
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Targeted multi-analyte UHPLC-MS/MS methodology for emerging contaminants in septic tank wastewater, sludge and receiving surface water†
Kai Wilschnack a,
Bess Homerb,
Elise Cartmellb,
Kyari Yatesa and
Bruce Petrie*a
a,
Bess Homerb,
Elise Cartmellb,
Kyari Yatesa and
Bruce Petrie*a
aSchool of Pharmacy and Life Sciences, Robert Gordon University, Aberdeen, AB10 7GJ, UK. E-mail: b.r.petrie@rgu.ac.uk; Tel: +44 (0)1224 262824
bScottish Water, 55 Buckstone Terrace, Edinburgh EH10 6XH, UK
First published on 5th January 2024
Abstract
Septic tanks treat wastewater of individual houses and small communities (up to 2000 people in Scotland) in rural and semi-urban areas and are understudied sources of surface water contamination. A multi-analyte methodology with solid phase extraction (SPE), ultra-sonic extraction, and direct injection sample preparation methods was developed to analyse a comprehensive range of emerging contaminants (ECs) including prescription and over-the-counter pharmaceuticals and related metabolites, natural and synthetic hormones, and other human wastewater marker compounds in septic tank influent and effluent, river water, suspended solids, and septic tank sludge by ultra-high-performance liquid chromatography coupled to tandem mass spectrometry (UHPLC-MS/MS). The number of quantifiable compounds in each matrix varied from 68 in septic tank wastewater to 59 in sludge illustrating its applicability across a range of matrices. Method quantification limits were 2.9 × 10−5–1.2 μg L−1 in septic tank influent, effluent and river water, with ≤0.01 μg L−1 achieved for 60% of ECs in all three water matrices, and 0.080–49 μg kg−1 in sludge. The developed method was applied to a septic tank (292 population equivalents) and the receiving river in the North-East of Scotland. Across all samples analysed, 43 of 68 ECs were detected in at least one matrix, demonstrating the method's sensitivity. The effluent concentrations suggest limited removal of ECs in septic tanks and a potential impact to river water quality for some ECs. However, further monitoring is required to better appreciate this. The developed methodology for a wide variety of ECs in a range of liquid and solid phases will allow, for the first time, a comprehensive assessment of ECs fate and removal in septic tanks, and their impact to surface water quality.
Introduction
Over the past years, a large variety of emerging contaminants (ECs), such as prescription or over-the-counter pharmaceuticals and related metabolites, natural and synthetic hormones, and other human wastewater marker compounds (e.g., caffeine), have been reported in various water sources worldwide in the ng to μg L−1 range.1–5 Due to their incomplete removal in conventional (biological) wastewater treatment, and ubiquitous presence in influent, treated wastewater discharges are considered the main entry source of ECs into the environment.6–8So far, research has focused on centralised wastewater treatment works (WWTWs) and their receiving surface waters.1,7,9–11 However, it is conservatively estimated that 9% of the Scottish population, are served by a public or privately owned septic tank.12–14 Septic tanks are typically located in rural and semi-urban areas and treat wastewater from individual houses and small communities (up to 2000 people in Scotland).4,14 In a watertight underground tank, often designed as a series of rectangular chambers, heavy solids settle as sludge to the bottom, while oil, grease and lighter solids float to the top.12 The sludge and scum need to be removed from the tank (typically every few months to every few years), and transported to a centralised WWTWs for further treatment.15 The septic tank effluent might be further treated, for example through subsoil infiltration systems, before being released into the ground or a nearby water body.12,16
Septic tank effluents can contain ECs in higher concentrations than in centralised WWTWs.13,17 For instance, Stanford and Weinberg17 reported the active ingredient in hormonal contraceptives 17α-ethinylestradiol up to 0.4 μg L−1 in a septic tank effluent serving a boarding school for girls, which is 4- to 400-times higher than centralised WWTWs influents.18 In a septic tank, ECs can be removed through the physical separation of the sludge and scum, when they are bound to particles or oil, and via anaerobic biodegradation.19 However, there is little information on the performance of septic tanks for the removal of ECs, and the effect of septic tank discharges to water quality. To this date, most studies focused on a few compounds only (maximum = 22),13,20–24 and there is a lack of multi-analyte methods for the analysis of ECs in septic tanks.
Most commonly, ECs are analysed by reversed-phase liquid chromatography coupled to tandem mass spectrometry as a highly sensitive and selective detector (LC-MS/MS).9–11 It is a suitable approach to determine low concentrations of ECs in the presence of other organics at comparatively high concentrations in complex environmental matrices, such as wastewater.9,11,19,25 Typically, solid phase extraction (SPE) is used to enrich, isolate and/or purify the target ECs, with reversed-phase hydrophilic-lipophilic balanced (HLB) polymeric sorbents being the most common.3,26,27 Although a wide range of compounds can be analysed with HLB sorbents, recoveries are low for very polar compounds such as the antidiabetic drug metformin.28–30 Hence, for very polar ECs, direct injection is proposed as a second sample preparation method.30 In wastewater, different ECs are present in a wide concentration range from low ng L−1 (e.g., ciprofloxacin) to high μg L−1 (e.g., metformin).29,31,32 As septic tanks are used by fewer people than centralised WWTWs, the variations in concentration and detection of ECs in effluents can be higher.13 The wide concentration range, poses a challenge for ‘SPE-only’ methods, as it requires the dilution and re-analysis of samples following data processing, when concentrations are above the calibration range.13,33 At the same time, method detection limits for ECs present at lower concentrations might not be reached by direct injection. Analysing each sample by direct injection and after SPE, allows the determination of a comprehensive range of ECs of different polarities over a wide concentration range without the need for further sample processing (e.g., dilution) and re-analysis.
Environmental samples are typically filtered prior to analysis to remove suspended solids. Due to the extra effort associated with analysing both matrices, most studies focus on the aqueous part of the sample only.7,34 However, ECs can adsorb to solid particulate matter, and desorb again once in the environment.1,35 Thus, analysing the aqueous part of the sample only leads to underestimation of the total concentration in the sample.11 Furthermore, in wastewater treatment, ECs can also adsorb to sludge, and for instance enter the environment when sludge is applied in agriculture.36 Most studies analysed ECs only in the liquid phase of septic tank effluent,20,21 and the receiving water bodies.13,22–24,37 Developing a multi-analyte method for the analysis of ECs in septic tank influent and effluent, including suspended solids, sludge, and the receiving surface water will allow a more accurate assessment of the performance of septic tanks for the removal of ECs and their effect to water quality. The most common methods for the extraction of ECs from solid environmental matrices, such as suspended solids or sludge, are microwave accelerated extraction (MAE), pressurised liquid extraction (PLE), and ultra-sonic extraction (USE).1,11,30,38 There is little difference found in the performance and extraction efficiency of the three methods.36,39,40 MAE and PLE are easier to automatise than USE. However, USE offers advantages due to low costs and easy operation for effective extraction of ECs from solid environmental samples.1,36
Therefore, the aim of the study was to develop a comprehensive multi-analyte methodology with SPE, USE, and direct injection as sample preparation methods to analyse a broad range of ECs in septic tank influent and effluent, river water, suspended solids, and septic tank sludge by ultra-high-performance liquid chromatography coupled to tandem mass spectrometry (UHPLC-MS/MS). The developed method was applied to a septic tank and the receiving surface water in a rural area in the North-East of Scotland.
Materials and methods
Materials
A total of 68 ECs (prescription or over-the-counter pharmaceuticals and related metabolites, natural and synthetic hormones, and other human wastewater marker compounds) were selected for method development (S1: Table S1†). The selection included those identified in prioritisation schemes by the European Union (EU) and the United Kingdom (UK),41–46 and those which posed the greatest threat to Scotland based on environmental risk assessment calculations (S2). Chemical names and properties of selected ECs and where they were obtained from are detailed in Tables S1 and S2.† Water was produced at ultra-pure quality in the laboratory (resistivity = 18.2 MΩ cm at 25 °C, PurA-Q18.2, LabPro, European Instruments, Oxford, UK), and methanol (HPLC grade, ≥99.9%) was purchased from Fisher Scientific (Loughborough, UK). Formic acid (≥99.0%, Fisher Scientific), ammonium formate (≥99.0%, Sigma Aldrich, Gillingham, UK), ammonium fluoride (NH4F, ≥99.99%, Sigma Aldrich), and ammonium hydroxide (NH4OH, 35%, Fison Instruments Ltd, Glasgow, UK) were used as mobile phase buffers and in ultrasonic extraction. Oasis HLB (60 mg, 3 mL; and 200 mg, 6 mL) SPE cartridges were purchased from Waters (Manchester, UK). Polytetrafluoroethylene (PTFE), cellulose acetate (CA), polyvinylidene fluoride hydrophilic (PVDF-HL), and polyvinylidene fluoride hydrophobic (PVDF) Q-Fil syringe filter (13 mm, 0.22 μm) from Greyhound (Birkenhead, UK) were received from Crawford Scientific Ltd (Strathaven, UK) and glass fibre filter (GF/F) discs (0.7 μm, 47 mm) were purchased from Fisher Scientific.Liquid samples (1 L septic tank influent, septic tank effluent, and river water), used during method development and validation were collected in the North-East of Scotland in polypropylene bottles in summer 2021. Samples were transported to the laboratory and frozen within 1 h after collection. The septic tank sludge (0.5 L) was collected in November 2021 with a custom-made polyvinylchloride sludge sampler (Fig. S1†), and frozen until processing.
Sample preparation of liquid samples
In a preliminary study, four different syringe filters were tested to minimize loss of ECs during the filtration step. Wastewater samples spiked with 60 ECs (available at the time of the experiment) were filtered through PVDF-HL, PTFE, CA, and PVDF syringes to determine any losses.The SPE method (Fig. 1) was developed based on a previous method for the analysis of septic tank effluent and river water.13 Initially, the samples were filtered under vacuum with a GF/F filter. Oasis HLB cartridges (3 mL, 60 mg) were conditioned under gravity with 2 mL methanol and 2 mL water for equilibration at a flow rate of 1 mL min−1. 50 mL wastewater, and 100 mL river water, were spiked with a 50 μL isotopic labelled surrogate working mix (c = 100 μg L−1), mixed and loaded onto the cartridges using vacuum at a flow rate of 5 mL min−1 and then dried for 20 min. The samples were eluted under gravity with 4 mL methanol at a flow rate of 1 mL min−1, and the solvent was evaporated at 40 °C under nitrogen.13 The dried residue was then redissolved in 500 μL water/methanol (95/5, v/v), and filtered through a PVDF-HL syringe filter prior to UHPLC-MS/MS injection. For direct injection, environmental samples were filtered through a PVDF-HL syringe filter, before 450 μL of the sample was spiked with 50 μL isotopic labelled surrogates (c = 100 μg L−1).
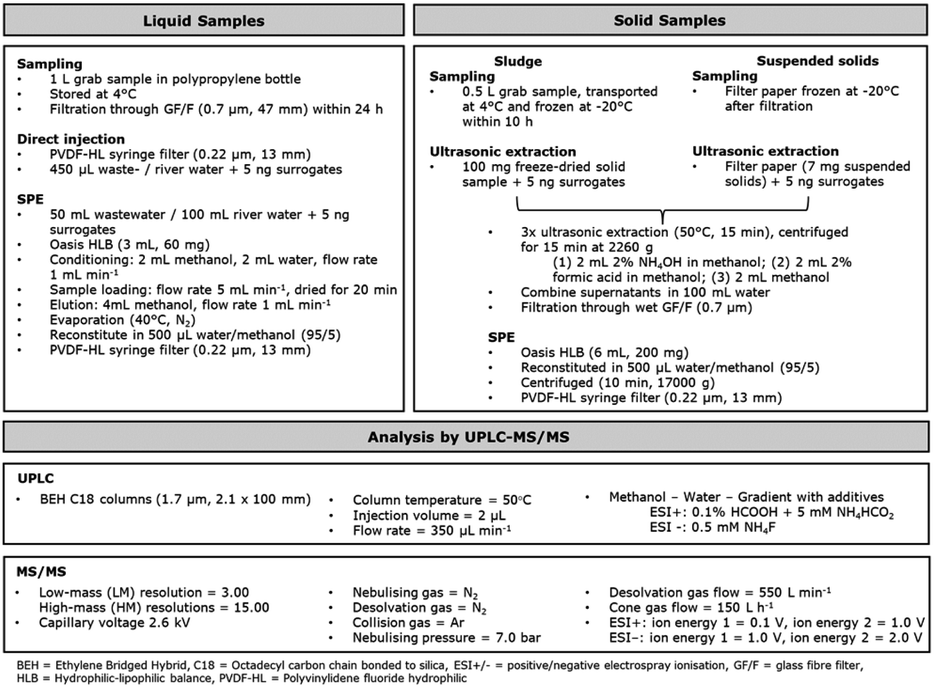 | ||
| Fig. 1 Overview of analytical workflow from sample preparation to analysis, for liquid and solid samples by ESI+ and ESI− methods. | ||
Extraction of solid matrices by ultra-sonic extraction
The sludge was frozen and freeze dried using a Heto Drywinner freeze dryer by Copley. The selected ECs were extracted from solid matrices with a Clifton Range ultra-sonic water bath (280 W, 50/60 Hz) using three extraction cycles similar to that described by Al-Khazrajy and Boxall.25 Briefly, 0.1 g of freeze-dried sludge (dry weight) was weighed into a 10 mL polypropylene centrifuge tube, spiked with 50 μL isotopically labelled surrogates (c = 100 μg L−1) and left overnight. In the first cycle, 2 mL of 2% NH4OH in methanol was added. The suspension was vortexed, ultra-sonicated for 15 min at 50 °C, and centrifuged at 2260 g for 15 min. The supernatant was collected in a 50 mL Duran® glass bottle. The extraction was repeated using 2 mL of 2% formic acid in methanol and then 2 mL of methanol. The combined supernatants were filtered through a wet GF/F disc and diluted with water to 100 mL (methanol < 5%). The extracts were cleaned up by Oasis HLB SPE cartridges (6 mL, 200 mg) following the same procedure as described for the extraction of liquid samples. For sludge samples, the reconstituted extract was centrifuged for 10 min at 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g prior to filtration through a PVDF-HL syringe filter.
000g prior to filtration through a PVDF-HL syringe filter.
Liquid chromatography tandem mass spectrometry
Samples were analysed with UHPLC-MS/MS using an ACQUITY UPLC system from Waters (Waters Corporation, Milford, MA) with a Xevo TQ-XS Triple Quadrupole Mass Spectrometer. Electrospray ionisation (ESI) was performed in both positive and negative modes with a capillary voltage of 2.6 kV, 3.00 low-mass (LM) resolutions, and 15.00 high-mass (HM) resolutions. The nebulising and desolvation gas was nitrogen, and the collision gas was argon. The gas temperature was 400 °C with a desolvation gas flow of 550 L min−1, and a nebulising pressure of 7.0 bar. The cone gas flow was 150 L h−1. The optimised ion energies were ion energy 1 = 0.1 V and ion energy 2 = 1.0 V in positive ionisation mode, and ion energy 1 = 1.0 V and ion energy 2 = 2.0 V in negative ionisation mode, respectively.Two different mobile phases were used for the analysis of basic and acidic compounds in positive and negative ionisation, respectively.30 Different additives to the mobile phase were tested. If not otherwise stated, the parameters were identical in both methods. Chromatographic separation was performed using reversed-phase ACQUITY UPLC Ethylene Bridged Hybrid (BEH) C18 columns (1.7 μm, 2.1 × 100 mm, Waters). The column temperature was kept constant at 50 °C. The injection volume was 2 μL and the flow rate was 350 μL min−1. A methanol–water-gradient along with additives was used as the mobile phase (S4: Table S3†). Additives were 5 mM ammonium formate and 0.1% formic acid in the positive ionisation method, and 0.5 mM NH4F in the negative ionisation method.
Instrumental performance
The instrumental performance was validated in terms of detection and quantification limits, linearity, intra- and inter-day precision, and accuracy. All samples were spiked with isotopically labelled analytes as surrogate to correct for matrix effects and analyte loss during sample preparation (c = 10 μg L−1 at injection).13The instrument detection (IDL) and quantification limits (IQL) for each analyte were determined by the lowest concentration with a signal-to-noise ratio (S/N) ≥ 3 or ≥ 10, respectively. Linearity was established through the injection of a range of standards between 0.05 and 100 μg L−1 (S8: eqn S3†).
Intra-day precision and accuracy were determined by injecting standards at concentrations of 1, 10, and 50 μg L−1 in triplicate within 24 h (S8: eqn S4 and S5†). This was repeated every 24 h over 3 days to establish inter-day precision and accuracy.
Method performance
The method performance was assessed for septic tank influent and effluent wastewater, river water, and sludge, for detection and quantification limits, matrix effects, absolute and relative recoveries, precision, and accuracy. Samples were prepared at three concentrations in triplicate. Spike concentrations were 1, 10, and 50 μg L−1 for direct injection of influent, effluent, and river water; 0.01, 0.1, and 0.5 μg L−1 for SPE of influent and effluent; 0.005, 0.05, and 0.25 μg L−1 for SPE of river water, and 50, 250, 500 μg kg−1 for sludge (S5: Table S4†). Prior to spiking with ECs, samples were spiked with isotopically labelled ECs only and analysed to determine the analyte concentrations in the environmental samples. Water samples were analysed by direct injection and SPE (S5: Table S4†).Absolute (RECabs) and relative recoveries (REC) were calculated following eqn (1) and (2) from peak areas (A) and area ratios (ar) of spiked and unspiked (US) samples and standards (std), respectively.
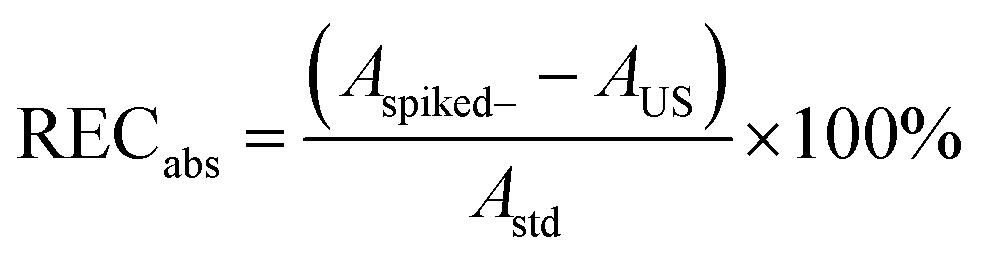 | (1) |
 | (2) |
The method detection (MDL) and quantification limits (MQL) were calculated for each analyte from the IDL and IQL, respectively, the recovery and concentration factor cF using eqn (3) and (4).
 | (3) |
 | (4) |
REC and cF were specific for each matrix and sample preparation method. cF was 0.9 for direct injection, 100 for septic tank influent and effluent in the SPE method, and 200 for river water in the SPE method. For solid matrices, cF is replaced with a conversion factor of 0.2 g mL−1, based on the extraction of 0.1 g sludge.
The relative standard deviation of the replicates was calculated for method precision. Accuracies were determined from the percentage deviation of the concentrations added to the samples from the calculated concentrations.
To ensure instrumental and method performance, blanks and quality control standards with concentrations of 1, 10, and 50 μg L−1 were injected before and after every batch of samples.
Application to a septic tank and receiving river
A septic tank and the receiving surface water in a rural area in the North-East of Scotland was investigated. The septic tank serves 292 population equivalents, with no tourist impact and around 8% non-household contribution.14 The nominal dilution of the septic tank discharge into the river was calculated (S6: eqn S1 and S2†). The receiving river mainly flows through agricultural land, with single houses and smaller villages along side. In the catchment area, 1% of land use is classified as urban.47 The largest settlement in the catchment area with a population of 3140 (mid-2020 estimate)48 is located roughly 7 km upstream of the studied septic tank. It is served by a secondary biological WWTW that discharges into the river.Sampling was conducted on the 10th of November 2021. Grab samples (1 L) were collected in polypropylene bottles at the influent and effluent point of the septic tank, in the river upstream and downstream of the septic tank discharge point at a minimum distance of five river widths, and from the sludge. Samples were transported to the laboratory at 4 °C. Liquid samples were filtered through 0.7 μm GF/F membrane filters within 24 h, processed as described previously, and analysed within 48 h. The filter papers were frozen at −20 °C until processing. The solids were extracted by ultra-sonic extraction following the previous description. All samples were prepared in duplicate.
Results and discussion
Liquid chromatography tandem mass spectrometry
All ECs were analysed using multiple reaction monitoring (MRM) transitions. The protonated ([M + H]+) or deprotonated molecular ion ([M − H]−) was monitored in ESI− and ESI+ mode, respectively. Following EU guidelines,49 two MRM transitions were monitored for most ECs (one in the case of isotopic labelled surrogates), using the fragment with the highest response for quantification and the fragment with the second highest response for confirmation. Ion ratios were monitored. In accordance with the literature, only one stable fragment was found for ibuprofen, gemfibrozil and lidocaine,3,30,50 which is considered semi-quantitative (optimised MS/MS parameters in S7: Table S5†).Following optimisation of MS/MS parameters for all compounds the chromatography methods were developed using a methanol–water-gradient with additives as the mobile phase and a reversed-phase BEH C18 column. Two different mobile phases were used, since basic and neutral compounds are best analysed in positive ionisation mode from acidic solutions, whereas acidic compounds are more efficiently analysed in negative ionisation mode from basic solutions.51 Different additives were tested to optimise separation, peak shape, and sensitivity. In the positive ionisation mode for the analysis of basic ECs, the use of 5 mM ammonium acetate with 0.1% formic acid was compared to using 5 mM ammonium formate and 0.1% formic acid. While the choice of ammonium salt generally had little effect on the chromatography, the peak shape improved substantially with ammonium formate in the mobile phase for metformin, guanylurea, and paracetamol. The highly polar drug metformin and its aerobic bacterial metabolite guanylurea are more suited to analysis by hydrophilic interaction chromatography (HILIC) columns,2,29 but satisfactory chromatography could be achieved under reversed phased conditions with ammonium formate as an additive.
In the negative ionisation mode, ammonium hydroxide (0.1%) and different concentrations of NH4F (0.1, 0.5 and 1 mM) in a methanol–water-gradient were considered to enable the analysis of estrogens together with acidic drugs.30,52 Overall, NH4F resulted in greater peak areas and sharper peaks than ammonium hydroxide. Improved sensitivity with NH4F might be due to the strong basicity of the fluoride anion, and hence increased deprotonation of ECs in the gas phase.53 Lower NH4F concentration increased the sensitivity for estrogens, with optimum concentrations being 0.1 mM. However, decreased sensitivity for ibuprofen was noted. Since estrogens are expected to be found in significantly lower concentrations in wastewater and river water compared to ibuprofen,6 0.1 mM NH4F was considered for further method development. However, in wastewater a contamination was present in the 17α-ethinylestradiol MS/MS spectrum at the same retention time. This was resolved from the 17α-ethinylestradiol peak by increasing the NH4F concentration to 0.5 mM. With the reversed-phase BEH C18 column, good separation, sensitivity, and peak shape was achieved for all compounds using a methanol–water-gradient along with 5 mM ammonium formate and 0.1% formic acid in the ESI+ method, and 0.5 mM NH4F in the ESI− method (Fig. 2).
 | ||
| Fig. 2 Chromatograms (quantification MRM) of septic tank effluent spiked at c = 62.5 μg L−1 and analysed by direct injection (A and C), and at c = 0.5 μg L−1 and analysed by SPE (B and D) (details in S5: Table S4†), analysed with the ESI+ (A and B) and ESI− (C and D) method. | ||
Instrument performance
The IDL and IQL were determined as the lowest concentration with a S/N ≥ 3 and ≥ 10 and ranged from 0.002 to 1 μg L−1, and from 0.005 to 5 μg L−1, respectively (S8: Table S6†). For the majority of compounds, IQL ≤ 0.5 μg L−1 was achieved. A wide range of IQLs is commonly observed in multi-analyte methods for compounds with a variety of physicochemical properties, and similar to what has been reported before.3,27,28,30Linearity was established through the injection of standards at concentrations between 0.05 and 100 μg L−1 (500 μg L−1 for paracetamol, ibuprofen, and metformin due to their higher concentrations in wastewater). A linear regression model was fitted (S8: eqn S3†), and the R2 was calculated. For the compounds without the isotopically labelled EC, a different deuterated surrogate was assigned (S8: Table S6†). The choice was based on retention time, structural similarity, and eventually linearity. The linear dependency was in range of 0.938 ≤ R2 ≥ 1.000 (S8: Table S6†). Approximately two thirds of the ECs, 52 compounds in the positive method and four compounds in the negative method, have R2 values ≥ 0.997. Atorvastatin and miconazole were calibrated externally using peak area as there was no suitable deuterated surrogate. Calibrations with R2 ≥ 0.991 were sufficient for accurate quantification, as indicated by the other instrumental performance criteria. Published studies for multi-analyte analysis of pharmaceuticals in wastewater accept R2 ≤ 0.990.11 Miconazole, clotrimazole, and climbazole, have R2 < 0.980, most likely due to the absence of suitable deuterated surrogate, and were analysed semi-quantitatively. Most compounds were linear over the whole concentration range from 0 to 100 μg L−1.
Intra- and inter-day accuracy and precision (S8: Table S7†) were determined by injecting three standards (c = 1 μg L−1, 10 μg L−1, and 50 μg L−1) three times within 24 h, and repeatedly every 24 h over three days (S8: eqn S4 and S5†). In multi-analyte methods, accuracies are generally expected to be within an ideal range of 90–110%, or within the accepted range 80–120%.10,28,54 A total of 63 compounds were accurate within the range of 90–110% in most samples above the IQL, with little or no difference between the intra- and inter-day accuracy (p > 0.05, S8: Table S7†). The remaining five compounds also have intra-day accuracies from 90% to 110% in most samples, but inter-day accuracies were 80% to 120% in most samples (0.004 ≥ p ≤ 0.046). As repeating the calibration every day is time-consuming, few ECs with inaccuracies are accepted in multi-analyte methods.28 QC standards were therefore injected with every batch to ensure accuracies stay within the accepted range. Calibrations were repeated after the mass spectrometer was turned off for an extended period of time, at least once a year, or if the QC data fell out with the performance data.
In general, relative standard deviations ≤ 10% are expected in the instrumental performance. However, higher standard deviations ≥ 20% are accepted for few ECs in multi-analyte methods, as long as other validation parameters are suitable.11,27 In the developed instrumental method, 50 ECs were very precise over all concentrations studied above the IQL with a relative standard deviation ≤ 10% except the occasional one concentration in the intra- and inter-day analysis. Of the remaining compounds, 15 had a relative standard deviation ≤ 20% over all three concentrations above the IQL. The remaining three ECS had relative standard deviation ≤ 10% in most samples. Overall, the method was very precise with relative standard deviations ≤ 10% for the majority of compounds.
The intra- and inter-day instrumental performance was high across the majority of ECs. In total, 94% of the compounds were precise and accurate with a suitable linear calibration using the area ratio. Atorvastatin was linear, precise and accurate using the peak area, and miconazole, clotrimazole and climbazole could be analysed on a semi-quantitative basis as they showed satisfactory accuracy and precision data.
Method performance
The most common syringe filter membrane used for ECs prior to UHPLC-MS/MS is PTFE.11,30,31,54 However, low recoveries have been observed for some ECs including erythromycin and gemfibrozil.55 Therefore, a range of syringe filters including PVDF-HL, PTFE, CA, and PVDF were investigated to minimize loss of ECs during the filtration step.Absolute recoveries were >75% for all four syringe filters for 49 ECs (S9: Table S8†). Similarly, Darwano et al.1 reported high recoveries for most analytes with little variation between different syringe filters. However, for clarithromycin, erythromycin, chlorpheniramine, cetirizine and citalopram poorer recoveries were found with PVDF, which is in line with what has been reported before for antibiotics including clarithromycin.56 For all five compounds recoveries were at least 20% higher in the other filters, with CA and PVDF-HL being more effective than PTFE. However, CA gave lower recoveries for amoxicillin, estrone, and 17β-estradiol than what was achieved with PTFE, PVDF, and PVDF-HL syringe filters (>80%). PVDF-HL syringe filters were the best compromise for the studied EC, giving recoveries >70% for the majority of ECs. The effective use of PVDF-HL syringe filters has, for example, also been reported by Wang et al.57 Low recoveries of approximately 10% were only found for fluoxetine, miconazole, and clotrimazole, and this was observed for all four syringe filters. All samples were filtered through PVDF-HL syringe filters prior to UHPLC-MS/MS injection.
To determine method performance, septic tank influent and effluent, river water, and sludge samples were spiked at three concentrations (S5: Table S4†). Water samples were analysed by direct injection and SPE. Calculations were not practical for 29 ECs in at least one sample, when the environmental concentration exceeded the spike concentration, most common at lowest spike concentrations in effluent SPE samples.
In direct injection samples, absolute recoveries were 23–209% in septic tank influent, 19–192% in septic tank effluent, and 19–186% in river water (S9: Table S9†). Most ECs have absolute recoveries from 25 to 125% (Fig. 3). Recoveries over 100% were due to signal enhancement. This highlights the requirement of the use of deuterated surrogates to correct for matrix effects and variations in the instrumental and method performance.
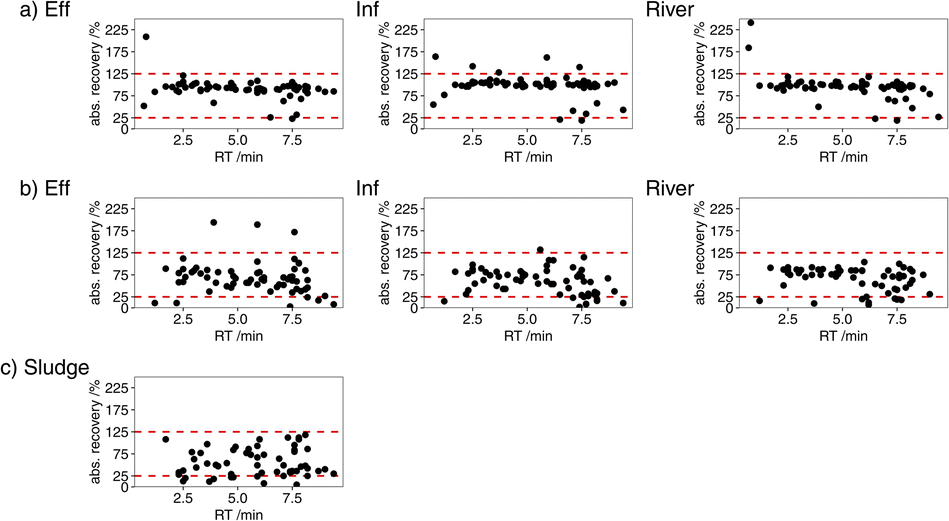 | ||
| Fig. 3 Absolute recoveries (%) in influent, effluent and river water analysed by (a) direct injection and (b) SPE, and in (c) sludge. | ||
For 41 ECs (63%), relative recoveries by direct injection were in the range of 90% to 110% in all three matrices, and in the range of 75% to 125% for a further 11 ECs (Fig. 4). The remaining 14 ECs have relative recoveries from 22 to 197%, most likely due to the absence of a suitable deuterated surrogate to account for matrix effects and analyte loss. Similar results have been reported by Oliveira et al.58 who found relative recovery from 20 to 230%, with the majority recoveries being in the range of 70–150% in the analysis of ECs in wastewater influent and effluent by direct injection LC-MS/MS. The direct injection MDLs were 3.3 × 10−3–3.0 μg L−1 in influent, were 4.1 × 10−3–3.7 μg L−1 in effluent, and 3.6 × 10−3–3.4 μg L−1 in river water. MQLs were 6.7 × 10−3–8.8 μg L−1 in influent, 8.1 × 10−3–14 μg L−1 in effluent, and 7.2 × 10−3–8.3 μg L−1 in river water (S9: Table S10†). While these MQLs were sufficient for the determination of high use compounds, such as metformin or paracetamol,29,31,32 hormones and antibiotics have predicted no-effect concentrations (PNEC) < 1 μg L−1 and are reported in freshwater at ng L−1. Hence, the use of a SPE method was necessary to determine all ECs at the relevant concentrations. In direct injection, 29 ECs were very precise over all three concentrations with a relative standard deviation ≤ 10% in influent, effluent and river water (S9: Table S11†). Of the remaining compounds, 29 ECs were precise with a relative standard deviation ≤ 20% over all three concentrations in all matrices. The remaining ECs were precise for most spiked concentrations and matrices. Accuracies within the range of 75–125% were observed for the majority of 54 ECs. Most remaining ECs were accurate for most concentrations and matrices. This is similar to the results reported by Rapp-Wright et al.31 for direct injection LC-MS/MS, and considering the complexitiy of matrices and the number of analytical steps involved, precision and accuracy were considered acceptable.
 | ||
| Fig. 4 Relative recoveries (%) in influent, effluent and river water analysed by direct injection and SPE, and in sludge, for the 66 ECs with assigned deuterated surrogate. | ||
Absolute recoveries following SPE were 0–194% in septic tank influent, 1–200% in septic tank effluent, and 0–122% in river water (Fig. 3). The measured absolute recoveries were in the range of what has been previously reported using LC-MS/MS to determine multiple ECs in wastewater.10,30,59 While the lack of selectivity of HLB allows the extractions of a wide range of analytes, matrix can be co-extracted and cause significant signal interference.10 Signal interference is typically reported to be high in multi-residue LC-MS/MS methods using ESI as ionisation method and HLB columns in SPE due to lack of selectivity.11,30,54 Lowest and no absolute recoveries from SPE were observed for the very polar compounds guanylurea, metformin, gabapentin, sulfanilamide, and amidotrizoic acid, and amoxicillin from river water (S9: Table S9†). HLB sorbents are known for their low recovery of very polar compounds,27,28,30e.g., Klančar et al.28 reported recoveries of 0.3% for metformin and 2.6% for gabapentin from river water. Due to the low absolute recoveries, guanylurea, metformin, gabapentin and amidotrizoic acid were determined by direct injection only. Relative recoveries for the remaining ECs analysed by SPE were 90–110% in all three matrices for 16 ECs and 75–125% for 17 ECs in all three matrices (Fig. 4). The remaining ECs had relative recoveries <75% or >125% in at least one water matrix. Similar relative recoveries have been reported by Anumol and Snyder in wastewater,37 and the results used in the determination of concentrations to account for differences in the behaviour of the deuterated surrogate and analyte. The MDLs for SPE were 5.4 × 10−5–0.073 μg L−1 in influent, 5.3 × 10−5–0.033 μg L−1 in effluent, and 2.9 × 10−5–0.40 μg L−1 in river water. MQLs were 1.5 × 10−4–0.096 μg L−1 in influent, 1.6 × 10−4–0.22 μg L−1 in effluent, and 6.6 × 10−5–0.50 μg L−1 in river water (S9: Table S10†). Including SPE in the method preparation allows the determination of ECs at the relevant concentrations. The precision of 58 ECs was high over all three concentrations in influent, effluent and river water with relative standard deviations ≤ 20%. The remaining ten ECs were precise over most concentrations and matrices (S9: Table S12†). Similar precision were obtained by Ofrydopoulou et al.27 The majority of ECs analysed by SPE had accuracies within the range of 75–125% for all concentrations above the MQL in influent, effluent and river water. Comparatively lower accuracies were found when the EC was present in the sample, e.g., sulfanilamide in the effluent, trimethoprim at the smallest spike concentration in river water, and citalopram in influent. Lower accuracies were also found for amoxicillin in river water, with a low absolute recovery, and for warfarin at 1 μg L−1 close to the MQL (S9: Table S12†).
The USE method for the extraction of sediments described by Al-Khazrajy and Boxall25 was modified to optimise extraction of the selected 68 ECs from sludge. To accommodate the higher concentrations of ECs in sludge compared to sediments,60 a smaller mass of 0.1 g was used. Furthermore, the clean-up step was adjusted to keep it as similar as possible to the SPE of liquid samples. However, a larger SPE cartridge (200 mL for sludge) was chosen to avoid blocking of the cartridge during sample loading. Furthermore, an additional centrifuge step prior to filtration through a PVDF-HL syringe filter was necessary. The method was successfully applied for the extraction of 59 out 68 ECs from sludge (S9: Table S9†). Due to the complexity of the environmental matrices, a different number of analytes is often reported for different matrices in multi-analyte methods.10,30 For example, the USE method is not suitable for very polar compounds, such as metformin, sulfanilamide and gabapentin with low absolute recoveries from SPE. Due to their high polarity they are more likely to stay in the water phase and less likely to be found in the sludge.61 For the remaining compounds absolute recoveries from sludge were 12–112% (Fig. 3). The majority of ECs had relative recoveries of 75–125% from sludge (Fig. 4). Low relative recoveries below 50% (e.g., diclofenac and sulfadiazine) and high relative recoveries over 150% (e.g., trimethoprim and estriol) were found for ECs when the deuterated surrogate behaved differently than the analyte. MDLs and MQLs were 0.025–7.4 μg kg−1 and 0.080–49 μg kg−1, respectively. However, only five ECs have MQLs > 10 μg kg−1 and only mebendazole has an MQL > 15 μg kg−1 (S9: Table S10†). Most ECs have accuracies within the range of 75–125% for all spike concentrations (S9: Table S12†). Lower accuracies were found for few ECs at one spike concentration, e.g., for sulfadiazine at 50 μg kg−1 and for hydroxyibuprofen at 500 μg kg−1. The precision of 53 ECs was high over all three spike concentrations with relative standard deviations ≤ 20%; the remaining six compounds have higher relative standard deviations at one concentration only.
The number of quantifiable compounds in each matrix varied from 68 in effluent to 59 in sludge, demonstrating the method's wide applicability.
Application to environmental matrices
The developed method was applied to samples collected from a septic tank in the North-East of Scotland at the influent and effluent point, from the sludge, and from the receiving river upstream and downstream of the septic tank's discharge point. Additionally, the suspended solids from the influent and effluent were analysed. At sampling time, the dilution factor of effluent into the river was 756.62Across all samples analysed, 43 ECs were detected at least once (Table 1). Fifteen ECs from six different groups (analgesics, antibiotics, anticonvulsants, antihistamines, β-blockers, wastewater discharge marker) were found in all matrices.
| Class | EC | Inf (μg L−1) | Eff (μg L−1) | Up (μg L−1) | Down (μg L−1) | SuS Inf (μg kg−1) | SuS Eff (μg kg−1) | Sludge (μg kg−1) |
|---|---|---|---|---|---|---|---|---|
| a nd, not detected; —, method not suitable. | ||||||||
| Anaesthetics | Lidocaine | 0.025 ± 0.0056 | 0.043 ± 0.0020 | nd | nd | (2.6 ± 0.14) × 103 | (1.1 ± 0.084) × 102 | nd |
| Analgesics | 3-Methoxyparacetamol | 8.2 ± 0.44 | 13 ± 0.85 | (2.3 ± 0.10) × 10−3 | 0.026 ± 0.0013 | (1.1 ± 0.064) × 102 | (1.6 ± 0.40) × 102 | 68 ± 8.3 |
| Diclofenac | 2.4 ± 0.10 | 0.58 ± 0.0047 | 0.023 ± 0.0024 | 0.016 ± 0.00097 | 78 ± 2.6 | 47 ± 3.2 | (6.2 ± 1.3) × 102 | |
| Hydroxyibuprofen | 2.7 ± 0.25 | 17 ± 0.56 | (2.9 ± 0.41) × 10−3 | 0.019 ± 0.0017 | nd | nd | nd | |
| Ibuprofen | 34 ± 1.8 | 26 ± 0.71 | 0.47 ± 0.083 | 0.63 ± 0.070 | (2.1 ± 0.067) × 104 | (6.1 ± 0.55) × 103 | (1.6 ± 0.26) × 103 | |
| Ketoprofen | nd | nd | nd | nd | nd | nd | nd | |
| Naproxen | 3.7 ± 0.079 | 26 ± 0.26 | (3.2 ± 0.12) × 10−3 | 0.032 ± 0.0017 | (5.1 ± 0.62) × 102 | (7.0 ± 0.88) × 102 | (3.0 ± 0.41) × 102 | |
| Paracetamol | (2.0 ± 0.12) × 102 | (2.9 ± 0.076) × 102 | 0.039 ± 0.0043 | 0.59 ± 0.053 | (1.4 ± 0.45) × 103 | (1.3 ± 0.18) × 104 | (3.6 ± 0.045) × 103 | |
| Antibiotics | 3-Desmethyltrimethoprim | 0.013 ± 0.0053 | 0.16 ± 0.014 | (1.8 ± 0.23) × 10−3 | (2.5 ± 0.30) × 10−3 | 78 ± 5.4 | 84 ± 10 | 4.8 ± 0.63 |
| Amoxicillin | nd | nd | nd | nd | — | — | — | |
| Ciprofloxacin | 10 ± 0.61 | 2.4 ± 0.43 | nd | nd | (3.8 ± 0.93) × 103 | (4.9 ± 0.59) × 102 | nd | |
| Clarithromycin | nd | nd | nd | nd | (3.1 ± 0.027) × 103 | (1.3 ± 0.13) × 103 | 13 ± 2.0 | |
| Erythromycin | 0.077 ± 0.020 | 0.14 ± 0.016 | nd | nd | nd | nd | — | |
| Ofloxacin | nd | nd | nd | nd | (1.2 ± 0.16) × 102 | 69 ± 15 | — | |
| Sulfadiazine | nd | nd | nd | nd | nd | nd | nd | |
| Sulfamethoxazole | 0.029 ± 0.0032 | 0.59 ± 0.029 | (3.1 ± 0.55) × 10−3 | (3.1 ± 0.56) × 10−3 | nd | 43 ± 3.7 | nd | |
| Sulfanilamide | 0.33 ± 0.031 | 0.13 ± 0.018 | nd | nd | — | — | — | |
| Trimethoprim | 0.014 ± 0.0047 | 0.25 ± 0.018 | (8.8 ± 1.8) × 10−4 | (1.1 ± 0.14) × 10−3 | 44 ± 3.0 | 63 ± 1.8 | 7.5 ± 0.67 | |
| α-Hydroxytrimethoprim | nd | (7.0 ± 2.8) × 10−3 | nd | nd | nd | nd | nd | |
| Anticoagulants | Warfarin | nd | nd | nd | nd | nd | nd | nd |
| Anticonvulsants | Carbamazepine | nd | (7.2 ± 0.43) × 10−4 | (2.0 ± 0.14) × 10−4 | (1.4 ± 0.097) × 10−4 | nd | nd | nd |
| Carbamazepine-10,11-epoxide | nd | nd | nd | nd | nd | nd | nd | |
| Gabapentin | 1.9 ± 0.30 | 7.4 ± 0.56 | nd | nd | — | — | — | |
| Lamotrigine | 0.97 ± 0.17 | 1.2 ± 0.055 | (3.0 ± 0.26) × 10−3 | (4.5 ± 0.27) × 10−3 | (4.3 ± 0.68) × 102 | (1.3 ± 0.15) × 103 | 70 ± 11 | |
| Primidone | nd | nd | nd | nd | nd | nd | nd | |
| Antidepressants | Citalopram | 0.19 ± 0.014 | 0.14 ± 0.0091 | nd | nd | (7.2 ± 1.5) × 102 | (1.3 ± 0.26) × 102 | (1.4 ± 0.088) × 102 |
| Desmethylcitalopram | 0.14 ± 0.0084 | 0.079 ± 0.0030 | nd | nd | (1.0 ± 0.22) × 103 | (1.2 ± 0.26) × 102 | (1.5 ± 0.067) × 102 | |
| Desmethylvenlafaxine | 0.028 ± 0.0015 | 0.21 ± 0.011 | nd | (6.3 ± 1.7) × 10−4 | 44 ± 7.2 | 47 ± 7.2 | 15 ± 3.3 | |
| Fluoxetine | 0.013 ± 0.0026 | 0.016 ± 0.0025 | nd | nd | (1.4 ± 0.54) × 102 | 68 ± 12 | nd | |
| Venlafaxine | 0.081 ± 0.0025 | 0.14 ± 0.0084 | nd | nd | (2.0 ± 0.12) × 102 | 66 ± 3.6 | 55 ± 10 | |
| Anti-diabetics | Guanylurea | nd | nd | nd | nd | — | — | — |
| Metformin | (2.2 ± 0.082) × 102 | (1.6 ± 0.067) × 102 | 0.85 ± 0.031 | 1.1 ± 0.038 | — | — | — | |
| Anti-fungals | Climbazole | nd | nd | nd | nd | nd | nd | nd |
| Clotrimazole | nd | nd | nd | nd | (5.1 ± 0.64) × 102 | (4.8 ± 0.89) × 102 | (1.7 ± 0.11) × 102 | |
| Fluconazole | nd | nd | nd | nd | nd | nd | nd | |
| Miconazole | nd | nd | nd | nd | nd | nd | 78 ± 25 | |
| Anti-helmintics | Mebendazole | nd | nd | nd | nd | nd | nd | nd |
| Antihistamines | Cetirizine | 1.2 ± 0.050 | 1.2 ± 0.046 | (3.4 ± 0.32) × 10−3 | (5.6 ± 0.36) × 10−3 | (1.0 ± 0.18) × 102 | (1.2 ± 0.14) × 102 | (1.1 ± 0.060) × 102 |
| Chlorpheniramine | nd | (1.8 ± 0.034) × 10−3 | nd | nd | 93 ± 4.0 | 39 ± 3.0 | 22 ± 6.0 | |
| Fexofenadine | 0.16 ± 0.0097 | 0.81 ± 0.018 | 0.013 ± 0.0014 | 0.012 ± 0.0016 | (2.6 ± 0.049) × 102 | (1.1 ± 0.088) × 102 | (3.4 ± 0.67) × 103 | |
| Anti-pruritic | Crotamiton | 0.74 ± 0.061 | 1.5 ± 0.13 | nd | nd | (2.2 ± 0.14) × 102 | (2.3 ± 0.51) × 102 | 89 ± 6.6 |
| Antiulcer | Ranitidine | nd | nd | nd | nd | nd | nd | nd |
| 4-Hydroxyomeprazole | 0.33 ± 0.023 | 0.43 ± 0.035 | nd | nd | (3.6 ± 0.19) × 102 | 45 ± 4.5 | (1.1 ± 0.027) × 102 | |
| Lansoprazole | nd | nd | nd | nd | — | — | — | |
| Omeprazole | nd | nd | nd | nd | — | — | — | |
| Benzodiazepines | Lorazepam | nd | nd | nd | nd | nd | nd | nd |
| Oxazepam | nd | nd | nd | nd | nd | nd | nd | |
| Temazepam | nd | 0.019 ± 0.0026 | nd | nd | nd | nd | nd | |
| Betablockers | Acebutolol | nd | nd | nd | nd | nd | nd | nd |
| Atenolol | (7.5 ± 0.60) × 10−3 | 0.088 ± 0.0067 | (3.8 ± 0.27) × 10−4 | (6.3 ± 0.16) × 10−4 | 59 ± 4.1 | 50 ± 12 | 53 ± 4.6 | |
| Bisoprolol | 0.23 ± 0.020 | 0.086 ± 0.0095 | (4.0 ± 2.5) × 10−4 | (3.6 ± 0.42) × 10−4 | 54 ± 10 | 41 ± 3.6 | 3.8 ± 0.36 | |
| Metoprolol | nd | nd | nd | nd | nd | nd | nd | |
| Propranolol | 0.43 ± 0.035 | 0.24 ± 0.029 | nd | nd | (6.2 ± 0.98) × 102 | (3.7 ± 0.77) × 102 | 46 ± 6.5 | |
| Salbutamol | (3.1 ± 0.27) × 10−3 | 0.011 ± 0.00030 | nd | nd | 11 ± 1.1 | 9.5 ± 1.0 | 5.7 ± 1.8 | |
| Sotalol | nd | nd | nd | nd | nd | nd | nd | |
| Chemotherapeutic | Ifosfamide | nd | nd | nd | nd | nd | nd | nd |
| Coccidiostat | Clopidol | nd | nd | nd | nd | nd | nd | nd |
| Hormones | 17β-Estradiol | nd | nd | nd | nd | nd | nd | nd |
| 17α-Ethinylestradiol | nd | nd | nd | nd | nd | nd | nd | |
| Estriol | 0.093 ± 0.0086 | 0.70 ± 0.013 | nd | nd | nd | nd | 16 ± 3.3 | |
| Estrone | 0.15 ± 0.0030 | 0.054 ± 0.0035 | nd | nd | (1.2 ± 0.14) × 102 | nd | 15 ± 1.4 | |
| Norethisterone | nd | nd | nd | nd | nd | nd | nd | |
| Lipid regulators | Atorvastatin | 2.1 ± 0.19 | 1.9 ± 0.090 | nd | nd | nd | nd | (3.9 ± 2.3) × 102 |
| Bezafibrate | nd | nd | nd | nd | nd | nd | nd | |
| Gemfibrozil | nd | nd | nd | nd | nd | nd | nd | |
| Wastewater discharge marker | Caffeine | (1.4 ± 0.12) × 102 | 41 ± 4.1 | 0.079 ± 0.025 | 0.19 ± 0.017 | (1.0 ± 0.17) × 104 | (9.2 ± 1.2) × 103 | (2.7 ± 0.35) × 103 |
| Cotinine | 1.1 ± 0.10 | 1.2 ± 0.086 | (1.1 ± 0.055) × 10−3 | (2.8 ± 0.14) × 10−3 | 85 ± 22 | (1.1 ± 0.12) × 102 | 31 ± 4.3 | |
| X-ray contrast | Amidotrizoic acid | nd | nd | nd | nd | — | — | — |
In the influent, 34 ECs were detected at concentrations from (7.5 ± 0.60) × 10−3 μg L−1 (atenolol) to (2.2 ± 0.082) × 102 μg L−1 (metformin). A wide concentration range is typically observed for different ECs in wastewater.29,31,32
The highest detection frequency was observed in the effluent, where 38 ECs could be quantified. ECs were found at concentrations lower (e.g., ciprofloxacin), similar to (e.g., venlafaxine) and higher (e.g., gabapentin) than in the influent. Some determined effluent concentrations were in the range of what is typically reported in the influent of centralised WWTWs; for instance, both influent and effluent concentrations of metformin, were found to be (2.2 ± 0.082) × 102 μg L−1 and (1.6 ± 0.067) × 102 μg L−1, respectively.2 This suggests that in contrast to the high removal efficiency in centralised WWTWs of over 90% from the liquid phase,2 metformin is not degraded in the septic tank. Furthermore, effluent concentrations of some compounds exceeded concentrations typically reported from centralised WWTWs. For example, the antipruitic drug crotamiton was present at (1.5 ± 0.13) μg L−1 in the effluent, higher than previously reported concentrations of 0.11–0.27 μg L−1 by Nakada et al.26 in the UK. On the other hand, effluent concentrations of ECs such as fexofenadine, cetirizine, ciprofloxacin and lidocaine were similar to what has been reported in centralised WWTWs.30,31 Further research is necessary to better understand the removal of different ECs in septic tanks.
In the river, 18 ECs were detected upstream and 19 downstream of the septic tank discharge point. The EC found at the highest concentration in the river, both upstream and downstream, was the anti-diabetic metformin at (0.85 ± 0.031) μg L−1 and (1.1 ± 0.038) μg L−1, respectively. Metabolites can potentially have a significant effect on the total concentration of ECs in the environment, e.g., both desmethylvenlafaxine and 3-desmethyltrimethoprim were detected at higher concentrations in the river than the parent compound. The contribution of the septic tank to the pharmaceutical concentrations in the river varied from no difference between upstream and downstream concentrations to a marked increase. The biggest contribution was found for paracetamol with an increase by a factor of 15 from (0.039 ± 0.0043) μg L−1 to (0.59 ± 0.053) μg L−1. Other sources that contribute to ECs concentrations in the river are the secondary WWTWs and additional private septic tanks. Further work focussing on ECs in rural Scotland is needed to understand the impact of septic tank discharges on rivers.
With 30 detected ECs, detection frequencies in the suspended solids were similar to the wastewater. For most ECs, the liquid phase is the main contributor to the total concentrations in the septic tank discharge. However, clotrimazole, clarithromycin and ofloxacin that were not detected in the water, were found in the suspended solids at concentrations up to (1.3 ± 0.13) × 103 μg kg−1 for clarithromycin in the effluent. This stresses the importance of analysing the solids when assessing the impact of wastewater discharges to the environment. Most ECs had similar concentrations in the suspended solids of the influent and effluent, showing a potential for removal of ECs in the septic tank through sludge formation and consequent reduction of the total suspended solids in the effluent.
The 30 ECs that were determined in the sludge sample were found at concentrations from 4 (bisoprolol) to 3617 μg kg−1 (paracetamol). A wide concentration range of ECs in digested sludge from centralised WWTWs was also reported by Aydın et al.63 at mean concentrations from 0.73 (sulfamethazine) to 147 μg kg−1 (clarithromycin), and a maximum concentration of 1496 μg kg−1 (clarithromycin). Higher levels of some ECs such as fexofenadine and diclofenac in the sludge versus the suspended solids may reflect an accumulation over time, whereas lower levels of other ECs such as caffeine, paracetamol and clarithromycin could be due to degradation in the sludge.36 Future research on the distribution of ECs between the liquid and solid phase could increase the understanding of the removal of different ECs through sorption or degradation.
The contribution of the septic tank to the pharmaceutical concentrations detected in the river varies from no difference between upstream and downstream concentrations to an increase by the factor 15. The observed effluent concentrations of some pharmaceuticals suggest less removal in septic tanks than in centralised WWTWs. Finally, the detection of 30 ECs in the suspended solids in the effluent stresses the importance of including solid analysis when analysing environmental samples to avoid underestimation of the total concentration in the sample.
Conclusions
A new multi-analyte method was developed for the accurate determination of a broad range of ECs in liquid and solid environmental matrices of varying complexity. Analysing septic tank influent and effluent, including suspended solids, sludge, and the receiving surface water allows an accurate assessment of the performance of septic tanks for the removal of ECs and their effect on water quality. Including suspended solids in the analysis of environmental samples minimises underestimating the total concentration of ECs.The reported effluent concentrations of some pharmaceuticals suggest less removal in septic tanks than in centralised WWTWs. Furthermore, the river sampling suggests that septic tanks have an impact on water quality for some ECs. Hence, a more robust sampling of septic tanks in Scotland is proposed to accurately determine their impact to the environment.
Author contributions
Kai Wilschnack: conceptualisation, data curation, formal analysis, investigation, methodology, validation, visualisation, writing – original draft; Bess Homer: conceptualisation, resources, writing – review & editing; Elise Cartmell: conceptualisation, resources, writing – review & editing; Kyari Yates: conceptualisation, supervision, writing – review & editing; Bruce Petrie: conceptualisation, funding acquisition, methodology, project administration, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by a joined studentship from Scottish Water and the Robert Gordon University. The authors would like to thank Anna Baran for organising the sampling, and to Sarah Gillman for her contributions. Thanks are also extended to Morgan Black and Kaja Rzepkowski for performing the sludge extractions.References
- H. Darwano, S. V. Duy and S. Sauvé, Arch. Environ. Contam. Toxicol., 2014, 66, 582–593 CrossRef CAS PubMed.
- M. Scheurer, A. Michel, H.-J. Brauch, W. Ruck and F. Sacher, Water Res., 2012, 46, 4790–4802 CrossRef CAS PubMed.
- N. A. Alygizakis, P. Gago-Ferrero, V. L. Borova, A. Pavlidou, I. Hatzianestis and N. S. Thomaidis, Sci. Total Environ., 2016, 541, 1097–1105 CrossRef CAS PubMed.
- Q. Gao, K. M. Blum, P. Gago-Ferrero, K. Wiberg, L. Ahrens and P. L. Andersson, Sci. Total Environ., 2019, 651, 1670–1679 CrossRef CAS PubMed.
- S. Letsinger, P. Kay, S. Rodríguez-Mozaz, M. Villagrassa, D. Barceló and J. M. Rotchell, Sci. Total Environ., 2019, 678, 74–84 CrossRef CAS PubMed.
- B. Petrie, R. Barden and B. Kasprzyk-Hordern, Water Res., 2015, 72, 3–27 CrossRef CAS PubMed.
- S. Comber, M. Gardner, P. Sörme, D. Leverett and B. Ellor, Sci. Total Environ., 2018, 613–614, 538–547 CrossRef CAS PubMed.
- D. White, D. J. Lapworth, W. Civil and P. Williams, Environ. Pollut., 2019, 249, 257–266 CrossRef CAS PubMed.
- M. Gros, M. Petrović and D. Barceló, Talanta, 2006, 70, 678–690 CrossRef CAS PubMed.
- K. Proctor, B. Petrie, R. Barden, T. Arnot and B. Kasprzyk-Hordern, Anal. Bioanal. Chem., 2019, 411, 7061–7086 CrossRef CAS PubMed.
- D. R. Baker and B. Kasprzyk-Hordern, J. Chromatogr. A, 2011, 1218, 7901–7913 CrossRef CAS PubMed.
- S. Richards, E. Paterson, P. J. A. Withers and M. Stutter, Sci. Total Environ., 2016, 542, 854–863 CrossRef CAS PubMed.
- S. Ramage, D. Camacho-Muñoz and B. Petrie, Chemosphere, 2019, 219, 191–201 CrossRef CAS PubMed.
- Scottish Water, List of Wastewater Treatment Works, Annual Return 2021, Intern information, 2021 Search PubMed.
- L. Gill, J. Mac Mahon, J. Knappe, S. Gharbia and F. Pilla, Desludging Rates and Mechanisms for Domestic Wastewater Treatment System Sludges in Ireland (2016-W-DS-26), Environmental Protection Agency, Dublin, 2018 Search PubMed.
- D. Dubber and L. Gill, Sustainability, 2014, 6, 1623–1642 CrossRef CAS.
- B. D. Stanford and H. S. Weinberg, Environ. Sci. Technol., 2010, 44, 2994–3001 CrossRef CAS PubMed.
- Y. F. Ting and S. M. Praveena, Environ. Monit. Assess., 2017, 189, 178 CrossRef PubMed.
- L. A. Schaider, K. M. Rodgers and R. A. Rudel, Environ. Sci. Technol., 2017, 51, 7304–7317 CrossRef CAS PubMed.
- S. N. Garcia, R. L. Clubbs, J. K. Stanley, B. Scheffe, J. C. Yelderman and B. W. Brooks, Chemosphere, 2013, 92, 38–44 CrossRef CAS PubMed.
- C. A. James, J. P. Miller-Schulze, S. Ultican, A. D. Gipe and J. E. Baker, Water Res., 2016, 101, 241–251 CrossRef CAS PubMed.
- J. A. Oppenheimer, M. Badruzzaman and J. G. Jacangelo, Water Res., 2012, 46, 5904–5916 CrossRef CAS PubMed.
- B. Subedi, N. Codru, D. M. Dziewulski, L. R. Wilson, J. Xue, S. Yun, E. Braun-Howland, C. Minihane and K. Kannan, Water Res., 2015, 72, 28–39 CrossRef CAS PubMed.
- E. Godfrey, W. W. Woessner and M. J. Benotti, Groundwater, 2007, 45, 263–271 CrossRef CAS PubMed.
- O. S. A. Al-Khazrajy and A. B. A. Boxall, Anal. Methods, 2017, 9, 4190–4200 RSC.
- N. Nakada, S. Hanamoto, M. D. Jürgens, A. C. Johnson, M. J. Bowes and H. Tanaka, Sci. Total Environ., 2017, 575, 1336–1348 CrossRef PubMed.
- A. Ofrydopoulou, C. Nannou, E. Evgenidou and D. Lambropoulou, J. Chromatogr. A, 2021, 1652, 462369 CrossRef CAS PubMed.
- A. Klančar, J. Trontelj and R. Roškar, Water, Air, Soil Pollut., 2018, 229, 192 CrossRef.
- R. Oertel, J. Baldauf and J. Rossmann, J. Chromatogr. A, 2018, 1556, 73–80 CrossRef CAS PubMed.
- B. Petrie, J. Youdan, R. Barden and B. Kasprzyk-Hordern, J. Chromatogr. A, 2016, 1431, 64–78 CrossRef CAS PubMed.
- H. Rapp-Wright, F. Regan, B. White and L. P. Barron, Sci. Total Environ., 2023, 860, 160379 CrossRef CAS PubMed.
- K. Styszko, K. Proctor, E. Castrignanò and B. Kasprzyk-Hordern, Sci. Total Environ., 2021, 768, 144360 CrossRef CAS PubMed.
- R. López-Roldán, M. L. de Alda, M. Gros, M. Petrovic, J. Martín-Alonso and D. Barceló, Chemosphere, 2010, 80, 1337–1344 CrossRef PubMed.
- L. Duan, Y. Zhang, B. Wang, G. Yu, J. Gao, G. Cagnetta, C. Huang and N. Zhai, Water Res., 2022, 216, 118321 CrossRef CAS PubMed.
- I. L. Costa Junior, C. S. Machado, A. L. Pletsch and Y. R. Torres, Int. J. Sediment Res., 2022, 37, 346–354 CrossRef.
- J. L. Malvar, J. L. Santos, J. Martín, I. Aparicio and E. Alonso, Microchem. J., 2020, 157, 104987 CrossRef CAS.
- T. Anumol and S. A. Snyder, Talanta, 2015, 132, 77–86 CrossRef CAS PubMed.
- A. Eaglesham, A. Scott and B. Petrie, Environ. Chem. Lett., 2020, 18, 2119–2126 CrossRef CAS.
- P. N. Carvalho, A. Pirra, M. C. P. Basto and C. M. R. Almeida, Anal. Methods, 2013, 5, 6503–6510 RSC.
- N. Dorival-García, A. Zafra-Gómez, F. J. Camino-Sánchez, A. Navalón and J. L. Vílchez, Talanta, 2013, 106, 104–118 CrossRef PubMed.
- European Commission, Commission Implementing Decision (EU) 2018/840 of 5 June 2018 Establishing a Watch List of Substances for Union-wide Monitoring in the Field of Water Policy Pursuant to Directive 2008/105/EC of the European Parliament and of the Council and Repealing Commission Implementing Decision (EU) 2015/495, 2018, vol. 141 Search PubMed.
- B. Ellor and M. J. Gardner, The National Chemical Investigations Programme 2015-2020, Monitoring Substances of Emerging Concern, UKWIR, vol. 2, 2018 Search PubMed.
- European Commission, Common Implementation Strategy for the Water Framework Directive and the Floods Directive: Voluntary Groundwater Watch List, 2019 Search PubMed.
- L. Gomez Cortes, D. Marinov, I. Sanseverino, A. Navarro Cuenca, M. Niegowska, E. Porcel Rodriguez and T. Lettieri, Selection of Substances for the 3rd Watch List under the Water Framework Directive. JRC121346, Publications Office of the European Union, 2020 Search PubMed.
- B. Ellor, G. Castle and C. Yates, The National Chemical Investigations Programme 2020–2022, Monitoring Substances of Emerging Concern, UKWIR, vol. 5, 2023 Search PubMed.
- K. Helwig, A. Aderemi, D. Donnelly, S. Gibb, L. Gozdzielewska, J. Harrower, R. Helliwell, C. Hunter, L. Niemi, E. Pagaling, L. Price, J. Roberts and Z. Zhang, CREW Research Summary: Pharmaceuticals in the Water Environment: Baseline Assessment and Recommendations, 2021 Search PubMed.
- UK Centre for Ecology & Hydrology, National River Flow Archive, https://nrfa.ceh.ac.uk/ Search PubMed.
- National Records of Scotland, Mid-2020 Population Estimates for Settlements and Localities in Scotland, https://www.nrscotland.gov.uk/statistics-and-data/statistics/statistics-by-theme/population/population-estimates/settlements-and-localities/mid-2020, accessed 17 October 2022 Search PubMed.
- 2002/657/EC: Commission Decision of 12 August 2002 Implementing Council Directive 96/23/EC Concerning the Performance of Analytical Methods and the Interpretation of Results (Text with EEA Relevance) (Notified under Document Number C(2002) 3044), 2002 Search PubMed.
- L. Boulard, G. Dierkes, M. P. Schlüsener, A. Wick, J. Koschorreck and T. A. Ternes, Water Res., 2020, 171, 115366 CrossRef CAS PubMed.
- M. Petrović, M. D. Hernando, M. S. Díaz-Cruz and D. Barceló, J. Chromatogr. A, 2005, 1067, 1–14 CrossRef PubMed.
- C. Ripollés, M. Ibáñez, J. V. Sancho, F. J. López and F. Hernández, Anal. Methods, 2014, 6, 5028–5037 RSC.
- O. Yanes, R. Tautenhahn, G. J. Patti and G. Siuzdak, Anal. Chem., 2011, 83, 2152–2161 CrossRef CAS PubMed.
- P. Paíga, L. H. M. L. M. Santos and C. Delerue-Matos, J. Pharm. Biomed. Anal., 2017, 135, 75–86 CrossRef PubMed.
- K. B. Bodle, M. R. Pernat and C. M. Kirkland, Water, Air, Soil Pollut., 2022, 233, 505 CrossRef CAS PubMed.
- C. Miossec, T. Mille, L. Lanceleur and M. Monperrus, Food Chem., 2020, 322, 126765 CrossRef CAS PubMed.
- J. Wang, L. Qi, C. Hou, T. Zhang, M. Chen, H. Meng, M. Su, H. Xu, Z. Hua, Y. Wang and B. Di, J. Pharm. Anal., 2021, 11, 739–745 CrossRef CAS PubMed.
- T. S. Oliveira, M. Murphy, N. Mendola, V. Wong, D. Carlson and L. Waring, Sci. Total Environ., 2015, 518–519, 459–478 CrossRef CAS PubMed.
- Y. Li, M. A. Taggart, C. McKenzie, Z. Zhang, Y. Lu, S. Pap and S. W. Gibb, J. Environ. Sci., 2021, 100, 18–27 CrossRef CAS PubMed.
- A. Azzouz and E. Ballesteros, Sci. Total Environ., 2012, 419, 208–215 CrossRef CAS PubMed.
- Y. Luo, W. Guo, H. H. Ngo, L. D. Nghiem, F. I. Hai, J. Zhang, S. Liang and X. C. Wang, Sci. Total Environ., 2014, 473–474, 619–641 CrossRef CAS PubMed.
- SEPA, SEPA Time series data service (API), https://timeseriesdoc.sepa.org.uk/ Search PubMed.
- S. Aydın, A. Ulvi, F. Bedük and M. E. Aydın, Sci. Total Environ., 2022, 817, 152864 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ay01201h |
| This journal is © The Royal Society of Chemistry 2024 |