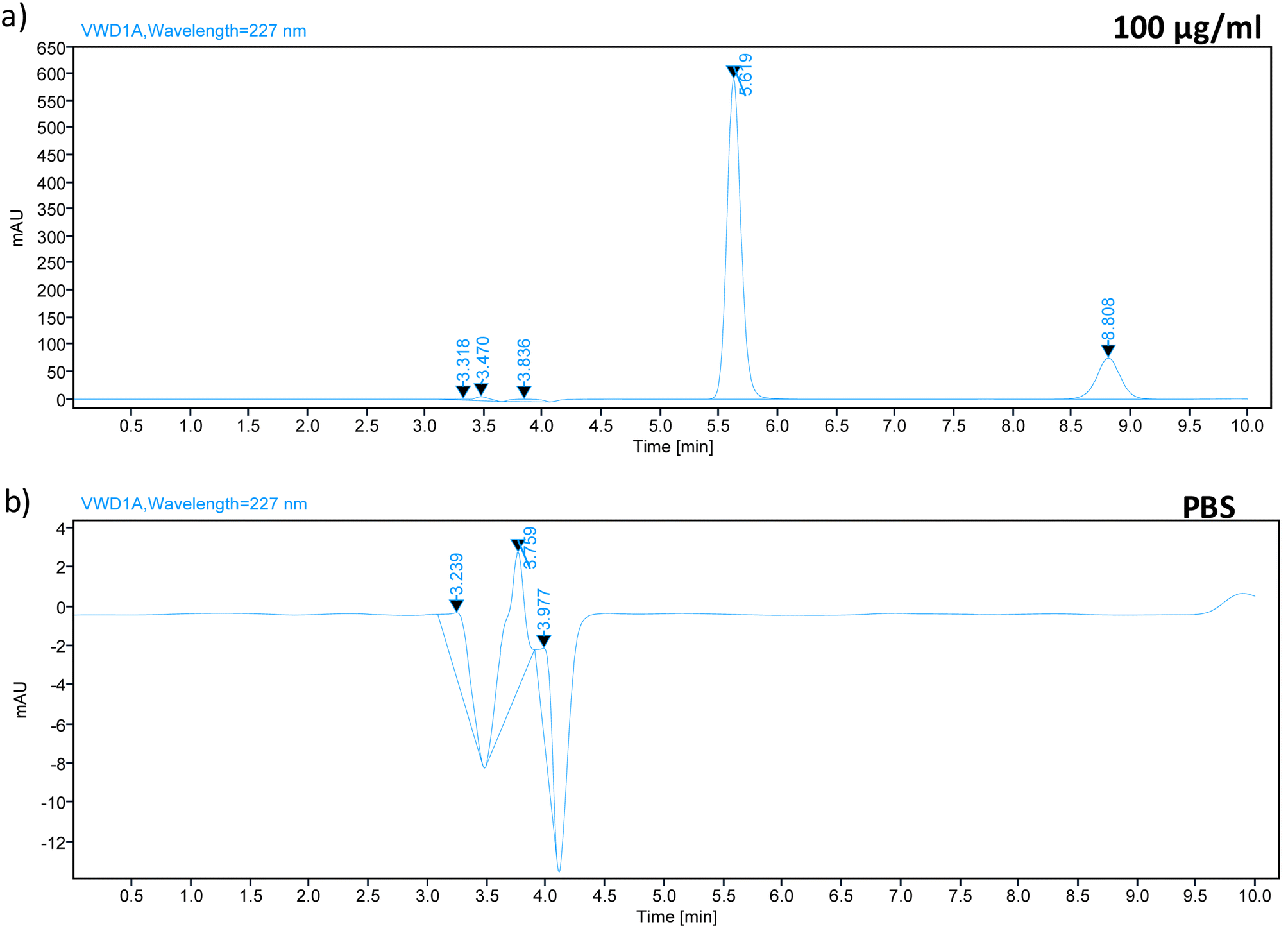
Carry-over was evaluated by running a blank PBS sample after injecting TZ–LD at the highest concentration of the calibration curve (Fig. 4). Chromatograms of the blank samples revealed no peaks at 5.7 min and 8.9 min, corresponding to the retention times of TZ and LD, respectively. This indicates the absence of any carry-over effect after injecting the high-concentration drug sample.
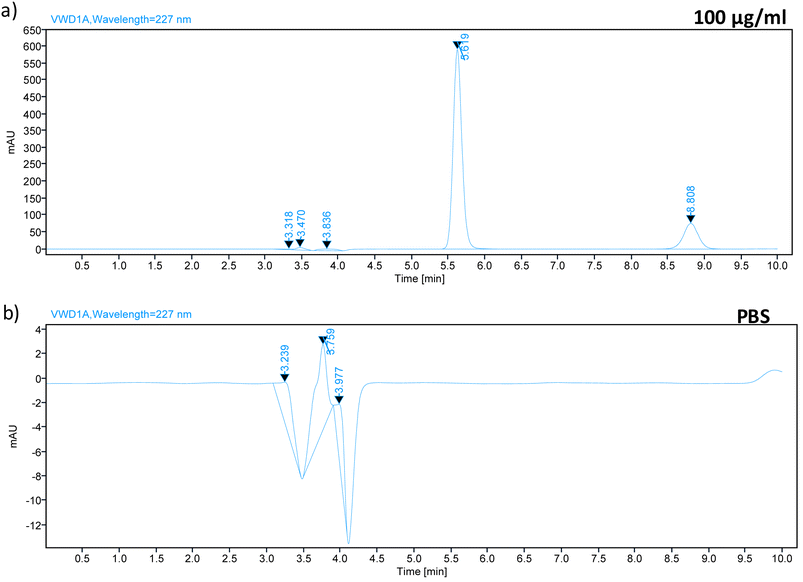 |
| | Fig. 4 Chromatograph spectrum of (a) 100 μg ml−1 sample of TZ–LD and chromatograph spectrum of (b) PBS, injected after the highest concentration (100 μg ml−1). | |
3.1.5. Dilution integrity.
Dilution integrity evaluation was conducted to assess the impact of dilution on the concentration of TZ–LD in PBS (pH 7.4). Dilution factors of 5 and 10 were applied for the test. The results indicated that the recovery percentages ranged from 101.89 ± 0.06% to 102.57 ± 4.14% for TZ, and from 99.58 ± 2.46% to 99.93 ± 0.75% for LD, following the determination of TZ–LD concentrations at these dilution factors. The results obtained are in line with ICH guidelines, suggesting an acceptable precision range of ±15%.53
3.1.6. Robustness.
Robustness is related to a method's capacity to remain unaffected by minor, deliberate changes in chromatographic parameters, providing an indication of its reliability under normal conditions. To ascertain the reliability of the validated method, a standard concentration of TZ–LD at 50 μg ml−1 was examined, assessing the percentage of recovery under different conditions i.e., column oven temperature (±0.5 °C), mobile phase composition (±0.2%), and flow rate (±0.01 ml min−1). Robustness data results are detailed in Table 6. Following the analysis of samples by HPLC, data was compared with that of the standard solution. It was observed that the TZ–LD method exhibited commendable robustness, with percentage recovery under different variation factors falling within the range of 98.97–101.36% for TZ and 96.27–100.13% for LD. These outcomes align with the ICH guidelines.53
Table 6 Robustness testing of TZ and LD, expressed in percentage of recovery at different variations: column temperature, flow rate and mobile phase composition
| Parameter |
Setting |
Tizanidine |
Lidocaine |
| Recovery (%) |
Retention time (min) |
Peak area (mAU) |
Recovery (%) |
Retention time (min) |
Peak area (mAU) |
| Column temperature (°C) |
29.5 |
100.08 |
5.6 |
2658.28 |
98.33 |
8.7 |
554.08 |
| 30 |
100 |
5.6 |
2656.20 |
100 |
8.7 |
563.49 |
| 30.5 |
99.95 |
5.6 |
2654.78 |
98.49 |
8.6 |
554.97 |
| Mobile phase composition (% v/v) |
57.9:42.1 |
101.36 |
5.6 |
2655.04 |
100.12 |
8.7 |
564.243 |
| 58:42 |
100 |
5.6 |
2656.20 |
100 |
8.7 |
563.49 |
| 58.1:41.9 |
98.97 |
5.6 |
2655.06 |
96.27 |
8.7 |
558.00 |
| Flow rate (ml min−1) |
0.69 |
99.96 |
5.6 |
2692.29 |
100.13 |
8.8 |
564.17 |
| 0.70 |
100 |
5.6 |
2656.20 |
100 |
8.7 |
563.49 |
| 0.71 |
99.96 |
5.5 |
2628.87 |
99.03 |
8.6 |
542.43 |
3.2. Stability studies
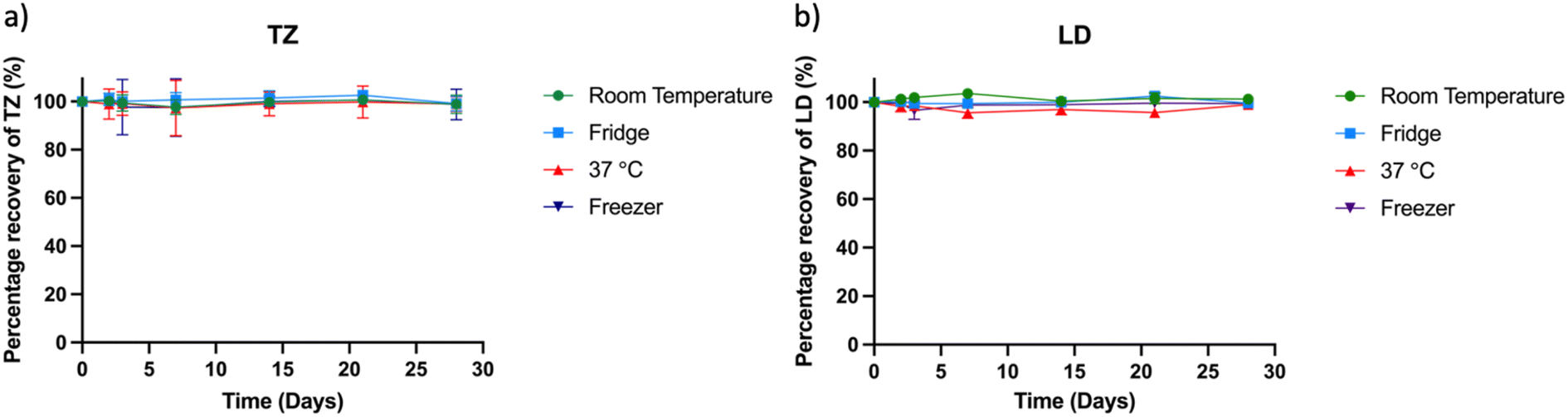
Stability studies play a pivotal role in estimating the shelf life of samples under controlled environmental conditions encompassing factors such as temperature, humidity, and light exposure. For successful release experiments, it is crucial that TZ and LD remain stable in solution for a specified duration (5 to 20 days) to enable accurate drug quantification. In this study, a drug concentration of 2.5 μg ml−1 in PBS was stored under varying conditions: room temperature, 37 °C incubator, refrigerator, and −20 °C freezer. Subsequently, samples of 1 ml were extracted and quantified using the HPLC method (Fig. 5). The drug's concentration in solution after 28 days was determined as 99.76 ± 0.89% at room temperature, 99.09 ± 2.4% at 37 °C, 101.42 ± 1.49% in the refrigerator, and 100.06 ± 1.08% at −20 °C, relative to the initial concentration at day zero. These results confirm the stability of the drugs across diverse conditions, aligning with existing literature on their stability attributes.40,62 Notably, while prior research demonstrated TZ's stability over extended periods (70 days) with consistently high purity results, this 30 days study timeframe is deemed sufficient for sample collection and quantification.
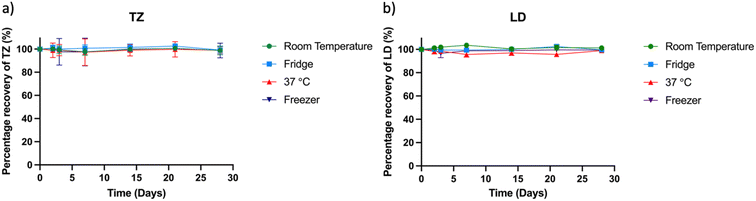 |
| | Fig. 5 Stability study showing the percentage of recovery of (a) TZ and (b) LD after 2, 3, 7 and 14 days. | |
3.3. Preparation of the implant device
As previously outlined, the implants were prepared using a blend of PCL polymer and two drugs, TZ and LD. The initial step involved fabricating films for each drug individually. To achieve this, both the drug and the polymer were dissolved in DCM, a solvent chosen for its elevated volatility and capacity to dissolve both components effectively, thereby enhancing drug loading capabilities. Following this, the solutions were placed in petri dishes under ambient lab conditions to enable the formation of solid films containing the drugs. This method, previously employed to develop membranes for implant devices, was adapted in this context to achieve a more favourable mixture within the VCM and an improved distribution of the drug within the implant. VCM is a technology that can be used for pre-formulation studies of implants and solid amorphous polymer–drug dispersions without using hot-melt extrusion.63–68 This allows the use of small samples. In this case the pre-mixture of drugs with an organic solvent will mimic the mixing of the drugs and polymers within the barrel of a hot-melt extruder. Notably, these films differ from controlled membranes as they integrate the drug component.17,69 The implants obtained exhibited dimensions of 2.9 mm in diameter and 10 mm in length for TZ, and 2.9 mm in diameter and 5 mm in length for LD. Subsequently, these implants were combined to create a cylindrical implant measuring 2.9 mm in diameter and 15 mm in length. In design and size this implant is similar to those described in prior research and to products available on the market.70–72 In the selection of polymers for implant formulation, PCL was chosen due to its biocompatible and biodegradable attributes. Notably, this polymer has been previously utilised in the preparation of long-acting implants for drug administration,70,73–76 rendering it a suitable choice for this formulation.
3.4. Application of the method to in vitro tizanidine/lidocaine release
Once the HPLC method had been established and validated, it was then employed to assess the release of TZ and LD using the previously described implants in an in vitro study. This drug release evaluation extended over a 30 days period. Implants containing TZ–PCL and LD–PCL were combined using the melting method (VCM) (Fig. 6a and b). Upon completion of the 30 days experiment, the total drug released amounted to 101.71 ± 5.51% for TZ and 85.30 ± 2.90% for LD (Fig. 6c). Remarkably, it was observed that nearly the entire quantity of drug was released in both cases. This observation implies that the melting process has the potential to facilitate controlled and extended drug release. In the realm of pharmaceutical treatments, minimising the number of implants is pivotal to reduce the impact of application. With attributes like a relatively compact size and the ability to provide controlled and sustained drug release, this implantable device holds promise for addressing chronic conditions while also offering local anaesthesia following implant insertion. Furthermore, system devices incorporating compressed pellets have previously been explored with other drugs, often incorporating a controlled membrane to cover these pellets, as seen in the work by Karunakaran et al.77 Their study focused on an implant composed of a polymeric tubular membrane made from different medical grade hydrophilic poly(tetramethylene oxide) based poly(ether urethane), housing four compressed tablets of cabotegravir. The dimensions of their implants were 47 mm in length, 3.6 mm in diameter, and 200 μm in thickness. In contrast, the present work introduces the development of an implant loaded containing two separate regions for each drug, providing 30 days of TZ release and 4 days of LD release. It is well known that implants can have local side-effects such as pain at the insertion site. This is due to the process used to insert the implant into the human body. Lidocaine is a local anaesthetic agent broadly used as an analgesic to relieve pain in a specific area of the body.78 In this way the system device developed will not only be focused on speciesism treatment, but will also aim to relieve pain in the area where the implant is allocated. Moreover, these implants were monolithic-type implants, which do not require the use of rate controlling membranes. The composition of implantable devices directly affects drug release profile.69,73,79,80 In order to optimise implant composition robust and sensitive quantification methodologies are required. This is especially important for devices such as this that contain more than one drug. Consequently, this validated method was effectively employed for the precise quantification of TZ and LD.
 |
| | Fig. 6 (a) Final implant containing LD and TZ, (b) SEM images of LD and TZ implants at ×50 (upper images) and ×500 (down images) magnification and (c) cumulative release of both drugs over 30 days. | |
4 Conclusions
This study introduced a simple, novel, and sensitive HPLC method with UV detection for the simultaneous quantification of TZ and LD. The analytical approach underwent rigorous validation in accordance with the ICH guidelines, confirming its efficiency, cost-effectiveness, specificity, and, notably, its high precision. A significant indicator of the method's sensitivity was its remarkable LOQ, which stood at 0.48 μg ml−1 for TZ and 0.92 μg ml−1 for LD. Notably, this HPLC technique offers several advantages compared to previously documented methods, including its simplicity, affordability, and utilisation of binary isocratic conditions. Accordingly, the duly validated HPLC technique was effectively applied to investigate the release of drugs through a long-acting formulation containing TZ and LD over a 30 days period in an in vitro study.
Author contributions
Camila J. Picco: conceptualization; data curation; investigation; methodology; project administration; validation; writing – original draft; writing – review & editing, Qonita Kurnia Anjani: conceptualization; investigation; methodology; software; writing – review & editing, Ryan F. Donnelly: supervision; writing – review & editing, Eneko Larrañeta: conceptualization; funding acquisition; project administration; supervision; writing – review & editing.
Conflicts of interest
The authors declare no conflicts of interest.
References
- G. Malanga, R. D. Reiter and E. Garay, Expert Opin. Pharmacother., 2008, 9, 2209–2215 CrossRef CAS PubMed.
- M.-L. Qi, P. Wang and L. Wang, Anal. Chim. Acta, 2003, 478, 171–177 CrossRef CAS.
-
N. L. of M. N. C. for B. I. PubChem, Tizanidine hydrochloride, https://pubchem.ncbi.nlm.nih.gov/compound/Tizanidine-hydrochloride#section=Computed-Properties, accessed 4 May 2023 Search PubMed.
- A. Kheder and K. P. S. Nair, Pract. Neurol., 2012, 12, 289 CrossRef PubMed.
- J. Ashton, Degener. Neurol. Neuromuscular Dis., 2011, 1, 15–23 CAS.
- E. Chang, N. Ghosh, D. Yanni, S. Lee, D. Alexandru and T. Mozaffar, Crit. Rev. Phys. Rehabil. Med., 2013, 25, 11–22 CrossRef PubMed.
- R. Langer, Science, 1990, 249, 1527–1533 CrossRef CAS PubMed.
- A. K. Dash and G. C. Cudworth, J. Pharmacol. Toxicol. Methods, 1998, 40, 1–12 CrossRef CAS PubMed.
- Y. Wang, L. Sun, Z. Mei, F. Zhang, M. He, C. Fletcher, F. Wang, J. Yang, D. Bi, Y. Jiang and P. Liu, Mater. Des., 2020, 186, 108336 CrossRef CAS.
- M. Gonzalez-Alvarez, Pharmaceutics, 2021, 13, 10–12 Search PubMed.
- R. Singh, M. J. Bathaei, E. Istif and L. Beker, Adv. Healthcare Mater., 2020, 9, 2000790 CrossRef CAS PubMed.
- Y. Onuki, U. Bhardwaj, F. Papadimitrakopoulos and D. J. Burgess, J. Diabetes Sci. Technol., 2008, 2, 1003–1015 CrossRef PubMed.
- J. C. Quarterman, S. M. Geary and A. K. Salem, Eur. J. Pharm. Biopharm., 2021, 159, 21–35 CrossRef CAS PubMed.
- S. A. Stewart, J. Domínguez-Robles, R. F. Donnelly and E. Larrañeta, Polymers, 2018, 10(12), 1379 CrossRef PubMed.
- E. Magill, S. Demartis, E. Gavini, A. D. Permana, R. R. S. Thakur, M. F. Adrianto, D. Waite, K. Glover, C. J. Picco, A. Korelidou, U. Detamornrat, L. K. Vora, L. Li, Q. K. Anjani, R. F. Donnelly, J. Domínguez-Robles and E. Larrañeta, Adv. Drug Delivery Rev., 2023, 199, 114950 CrossRef CAS PubMed.
- J. J. Water, A. Bohr, J. Boetker, J. Aho, N. Sandler, H. M. Nielsen and J. Rantanen, J. Pharm. Sci., 2015, 104, 1099–1107 CrossRef CAS PubMed.
- C. J. Picco, J. Domínguez-Robles, E. Utomo, A. J. Paredes, F. Volpe-Zanutto, D. Malinova, R. F. Donnelly and E. Larrañeta, Drug Delivery, 2022, 29, 1038–1048 CrossRef CAS.
- Y. Maslii, I. Bezruk, A. Materiienko, O. Ruban, L. Ivanauskas and M. Velia, Chemija, 2023, 34 DOI:10.6001/chemija.v32i2.4433.
- A. Malenovic, M. Medenica, D. Ivanovic, B. Jancic and S. Markovic, Il Farmaco, 2005, 60, 157–161 CrossRef CAS PubMed.
- V. Sukumar, H. K. Chanduluru and S. Chinnusamy, J. Taibah Univ. Sci., 2023, 17 DOI:10.1080/16583655.2023.2252593.
- T. Mehmood, S. Hanif, F. Azhar, I. Ali, A. Alafnan, T. Hussain, A. Moin, M. A. Alamri and M. A. Syed, Int. J. Mol. Sci., 2022, 24, 440 CrossRef PubMed.
- A. Shaukat, K. Hussain, N. I. Bukhari and N. Shehzadi, J. Res. Pharm., 2022, 26(3), 609–616 CAS.
- M. Gamal, I. A. Naguib and R. M. Abdelfatah, Arch. Pharm., 2021, 354, e2100131 CrossRef PubMed.
- D. B. Khudhair and W. K. Ali, Int. J. Drug Delivery Technol., 2021, 11, 409–413 Search PubMed.
- V. Mulabagal, M. Annaji, S. Kurapati, R. P. Dash, N. R. Srinivas, A. K. Tiwari and R. J. Babu, Biomed. Chromatogr., 2020, 34, e4751 CrossRef CAS PubMed.
- P. Bhusal, M. Sharma, J. Harrison, G. Procter, G. Andrews, D. S. Jones, A. G. Hill and D. Svirskis, J. Chromatogr. Sci., 2017, 55, 832–838 CAS.
- E. R. Júnior, M. V. Lopes, B. Bentley, J. M. Marchetti and J. M. Marchetti, Rev. Bras. Cienc. Farm., 2002, 38 DOI:10.1590/S1516-93322002000100011.
- M. Thadasack, L. Chaunier, H. Rabesona, L. Viau, M. De-Carvalho, G. Bouchaud and D. Lourdin, Int. J. Pharm., 2022, 629, 122349 CrossRef CAS PubMed.
- L. Kang, H. W. Jun and J. W. McCall, J. Pharm. Biomed. Anal., 1999, 19, 737–745 CrossRef CAS PubMed.
- S.-Y. Liang, F. Shi, Y.-G. Zhao and H.-W. Wang, Molecules, 2022, 27, 5509 CrossRef CAS PubMed.
- M. Czapiewska and P. Bilski, Farm. Pol., 2021, 77, 339–348 CrossRef.
- L. L. Brkich, P. A. Markin, N. E. Moskaleva, N. V. Pyatigorskaya, G. E. Brkich, A. V. Panov and M. A. Grin, Pharm. Chem. J., 2020, 54, 69–72 CrossRef CAS.
- A. Doganay, B. Koksel, S. O. Gundogdu and Y. Capan, J. Chromatogr. Sci., 2018, 56(10), 903–911 CAS.
- S. Rathor and A. Sherje, Indian Drugs, 2021, 58, 50–55 Search PubMed.
- M. Al-Ghazawi, M. Alzoubi and B. Faidi, Int. J. Clin. Pharmacol. Ther., 2013, 51, 255–262 CrossRef CAS PubMed.
- B. N. Nalluri, J. Appl. Pharm. Sci., 2012, 2(7) DOI:10.7324/JAPS.2012.2714.
- V. Somasekhar and D. Gowrisankar, Asian J. Chem., 2011, 23, 1651–1654 CAS.
- V. V. Vaidya, G. R. Singh, M. P. Choukekar and M. B. Kekare, E-J. Chem., 2010, 7, 260–264 CrossRef CAS.
- F. A. Siddiqui, M. S. Arayne, N. Sultana and F. Qureshi, J. AOAC Int., 2011, 94(1), 150–158 CrossRef CAS PubMed.
- N. Kaul, S. R. Dhaneshwar, H. Agrawal, A. Kakad and B. Patil, J. Pharm. Biomed. Anal., 2005, 37, 27–38 CrossRef CAS PubMed.
- M. Zaman, M. Hanif, N. U. H. Khan, A. Mahmood, M. N. Qaisar and H. Ali, Acta Chromatogr., 2019, 31, 173–178 CrossRef CAS.
- M. Zaman, M. Hanif and H. Murtaza, Acta Pol. Pharm. – Drug Res., 2018, 75, 851–859 CrossRef CAS PubMed.
- P. B. Dudhe, S. Jadhav, V. Sawarkar and M. A. Nagras, Int. J. PharmTech Res., 2013, 5, 1212–1216 CAS.
- R. Siva Kumar, P. Senthil Nathan, P. Kumar Nallasivan, W. D. Sam Solomon and R. Venkatnarayanan, J. Pharm. Res. Health Care, 2010, 2, 84–94 CAS.
- A. L. Rao and D. P. Varma, Asian J. Chem., 2010, 22, 95–99 CAS.
- L. Sivasubramanian, Int. J. ChemTech Res., 2009, 1, 96–102 CAS.
- P. R. Battu, Int. J. ChemTech Res., 2009, 1, 499–501 CAS.
- G. Talele, R. Fursule, S. Surana and A. Bhavsar, Indian J. Pharm. Sci., 2006, 68, 641 CrossRef.
- D. N. Venkatesh, S. D. Shanmugakumar, AsfiaNajam, P. Parameshwar, K. Mahendar, B. Divya and E. Narsingam, Rasayan J. Chem., 2023, 16, 422–427 CrossRef CAS.
- S. Raza, M. Akhlaq, R. Habib, M. Ramzan, J. Mudassir, M. Z. Danish, M. Mukhtiar, A. Hussain and S. F. Khan, Lat. Am. J. Pharm., 2018, 37, 2154–2158 CAS.
- K. Dashora, G. Garg, S. Saraf and S. Saraf, Biosci., Biotechnol. Res. Asia, 2006, 3, 475–476 CAS.
- S. del Río-Sancho, V. Merino, A. López-Castellano and Y. N. Kalia, J. Chromatogr. Sci., 2016, 54, 790–795 Search PubMed.
-
Validation of Analytical Procedures Q2(R2), accessed 22/12/2023, https://www.ema.europa.eu/en/ich-q2r2-validation-analytical-procedures-scientific-guideline.
- Q. K. Anjani, A. H. Bin Sabri and R. F. Donnelly, Anal. Methods, 2022, 14, 125–134 RSC.
- Q. K. Anjani, E. Utomo, J. Domínguez-Robles, U. Detamornrat, R. F. Donnelly and E. Larrañeta, Molecules, 2022, 27(6), 1759 CrossRef CAS PubMed.
- D. R. Jenke, J. Liq. Chromatogr. Relat. Technol., 1996, 19, 737–757 CrossRef CAS.
- Q. K. Anjani, S. Demartis, F. Volpe-Zanutto, H. Li, A. H. Bin Sabri, E. Gavini and R. F. Donnelly, Pharmaceutics, 2023, 15(2), 408 CrossRef CAS PubMed.
-
FDA and CDER, Bioanalytical Method Validation Guidance for Industry Biopharmaceutics Bioanalytical Method Validation Guidance for Industry Biopharmaceutics Contains Nonbinding Recommendations, 2018 Search PubMed.
- Q. K. Anjani, A. H. Bin Sabri, M. B. McGuckin, H. Li, K. A. Hamid and R. F. Donnelly, AAPS PharmSciTech, 2022, 23(7), 273 CrossRef CAS PubMed.
-
Control pH during Method Development for Better Chromatography Technical Overview, https://www.agilent.com/Library/technicaloverviews/Public/5990-9984EN.pdf.
- Z. Muhammad, H. Muhammad and M. Hassan, Acta Pol. Pharm., 2018, 75, 851–859 CrossRef PubMed.
- C. Gobetti, M. E. Balsan, M. V. Ayres, S. H. O. de Almeida, E. de Saldanha Simon, N. R. Wingert, T. P. Oppe and C. V. Garcia, AAPS PharmSciTech, 2020, 21, 210 CrossRef CAS PubMed.
- J. Matić, M. Stanković-Brandl, H. Bauer, J. Lovey, S. Martel, C. Herkenne, A. Paudel and J. Khinast, Int. J. Pharm., 2023, 631, 122469 CrossRef PubMed.
- J. R. Jørgensen, W. Mohr, M. Rischer, A. Sauer, S. Mistry, A. Müllertz and T. Rades, Int. J. Pharm., 2023, 632, 122564 CrossRef PubMed.
- A. Krummnow, A. Danzer, K. Voges, S. Dohrn, S. O. Kyeremateng, M. Degenhardt and G. Sadowski, Pharmaceutics, 2022, 14(9), 1904 CrossRef CAS PubMed.
- J. R. Jørgensen, W. Mohr, M. Rischer, A. Sauer, S. Mistry, T. Rades and A. Müllertz, Eur. J. Pharm. Biopharm., 2023, 188, 26–32 CrossRef PubMed.
- K. Kayser, M. Monschke and K. G. Wagner, AAPS PharmSciTech, 2022, 23(6), 176 CrossRef CAS PubMed.
- G. Shadambikar, T. Kipping, N. Di-Gallo, A. G. Elia, A. N. Knüttel, D. Treffer and M. A. Repka, Pharmaceutics, 2020, 12, 1–17 CrossRef PubMed.
- A. Korelidou, J. Domínguez-Robles, E. R. Magill, M. Eleftheriadou, V. A. Cornelius, R. F. Donnelly, A. Margariti and E. Larrañeta, Biomater. Adv., 2022, 139, 213024 CrossRef PubMed.
- C. J. Picco, E. Utomo, A. McClean, J. Domínguez-Robles, Q. K. Anjani, F. Volpe-Zanutto, P. E. McKenna, J. G. Acheson, D. Malinova, R. F. Donnelly and E. Larrañeta, Int. J. Pharm., 2023, 631, 122477 CrossRef CAS PubMed.
- S. Rowlands and S. Searle, Open Access J. Contracept., 2014, 5, 73–84 CrossRef.
- S. A. Stewart, J. D. Robles, V. J. Mcilorum, E. Mancuso, D. A. Lamprou, R. F. Donnelly and E. Larrañeta, Pharmaceutics, 2020, 12(2), 105 CrossRef CAS PubMed.
- S. A. Stewart, J. Domínguez-Robles, E. Utomo, C. J. Picco, F. Corduas, E. Mancuso, M. N. Amir, M. A. Bahar, S. Sumarheni, R. F. Donnelly, A. D. Permana and E. Larrañeta, Int. J. Pharm., 2021, 607, 121011 CrossRef CAS PubMed.
- T. Patrício, M. Domingos, A. Gloria and P. Bártolo, Procedia CIRP, 2013, 5, 110–114 CrossRef.
- J. Holländer, N. Genina, H. Jukarainen, M. Khajeheian, A. Rosling, E. Mäkilä and N. Sandler, J. Pharm. Sci., 2016, 105, 2665–2676 CrossRef PubMed.
- L. Lei, X. Liu, Y. Y. Shen, J. Y. Liu, M. F. Tang, Z. M. Wang, S. R. Guo and L. Cheng, Eur. J. Pharm. Biopharm., 2011, 78, 49–57 CrossRef CAS PubMed.
- D. Karunakaran, S. M. Simpson, J. T. Su, E. Bryndza-Tfaily, T. J. Hope, R. Veazey, G. Dobek, J. Qiu, D. Watrous, S. Sung, J. E. Chacon and P. F. Kiser, J. Controlled Release, 2021, 330, 658–668 CrossRef CAS PubMed.
-
G. B. Beecham, T. A. Nessel and A. Goyal, Lidocaine, StatPearls, Treasure Island (FL), 2023 Search PubMed.
- N. K. Martin, J. Domínguez-Robles, S. A. Stewart, V. A. Cornelius, Q. K. Anjani, E. Utomo, I. García-Romero, R. F. Donnelly, A. Margariti, D. A. Lamprou and E. Larrañeta, Int. J. Pharm., 2021, 595, 120243 CrossRef CAS PubMed.
- E. Utomo, J. Domínguez-Robles, N. Moreno-Castellanos, S. A. Stewart, C. J. Picco, Q. K. Anjani, J. A. Simón, I. Peñuelas, R. F. Donnelly and E. Larrañeta, Int. J. Pharm., 2022, 624, 122061 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  and
Eneko
Larrañeta
and
Eneko
Larrañeta