
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDifferentiation of oligosaccharide isomers by direct infusion multidimensional mass spectrometry†
Enoch
Amoah
,
Taghi
Sahraeian
,
Ayesha
Seth
and
Abraham K.
Badu-Tawiah
 *
*
Department of Chemistry and Biochemistry, The Ohio State University, Columbus, Ohio 43210, USA. E-mail: badu-tawiah.1@osu.edu
First published on 1st October 2024
Abstract
Oligosaccharides demonstrate many bioactivities with applications in the pharmaceutical, cosmetic, and food industries. They also serve as biomarkers for various diseases including cancer and glycogen storage disorders. These make the structural characterization of oligosaccharides very important. Unfortunately, the structural diversity found in saccharides make their characterization challenging, necessitating the development of sophisticated instrumentation to enable isomer differentiation. Herein, we report the ability of halide (Cl− and Br−) adducts to enable direct differentiation of oligosaccharide isomers using conventional collision-induced dissociation (CID) tandem MS (MS/MS). The halide adducts were generated by direct infusion nano-electrospray ionization (nESI). For the first time, this traditional nESI CID MS/MS platform was used to differentiate stereoisomers of trisaccharides (cellotriose β(1 → 4) and maltotriose α(1 → 4), tetrasaccharides (cellotetraose and maltotetraose), and pentasaccharides (cellopentaose and maltopentaose)). In addition, the MS/MS of halide adducts enabled the differentiation of positional, structural, and linkage isomers from a total of 14 oligosaccharides. The isomer differentiation was realized by the generation of distinct diagnostic fragment ions in CID. We also performed principal component analysis using the entire range of MS/MS fragment ion profiles and found that negative-ion mode halide adduction provided more effective isomer differentiation compared with positive-ion mode sodium adduction. Finally, we demonstrated complex mixture analysis by spiking all 14 oligosaccharides into raw urine, of which we successfully distinguished species based on molecular weight (first dimension) and CID MS/MS fragmentation patterns as the second dimension separation. This work effectively showcases the potential to use direct infusion nESI-MS/MS to characterize synthetic oligosaccharide isomers in unpurified reaction mixture as well as from biofluids for diagnostic purposes.
Introduction
Diversity in saccharide structure is not only due to different monosaccharide composition. The diversity arises also from the way the monosaccharides are linked, including bond orientation (i.e., stereoisomers α, β), order of linkage, and position of the glycosidic bonds. This makes saccharide characterization very challenging. In this work, we seek to broaden the scope of the traditional collision-induced dissociation (CID) tandem mass spectrometry (MS/MS) analytical technique toward the characterization of complex oligosaccharide isomers.Oligosaccharides with 3–10 monosaccharide units have many health benefits, which include being non-digestible, that allow them to serve as prebiotics for important bacteria species such as lactobacilli and bifidobacterium.1–4 This function can limit pathogenic activities in the colon, enhance mineral absorption in the intestine, decrease blood glucose levels, as well as reduce serum lipid levels.1,3–12 As a result, oligosaccharides have recently received significant attention as potential biomarkers for disease diagnosis, especially in the field of cancer research and carbohydrate-based vaccines.13–17 Oligosaccharides have also found use in the agricultural, food, cosmetic, and pharmaceutical industries.6,8
Bioactive oligosaccharides exist in small quantities from natural sources such as plants, honey, dairy products, yeast, fungi, algae, and bacteria.1,4 As a consequence, the cost of extracting oligosaccharides from natural sources is high. Therefore, they are synthesized enzymatically in bulk.3,4,7 Thus, an analytical method capable of directly analyzing minute volumes of complex samples, either in the form of biofluid or unpurified reaction mixture, would be of great significance. Because of their structural diversity and lack of distinct fragmentation pathways, recent advancements in mass spectrometry (MS) methods targeting saccharide isomer characterization have employed exotic dissociation techniques such as electron capture and electron transfer dissociation, infrared photodissociation as well as ultraviolet photodissociation.18–23 Few studies have exploited the use of the CID fragmentation technique for oligosaccharides analysis although this MS/MS method is available on almost all commercial mass spectrometers. These reports show that the CID MS/MS of oligosaccharides mainly yield glycosidic cleavages, which provide less useful information for isomer differentiation.24–26 We believe simplicity is key to transforming technology for the hands of people that need it; in this case enabling the routine use of ordinary mass spectrometers in biomedicine or in characterizing complex synthetic products.
A simple but effective method for characterizing oligosaccharide structure is important because the subtle differences in monosaccharide sequence and bond position/linkage leads to distinct bioactivity of the resultant isomers, which have found their way into everyday life. For example, cellotriose, isomaltotriose, maltotriose, and nigerotriose are trisaccharide isomers composed of the same monosaccharide units (glucose) but differ only by their glycosidic linkages (Scheme 1). While cellotriose forms β(1 → 4) linkages, isomaltotriose, maltotriose, and nigerotriose form α(1 → 6), α(1 → 4) and α(1 → 3) linkages, respectively. As a consequence of these differences in glycosidic linkages, cellotriose acts as a prebiotic whereas isomaltotriose and maltotriose are used as a low glycemic sweetener and fermentable sugar in brewing,27 respectively.
 | ||
| Scheme 1 Structures of trisaccharide isomers: (a) structural isomers, (b) positional isomers and (c) linkage isomers. | ||
While cellotriose, isomaltotriose, maltotriose, and nigerotriose contain the same monosaccharide unit, gentianose, melezitose, and 1-kestose are trisaccharide isomers that have a heterogeneous monosaccharide make-up and linkages. Gentianose and melezitose consist of two glucose units and a fructose unit forming β(1 → 6), α(1 → 2) and α(1 → 3), β(2 → 1) hetero-linkages, respectively. On the other hand, 1-kestose consists of two fructose units and a glucose unit forming two β(2 → 1) homo-linkages. Here too, the structural differences lead to different bioactivities: gentianose has antioxidant properties, melezitose is the main cause of honeydew flow disease,28 and 1-kestose can suppress diabetes by improving glucose tolerance.29 Collectively, these seven oligosaccharides (Scheme 1) form a complex structural isomer mix, which would traditionally require analytical chromatographic separation before effective differentiation and characterization by MS.
The simple but highly effective MS method developed in this study involves a direct infusion of 5 μL of the complex sample via nano-electrospray ionization (nESI) without prior separation. MS is suitable for such efforts because it is highly sensitive, providing the benefits of requiring less analysis time and lower levels of sample purity and quantity. Some studies have used different ionization methods for saccharide analysis by MS30–35 but direct characterization of oligosaccharide isomers without front-end liquid chromatography and ion mobility is rare.36–50 Additionally, most MS studies involving differentiation of saccharides utilize positive-ion mode analysis. Group I metal adduction (e.g., Na+ adducts) is the most used approach for positive-ion mode analysis of saccharides. When subjected to CID, Na+ adducts of oligosaccharides fragment mainly through glycosidic bond cleavages (Scheme 2a)51,52 to give a low abundance of ions, limiting the ability to differentiate structural, positional, and linkage isomers. This limited fragmentation is due to the high binding capacity of Na+ toward saccharides, making the Na+ ion less mobile and thus creating few charged centers. This charge stabilization dictates less fragmentation. Contrast this to protonated species (although not typically favored for oligosaccharides), which could undergo both glycosidic and cross-ring cleavages because of the presence of multiple charge centers.53,54 To obtain detailed fragmentation that can lead to isomer differentiation via the use of Na+ adducts, without prior separation, multiple-stage tandem MS (MSn) is required. The challenge is that analysis time increases with each added stage of the tandem MS experiment, which in turn increases the volume of sample needed. Also, it has been observed that the sequence of fragmentation in MSn that can lead to diagnostic ion formation is distinct for different isomers, making it difficult to write a common algorithm that could be applied for rapid analysis.
 | ||
| Scheme 2 Possible fragmentation pathways observed for (a) left, sodium adducts of oligosaccharides, which predominantly give glycosidic (1) bond cleavages and (b) right, ammonium and halide adducts of oligosaccharides, both providing glycosidic (1) and cross-ring (2) cleavages. (c) Representative figure showing some of the commonly observed fragmentation patterns in trisaccharides. | ||
We envisioned that NH4+ adducts of oligosaccharides should intrinsically yield protonated species upon CID MS2via the loss of ammonia, which can subsequently induce both glycosidic and cross-ring cleavages (Scheme 2b). In our previous study,55 we showed that halide adducts (e.g., Cl− and Br−) of disaccharides provide diagnostic fragment ions for structural isomers in a single-stage tandem MS experiment. It became clear to us that the use of ammonium halide salts (NH4Cl and NH4Br) could be an effective way to generate both positive and negative ions in a single direct-infusion nESI experiment followed by isomer differentiation upon CID MS/MS in both positive- and negative-ion modes. Cole and coworkers introduced the use of ammonium halide salts to facilitate the formation of [M − H]− species when the halide adduct was subjected to in-source fragmentation.56 To the best of the authors’ knowledge, the use of halide adducts to differentiate various isomers of oligosaccharides in conventional CID MS/MS has not been demonstrated. We successfully showcase the application of the direct infusion nESI-MS/MS method for the differentiation of isomers of trisaccharides (cellotriose, isomaltotriose, maltotriose, gentianose, melezitose, nigerotriose, and 1-kestose), tetrasaccharides (cellotetraose, isomaltotetraose, maltotetraose, stachyose), and pentasaccharides (isomaltopentaose, maltopentaose, and verbascose). In a comprehensive saccharide mixture (14 sugars) prepared in urine, the presence of different oligosaccharides could be inferred in MS using molecular weight, but isomeric species packed into a single mass-to-charge (m/z) peak were deciphered in a second-dimensional experiment using diagnostic fragment ions derived from MS/MS of the chloride adducts, without prior separation.
Experimental section
Mass spectrometry
The data were collected using a Thermo Fisher Scientific Velos Pro ion-trap mass spectrometer at a full MS range (San Jose, CA, USA). The following MS parameters were employed: 3 microscans, 100 ms ion injection time, and 250 °C inlet capillary temperature. Spectra collection was done for at least 30 s. A constant distance of 5 mm was kept between the nESI tip and the MS inlet. All data collection and processing were done using Thermo Fisher Scientific Xcalibur 2.2 SP1 software. Identification and characterization of oligosaccharide isomers were achieved using tandem MS with collision-induced dissociation (CID) at 30% manufacturer's unit and 1.5 Th (mass/charge units) for an isolation window of normalized collision energy.Chemicals and reagents
Cellotriose, gentianose, 1-kestose, isomaltotetraose, maltotetraose, verbascose, cellopentaose, maltopentaose, and stachyose were purchased from Biosynth International Inc. Nigerotriose was obtained from Neogen Corporation (Lansing, MI, USA) Cellotetraose and isomaltotriose were obtained from VWR International (Radnor, PA, USA). Maltotriose and melezitose were purchased from Sigma-Aldrich (St Louis, MO, USA). Ammonium bromide and ammonium chloride were also purchased from Sigma-Aldrich (St Louis, MO, USA).Preparation of urine samples
A pooled human urine sample was purchased from Innovative Research (Novi, MI, USA). 5 mL of this sample was centrifuged using a Microsep™ Advance 3k MW cutoff centrifugal filter (Pall Corp., Ann Arbor, MI, USA) at ∼15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 15 minutes. 20 μL of the filtered urine sample was dissolved in 80 mL of 100% water containing 50 μM each of the 14 oligosaccharides.
000 rpm for 15 minutes. 20 μL of the filtered urine sample was dissolved in 80 mL of 100% water containing 50 μM each of the 14 oligosaccharides.
Results and discussion
Direct-infusion nano-electrospray ionization (nESI)
A theta glass capillary was employed for non-contact nESI to deliver/ionize all sample solutions toward the mass spectrometer (Fig. 1a). This setup consists of a silver (Ag) metal electrode and a theta borosilicate (disposable) glass capillary (O.D. 1.5 mm) pulled to a sharp tip using a micropipette puller (Model P-97, Sutter Instrument Co., Novato, CA, USA). In the non-contact mode, the Ag electrode is not in physical contact with sample solution or reagent in the theta capillary, creating an air gap between the reagent solution and the Ag electrode. The non-contact mode prevents Joule heating and also facilitates relatively lower sample consumption. We placed 5 μL of oligosaccharide sample (in urine or in water) in one barrel of the pulled borosilicate theta capillary while 5 μL of aqueous solution of the ammonium halide salt (NH4Cl or NH4Br) was placed in the other barrel as illustrated in Fig. 1a. To initiate electrospray, 1.5 kV direct current (DC) voltage was applied to the Ag electrode, which induces an electric field that charges the reagent solution electrostatically, facilitating charged microdroplets to be released from the tip of the pulled theta capillary. We found that the result is the same regardless of the barrel to which the DC voltage is applied. That is, the release of charged microdroplets is achieved from both barrels allowing in situ mixing of reagent with the analyte solution followed by adduct formation between the oligosaccharide and halide in negative-ion mode and oligosaccharide and ammonium in positive-ion mode. The theta capillary was stationed 5 mm away from the mass spectrometer inlet. This enabled direct transfer of the charged microdroplets containing the adducts to the mass spectrometer for subsequent characterization via MS/MS. All analyses were performed in the non-contact mode of the nESI platform. | ||
| Fig. 1 (a) Schematic representation of the non-contact nESI MS setup using a theta capillary. Ammonium chloride (1 mM, 5 μL) was placed in one side of the theta capillary to which a spray voltage of −1.5 kV was applied. The sample to be analyzed (e.g., raw urine, <5 μL) was placed in the other half of the theta capillary. (b) Representative negative-ion mode mass spectrum showing chloride adducts formed from spraying gentianose (100 μM) and ammonium chloride from the theta capillary. | ||
Formation of oligosaccharide adducts
Abundant evidence exists suggesting that compounds with polar functionalities can undergo anion attachment to form negative ions.55–59 This expectation is readily realized when oligosaccharides are electrosprayed in the presence of Cl− and Br− sources. For example, Fig. 1b shows the full mass spectrum recorded when the aqueous solutions of NH4Cl and gentianose (100 μM) were sprayed from separate barrels of the theta capillary. It can be observed that the trisaccharide gentianose (MW 504 Da) forms diagnostic adducts with 35Cl:37Cl in an isotope ratio of 3:1 at m/z 539 and 541, respectively, confirming the formation of an oligosaccharide-chloride adduct in negative-ion mode MS. The full MS also showed the formation of a minor peak at m/z 503, which represents deprotonated gentianose. Other trisaccharides analyzed in a similar manner formed abundant adducts with Cl− in the full MS. The oligosaccharides were analyzed at a sample concentration of 100 μM except for raw urine analysis where 50 μM was used. We also observed effective adduct formation of the sugars at a concentration as low as 0.5 μM and with a sample volume of 2 μL (Fig. S1g†).
Differentiation of trisaccharide isomers
We began the analysis with trisaccharide isomers cellotriose β(1 → 4), isomaltotriose α(1 → 6), maltotriose α(1 → 4), and nigerotriose α(1 → 3), which consist of the same monosaccharide units (glucose) but with different linkages of the glycosidic bonds. CID MS/MS of the trisaccharide/halide adducts enabled the differentiation of these four positional isomers (MW 504 Da) with ease. Tandem MS analysis of m/z 539 [M + Cl]− of each of the trisaccharides provided distinct diagnostic ions for differentiation of the isomers (Fig. 2a–d). As expected for species that bind less strongly than Na+, multiple charge centers become available with Cl− adduction, resulting in both glycosidic bond and cross-ring breakages. The example for maltotriose is shown in Fig. 2a where CID tandem MS at m/z 539 registered two diagnostic ions (m/z 455 and 473) that were not detected during the analysis of the other positional isomers (cellotriose, isomaltotriose and nigerotriose). In this case, the chloride adduct of maltotriose first lost HCl (MW 36 Da) to form the deprotonated species (M − H)− at m/z 503. This deprotonated species in turn undergoes cross-ring breakage with neutral loss of H2CO (MW 30 Da) to give a diagnostic ion at m/z 473. Subsequent loss of H2O from the ion at m/z 473 leads to the formation of another diagnostic ion at m/z 455. These diagnostic ions from maltotriose were not found to be present in the tandem MS of all other trisaccharide isomers analyzed, including linkage and positional isomers. Similarly, MS/MS analysis of nigerotriose α(1 → 3) provided two unique diagnostic ions at m/z 395 and 191, which are due to loss of C3H6O3 (MW 90 Da) from ions at m/z 485 and 281, respectively (Fig. 2b). The tandem MS analysis of isomaltotriose α(1 → 6) shows a similar fragmentation pattern to that of maltotriose α(1 → 4) but produces a distinct diagnostic ion that was absent in the tandem MS fragmentation of the other trisaccharide isomers (Fig. 2c). That is, isomaltotriose first lost HCl from the chloride-adducted precursor ion (m/z 539) to form the deprotonated (M − H)− species at m/z 503. Further fragmentation from the deprotonated isomaltotriose produces a diagnostic ion at m/z 387, after losing C2H4O2 and two molecules of CO. | ||
| Fig. 2 Negative-ion mode tandem MS/MS analysis of (a) maltotriose, (b) nigerotriose, (c) isomaltotriose, (d) cellotriose, all at m/z 539. | ||
Similarly, the analysis of cellotriose provided a unique diagnostic ion at m/z 297 to enable it to be differentiated from the other positional isomers (Fig. 2d). In this instance, the trisaccharide first undergoes glycosidic bond breakage from the deprotonated cellotriose leading to the formation of an ion fragment at m/z 341. Subsequently, the m/z 341 fragment ion loses CO2 (MW 44 Da) via cross-ring cleavage to afford the diagnostic ion at m/z 297. It is important to note that cellotriose and maltotriose are stereoisomers that differ only in the slight orientation of their atoms, thus, cellotriose has a β(1 → 4) structure while maltotriose has an α(1 → 4) structrure. Our method was still able to differentiate these two stereoisomers without any difficulty. We also observed that generally, the trisaccharides with α-configurations (mainly isomaltotriose and maltotriose) lost C2H4O2 from the deprotonated trisaccharide to form an ion with relatively higher MS signal intensity at m/z 443 that was almost absent from the mass spectra of β-configurations of the stereoisomers. Similar patterns are observed when the α-configured trisaccharides lose 2C2H4O2 from the deprotonated species to form an ion fragment at m/z 383. This behavior is also observed with bromide adduction. Hence, additional differentiation is available through comparison of MS/MS ion profiles and not just specific diagnostic ions, which can facilitate the development of efficient algorithms for straightforward analysis of data of other saccharides isomers.
The tandem MS analyses of bromide adducts provide complementary information to trisaccharide isomer differentiation when compared with data derived from chloride adducts. The full MS data of all the isomers formed intense peaks of 1:1 isotope distribution at m/z 583 and m/z 585, indicating the formation of bromide-trisaccharide adducts. A representative mass spectrum is shown in Fig. S2a.† The spectrum for bromide adducts contain traces of chloride adducts, which could be due to the ubiquitous nature of chloride salts. Nonetheless, the tandem MS analyses of bromide adducts provided unique diagnostic ions that complement the diagnostic ions obtained from the fragmentation patterns of chloride-adducted oligosaccharides. For example, MS/MS analysis shows that nigerotriose produced three diagnostic ions at m/z 395, 215, and 191. Cellotriose afforded two significant diagnostic ions at m/z 467 and 203 with high signal intensities. The tandem MS of maltotriose produced a diagnostic ion at m/z 493. Similarly, isomaltotriose fragments give two diagnostic ions at m/z 375 and 293 (Fig. S2b and c†). The combination of results from tandem MS of both chloride and bromide adducts provide higher confidence in the differentiation of trisaccharide isomers via CID tandem MS.
Analysis of mixtures of trisaccharide positional isomers
To ensure that our method can differentiate the positional isomers (cellotriose, isomaltotriose, maltotriose, and nigerotriose) in a complex mixture, a 100 μM standard of the four trisaccharides was prepared in a single solution and sprayed alongside the solution of ammonium chloride. Fig. S3† represents the full MS, which registered the presence of chloride-adducted trisaccharides at m/z 539. The tandem MS analysis of m/z 539 clearly shows the distinct diagnostic ions at m/z 473 and 455 indicating the presence of maltotriose. The diagnostic ions at m/z 395 and 191 also show the presence of nigerotriose. The spectrum also registered diagnostic ions at m/z 387 and 297 attributed to isomaltotriose and cellotriose, respectively. Tandem MS analysis of bromide adducts of the same mixture of the positional isomers was also performed. The expected unique diagnostic ions associated with each trisaccharide were identified, similarly to that observed during the individual analysis of the isomers. The full MS and tandem MS of bromide adducts are summarized in Fig. S3c and d,† respectively. Here, the presence of fragment ions at m/z 467 and 203 clearly show the unique diagnostic ions for cellotriose without any scrambling. Likewise, unique diagnostic ions registered at m/z 395 and 215 indicate the presence of nigerotriose. The diagnostic ions at m/z 493 and 293 also confirm the presence of maltotriose and isomaltotriose in the mixture, respectively.Analysis of other trisaccharide isomers
We extended the method from positional isomers to the analysis of gentianose and melezitose, which are linkage isomers. They both consist of two glucose units and a fructose unit, but gentianose forms β(1 → 6), α(1 → 2) linkages while melezitose forms α(1 → 3), β(2 → 1). We included structural isomer 1-kestose in this list, but it is made of two fructose units and glucose and forms a homo-linkage β(2 → 1). As expected, each of these trisaccharides registered a high-intensity peak at m/z 539 corroborating the formation of the trisaccharide-chloride adducts [M + Cl]− in the full MS (Fig. S1†). Tandem MS analyses were performed on the chloride-adducted precursor ions of each trisaccharide to examine their fragmentation patterns. Analysis of the fragmentation pattern of 1-kestose indicates that the precursor ion at m/z 539 first loses HCl to form the deprotonated 1-kestose at m/z 503. The deprotonated product ion undergoes glycosidic bond cleavage through a neutral loss of C6H12O6 (MW 180 Da) to give a fragment ion at m/z 323. Subsequent neutral loss of H2O and H2CO leads to the formation of a diagnostic ion at m/z 275 (Fig. S4a†). Further loss of C2H4O2 (MW 60 Da) from the ion at m/z 275 affords another diagnostic ion at m/z 215 that was also not observed in any other isomer analyzed. Unlike 1-kestose, melezitose is a hetero-linked isomer, which produces a distinct diagnostic ion at m/z 257. The loss of C6H12O6 from the fragment ion at m/z 503 (M − H)−via cross-ring cleavages provided the ion at m/z 323. Further loss of H2CO and 2H2O from the fragment ion at m/z 323 afforded the diagnostic ion observed at m/z 257 (Fig. S4b†). For gentianose, another hetero-linked isomer, a diagnostic ion at m/z 353 is observed. This diagnostic ion at m/z 353 resulted from the neutral loss of C2H4O2 (MW 60 Da) and C3H6O3 (MW 90 Da) from the deprotonated gentianose (Fig. S4c†). Here also, notice that gentianose and melezitose have the same make up of monosaccharides but differ only by their linkages and our method-enabled differentiation.In a similar fashion, the fragmentation patterns of bromide adducts were pursued. MS/MS analyses of the bromide adducts at m/z 583 showed that each of the oligosaccharides affords unique diagnostic ions. 1-Kestose fragments give diagnostic ions at m/z 275. The tandem MS analysis of gentianose afforded distinct diagnostic ions at m/z 521 and 447. Interestingly, tandem MS analysis of m/z 583 from melezitose yielded no unique ion that enables differentiation of melezitose from other trisaccharides via bromide adduction. Nonetheless, the complete differentiation of the isomer from others could equally be achieved with chloride adduction as explained earlier (Fig. S5†). Complete ion profiles could also be used to differentiate the isomers from Br− adducts.
Analysis of a mixture of all seven structural trisaccharide isomers
Following the remarkable results obtained from the analysis of individual trisaccharides, the seven trisaccharides, cellotriose, gentianose, isomaltotriose, 1-kestose, maltotriose, melezitose, and nigerotriose were mixed to form a 100 μM solution. The mixture was then sprayed with ammonium chloride salt from separate barrels of the theta capillary. The full MS registered a high intensity peak at m/z 539 representing the Cl− adduct of all seven trisaccharides. Tandem MS analysis of the signal at m/z 539 confirmed the presence of each isomer in the mixture without significant suppression effects. For example, the signals at m/z 473 and 455 in the tandem MS/MS analysis clearly confirms the presence of maltotriose in the mixture. Similarly, signals at m/z 395 and 191 registered the presence of nigerotriose. Likewise, the peak at m/z 387 indicates the presence of isomaltotriose while those at m/z 275 and 215 confirmed the presence of 1-kestose. The observation of peaks at m/z 297 and 353 in the mass spectrum also confirms the presence of cellotriose and gentianose, respectively, while that at m/z 257 shows the presence of melezitose in the mixture (Fig. S6†).Positive-ion mode analysis of trisaccharide isomers
One of the most used approaches for analyzing saccharides is via positive-ion mode MS. This method is mainly done through the formation of adducts such as potassium or sodium. As such, we sought to investigate if an Na+ adduct in the positive-ion mode can be used to differentiate the trisaccharide isomers. Additionally, we were able to generate ammonium adducts in the positive-ion mode via the use of NH4Cl or NH4Br. Therefore, we also investigated the potential to observe distinct fragment ions from NH4+ adducts of the trisaccharides.The full MS of the positive-ion mode showed formation of competitive adducts between ammonium and sodium ions at m/z 522 and 527, respectively (Fig. S7†). Interestingly, the ammonium adducts provided several significant diagnostic ions for characterization of the isomers. In the tandem MS analyses, both glycosidic bond and cross-ring breakages were observed. This was in accordance with our expectation that the NH4+ ion binds less strongly to the saccharides and hence can induce significant fragmentation upon CID MS/MS after the ammonia gas evaporates to leave the proton on the saccharide, which is labile. Tandem MS analysis of maltotriose produced three unique diagnostic ions observed at m/z 441, 385, and 367 (Fig. S8a†). The ammonium adduct (M + NH4+) first undergoes neutral loss of NH3 (17 Da) to form an ion at m/z 505. Subsequent loss of 2H2O (36 Da) and CO (28 Da) through cross-ring breakage leads to the formation of a fragment ion at m/z 441. In a similar fashion, neutral loss of C4H8O4 (120 Da) from the fragment ion at m/z 505 leads to the formation of an ion at m/z 385. Loss of H2O from m/z 385 produces the diagnostic ion at m/z 367. Isomaltotriose was found to produce two diagnostic ions at m/z 363 and 349 through cross-ring cleavages (Fig. S8b†). The diagnostic ion at m/z 363 is due to sequential loss of H2O, CO, and C2H4O2 from the fragment ion at m/z 505 while the ion at m/z 349 resulted from neutral loss of C3H6O3 (90 Da) from the fragment ion at m/z 505 to form an ion at m/z 415, which subsequently loses 2H2O and H2CO.
Likewise, cellotriose afforded three distinct diagnostic ions at m/z 432, 306, and 237 (Fig. S8c†). The diagnostic ion at m/z 432 is due to cross-ring cleavage of C3H6O3 (90 Da) from the ammonium-adducted cellotriose, while loss of C3H6O3 and 2H2O from the ion at m/z 432 resulted in the formation of the ion at m/z 306. Similarly, the release of C3H6O3 and CO from the fragment ion at m/z 355 produced the ion at m/z 237. Tandem MS analysis of other isomers also produced distinct diagnostic ions that were not present in the tandem MS analysis of others. For example, 1-kestose produced a diagnostic ion at m/z 193 (Fig. S8d†) while melezitose afforded diagnostic ions at m/z 267 (Fig. S8e†), and gentianose at m/z 243 and 215 (Fig. S8f†). In contrast, tandem MS analysis of adducts formed by the Na+ ion did not yield any significant diagnostic ions to enable complete characterization and differentiation of all isomers (Fig. S9†).
Principal component analysis of trisaccharide isomers
Following the successful differentiation of the trisaccharide isomers via distinct tandem MS diagnostic peaks, we further explored the characterization of the isomers using principal component analysis (PCA) and heatmap analysis. PCA and heatmap analysis of the isomers consider the entire MS/MS profile of each sugar. Fig. 3 shows the PC1 and PC2 score plots acquired by performing PCA with autoscaling of data derived from the isomers for tandem MS profiling in both negative- and positive-ion modes. We used MetaboAnalyst online software for this analysis. Each trisaccharide isomer is represented by a color-coded filled circle. In Fig. 3a, PC1 shows the differences that exist between the trisaccharide isomers, which are reflected on the PC2. It can be seen from the score plots that the mono-linked trisaccharide isomers (i.e., cellotriose, isomaltotriose, and maltotriose) are separated in the negative-ion mode tandem MS (Fig. 3a), which was not observed in the positive-ion mode MS (Fig. 3b). Again, notice that cellotriose and maltotriose are stereoisomers and yet their fragment ion profiles are very different (Fig. 4) in addition to having distinct diagnostic ions (Fig. 2). It can also be observed that well-separated mono-linked isomers caused the hetero-linked isomers (1-kestose, gentianose, and melezitose) to be clustered (Fig. 3a). To gain insight into how the hetero-linked isomers are separated, we performed PCA on these three isomers. Fig. S10,† shows the PCA results of the hetero-linked isomers in both negative- and positive-ion mode tandem MS. Here too, the negative-ion mode tandem MS based on the Cl− adduct provides relatively better differentiation of the isomers. From the heatmap analysis, we can observe a correlation between the various ions generated from the tandem MS and the individual isomers as well as variation among the isomers. This observation also provides similar insights as observed from PCA (Fig. S11†). Combination of the MS/MS profile with diagnostic fragment ions for each isomer provides a strong basis for isomer differentiation. These results are very important because they confirm the capabilities of halide adduction of saccharides in differentiating isomers using traditional CID tandem MS, without prior separation or instrument modification. | ||
| Fig. 3 Plots of PCA scores obtained for (a) negative-ion mode tandem MS and (b) positive-ion mode tandem MS, of trisaccharide isomers. | ||
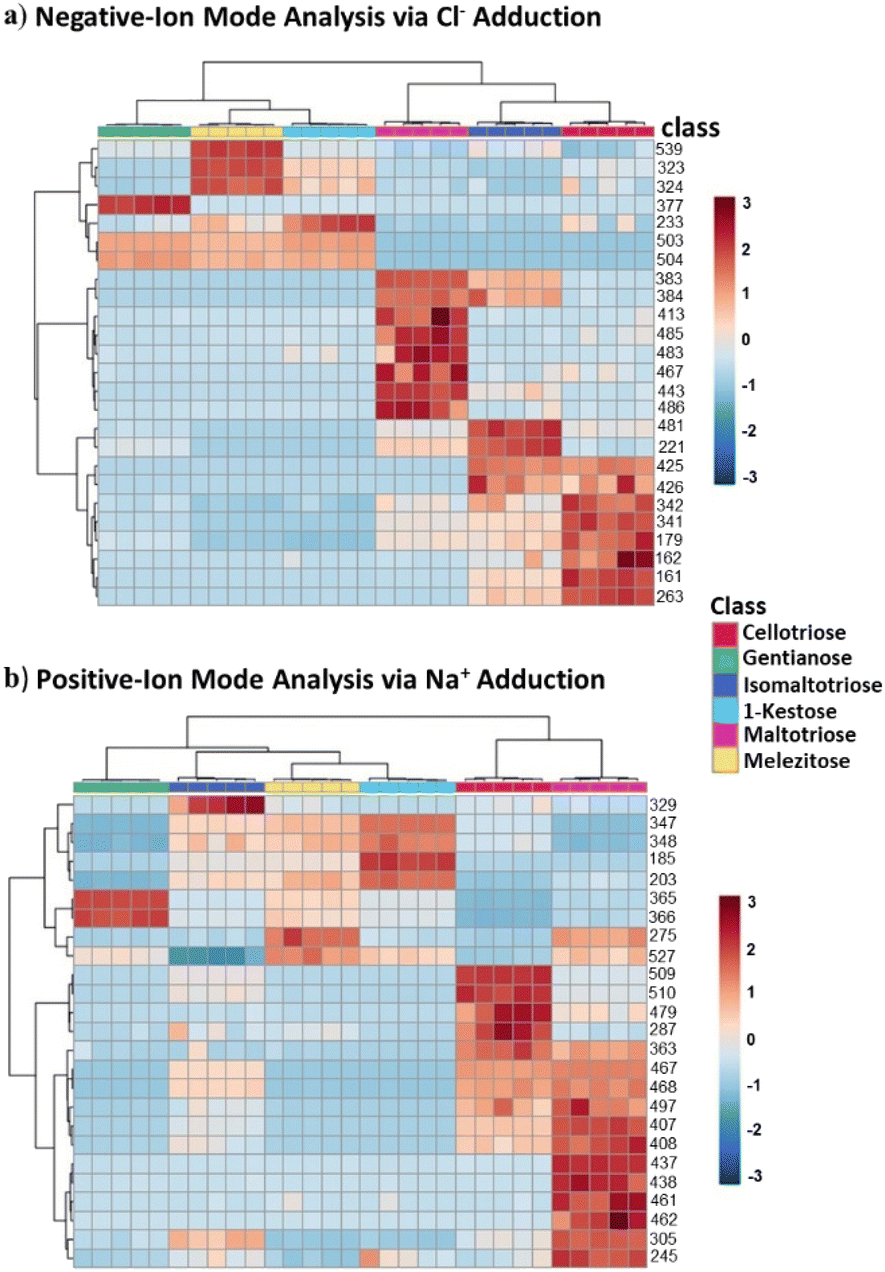 | ||
| Fig. 4 Heatmap results obtained for (a) negative-ion mode tandem MS and (b) positive-ion mode tandem MS of trisaccharide isomers, i.e. cellotriose, gentianose, isomaltotriose, 1-ketose, maltotriose, melezitose. | ||
Tetrasaccharide isomers
We further analyzed tetrasaccharide (MW 666 Da) positional isomers namely, cellotetraose, isomaltotetraose, maltotetraose, and stachyose (Scheme 3a). Cellotetraose is a β(1 → 4)-linked tetrasaccharide while isomaltotriose and maltotetraose are α(1 → 6)- and α(1 → 4)-linked tetrasaccharides, respectively, all consisting of four glucose units. Stachyose on the other hand consists of two galactose, a glucose and a fructose monosaccharide units connected through an α(1 → 6) linkage. Fig. S12† shows a representative full MS where a high intensity peak at m/z 701 is registered indicating the formation of a sugar-chloride adduct. Also formed in relatively low abundance is the peak at m/z 665, which is the deprotonated tetrasaccharide. Tandem MS analysis was performed on m/z 701 (Cl− adducts) for each tetrasaccharide (Fig. S13†). Careful observation of the fragmentation pattern of cellotetraose reveals that the tetrasaccharide fragments give two diagnostic ions registered at m/z 567 and 405. Stachyose registered a unique diagnostic ion peak at m/z 539 that was not found in other isomers upon CID fragmentation. Likewise, maltotetraose was also found to fragment to produce a diagnostic ion recorded at m/z 569. Isomaltotetraose showed a distinct diagnostic ion at m/z 575. Further analyses were performed via bromide adduction. Here, the tetrasaccharides formed an adduct at m/z 745 in the full MS (Fig. S14†). Tandem MS analysis of the adduct at m/z 745 revealed remarkable diagnostic ions for each sugar. Stachyose produced a diagnostic ion with a peak of very high intensity at m/z 583 and other peaks at m/z 565, 543, and 463 that were not present in any of the other isomers. Cellotetraose also registered three distinct ions at m/z 715, 523, and 365 that were not present in the CID fragmentation of the other positional tetrasaccharides. In the MS/MS analysis of isomaltotetraose, a unique ion was found at m/z 575. Similarly, these ions were not found in the tandem MS analysis of any other tetrasaccharide analyzed. Maltotetraose also afforded a diagnostic ion at m/z 569. Notice again that cellotetraose and maltotetraose are stereoisomers but our method enables direct differentiation of the two with no difficulties in both Cl− and Br− adduction. Again, we found that 1 → 4-linked isomers such as cellotetraose and maltotetraose produced a unique ion at m/z 425 that was not found in the 1 → 6-linked isomers. It is also interesting to note that in addition to the ion at m/z 425 observed in only 1 → 4-linked isomers, an ion at m/z 587 was observed in higher abundance as compared to the 1 → 6-linked isomers. Like before, we also analyzed the mixture of the four tetrasaccharide isomers in a single solution via both Cl− and Br− adduction. Tandem MS analysis in each case enabled complete differentiation of the isomers in the mixture without any suppression effects (Fig. S15†).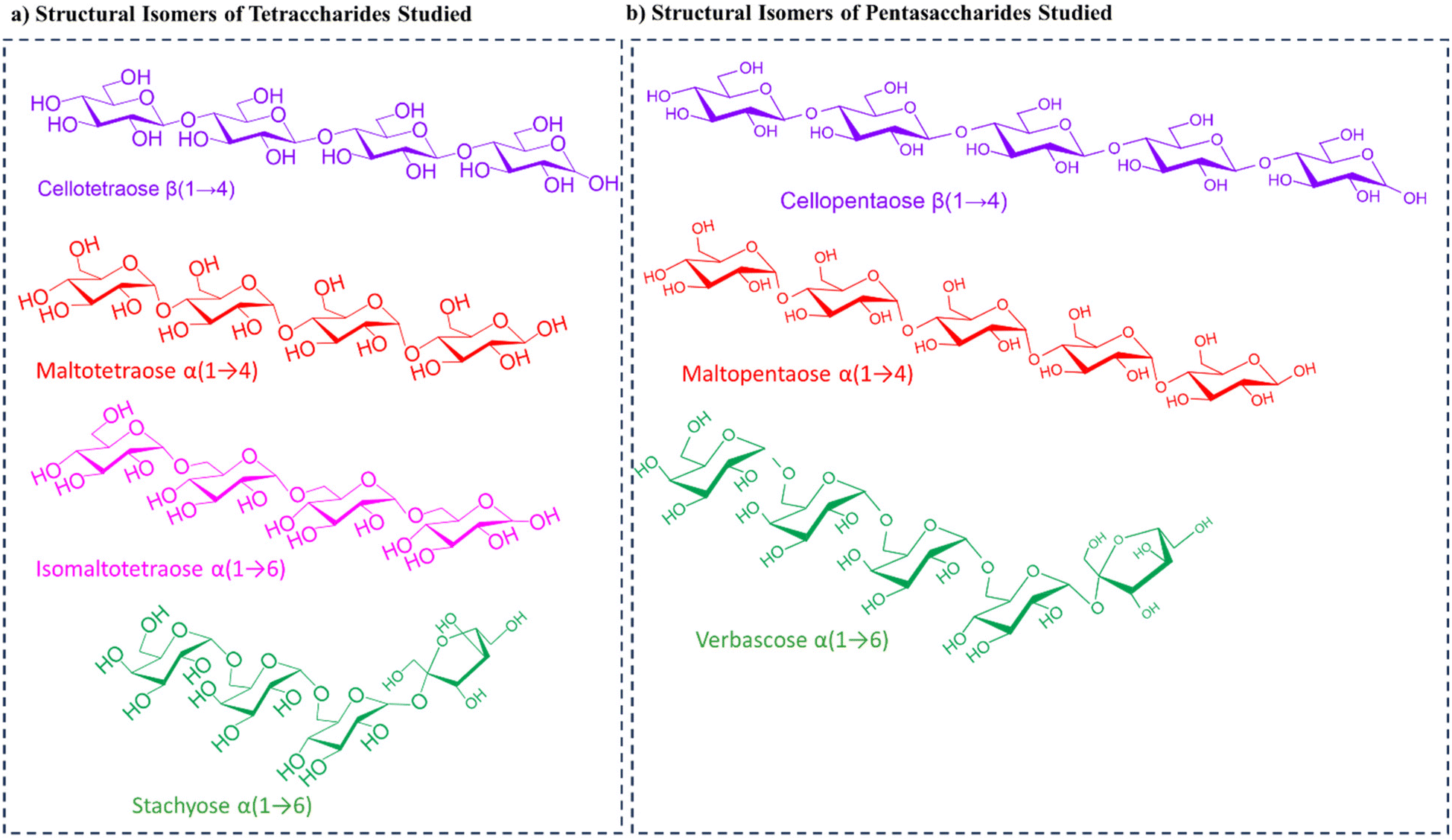 | ||
| Scheme 3 Structures of (a) tetrasaccharide and (b) pentasaccharide structural isomers studied. | ||
Pentasaccharide isomers
Finally, three pentasaccharide isomers (MW 828 Da) were analyzed using both chloride and bromide adduction. These isomers are cellopentaose, maltopentaose, and verbascose (Scheme 3b). Herein, the full MS shows signals at m/z 863 (Fig. S16a†) and 907 (Fig. 5a), indicating the formation of Cl− and Br− adducts, respectively. Comparison of the tandem MS from m/z 863 for Cl− adducts (Fig. S16b and c†) reveals that verbascose α(1 → 6 linked) afforded four unique ions at m/z 701, 645, 483, and 413. From a similar perspective, maltopentaose α(1 → 4 linked) produced three distinct diagnostic ions at m/z 647, 629, and 467 while cellopentaose β(1 → 4 linked) showed one diagnostic ion at m/z 731. MS/MS analysis of the Br− adducts of each sugar further produced unique ions to facilitate isomer differentiation (Fig. 5b and c). For example, the MS/MS analysis of verbascose produced four unique ions at m/z 827, 745, 727, and 413. Likewise, maltopentaose rendered four diagnostic ions at m/z 787, 685, 611, and 407. In a similar manner, cellopentaose provided three ions at m/z 731, 645, and 483 that were absent from the rest of the isomers. It is interesting how both maltopentaose and cellopentaose consist of the same monosaccharide units but differ only in the spatial arrangement of their atoms yet fragment to produce different diagnostic ions. This shows the sensitivity of our method and its ability to enable differentiation of saccharide isomers with direct infusion MS/MS. It is also noteworthy that here too we can differentiate 1 → 4-linked from 1 → 6-linked isomers using their tandem MS spectra. That is, 1 → 4-linked isomers produced signals at m/z 707 and 587 in higher abundance that were not observed in the spectrum of the 1 → 6-linked isomer. In the same fashion, analysis of a mixture of the pentasaccharide isomers was performed with both Cl− and Br− adduction. The tandem MS analysis in each case enabled differentiation of the isomers in the mixture with ease (Fig. S17†). | ||
| Fig. 5 (a) Negative-ion mode mass spectrum showing bromide adducts formed from spraying verbascose and ammonium bromide from the theta capillary. Negative-ion mode tandem MS analysis of (b) verbascose, (c) maltopentaose, and (d) cellopentaose. | ||
Effect of chain length on sensitivity of adduct formation
While the current method focused on oligosaccharide differentiation, we sought to evaluate the sensitivity of the method for the different oligosaccharide chain lengths investigated. Signal levels (Fig. S18†) for [M + Cl]− in the full MS reduced slightly with increasing oligosaccharide chain length (from 106 for trisaccharides to 105 for pentasaccharides). However, in the most useful mode of MS/MS, longer chain oligosaccharides produce high signal levels: 103 for cellotriose (Fig. S2b†), 104 for cellotetraose (Fig. S13a†), and 105 for cellopentaose (Fig. S16d†). In addition to higher signal levels in MS/MS, the longer-chain oligosaccharides typically produced multiple diagnostic ions for isomer differentiation. These results highlight the need to use the appropriate mode of analysis when isomer quantification is the objective.Analysis of all 14 oligosaccharide isomers in a complex urine matrix
We evaluated the ability of our direct infusion method to detect oligosaccharides in raw urine using a multi-dimensional MS analysis. We spiked all 14 oligosaccharides (50 μM each) into raw urine and analyzed the sample via Cl− adduction in negative-ion mode. The mixture of oligosaccharides consisted of seven isomers of trisaccharides, four isomers of tetrasaccharides, and three isomers of pentasaccharides. Fig. 6a shows the first dimension of this experiment where the 14 oligosaccharides were detected based on their molecular weights. In this case, the full MS spectrum showed three distinct peaks at m/z 539 for the seven trisaccharide isomers (cellotriose, isomaltotriose, maltotriose, gentianose, melezitose, nigerotriose, and 1-kestose), m/z 701 for the four tetrasaccharides (cellotetraose, isomaltotetraose, maltotetraose, and stachyose), and m/z 863 for the three pentasaccharide isomers (isomaltopentaose, maltopentaose, and verbascose). The negative-ion mode was used for this complex mixture analysis because, for raw urine, we expected multiple distribution of the ion signals for different species in the positive-ion mode (sodium, potassium, and ammonium adducts), which will make data interpretation difficult. Indeed, the negative-ion mode provided clear signals, mostly for the chloride adducts, and in proportions found in the solution phase. That is, the seven trisaccharide isomers produced peaks of higher ion intensity at m/z 539 compared with the four tetrasaccharide isomers at m/z 701, followed by the three pentasaccharide isomers at m/z 863. The second-dimension experiment involved three tandem MS analyses performed at m/z 39, m/z 701 and m/z 863 for structural characterization of the respective isomeric species. The second-dimensional MS/MS for the trisaccharides is shown in Fig. 6b, which revealed the expected diagnostic ions for maltotriose (m/z 473 and 455), isomaltotriose (m/z 387), 1-kestose (m/z 275 and 215), and nigerotriose (m/z 395 and 191). Melezitose was confirmed by the presence of a signal at m/z 257 while cellotriose and gentianose registered signals at m/z 297 and 353, respectively, all derived from tandem MS of the single adduct at m/z 539. Likewise, characterization of the four tetrasaccharide isomers packed into the peak at m/z 701 was achieved via a single CID MS/MS experiment, which showed the presence of specific diagnostic ions indicative of all four tetrasaccharide isomers (Fig. 6c). For example, the peak at m/z 575 shows the presence of isomaltotetraose while peaks at m/z 569 and 539 are attributed to the presence of maltotetraose and stachyose, respectively. Similarly, the peaks at m/z 567 and 405 are due to the presence of cellotetraose. In the tandem MS analysis of the signal at m/z 863, all three pentasaccharide isomers were identified without any suppression from other oligosaccharides present in the mixture (Fig. 6d). The pentasaccharide verbascose was identified by signals at m/z 701 and 413. Likewise, signals at m/z 647 and 629 confirmed the presence of maltopentaose, while cellopentaose was registered at m/z 731, as expected. Also, comparing the fragmentation pattern of all oligosaccharides analyzed in this study, we observed that almost all diagnostic ions observed were generated via cross-ring breakage (Fig. S19†). Diagnostic ions observed for all oligosaccharides are summarized in Table 1. In addition, schematic representations of the neutral losses for each isomer leading to the formation of diagnostic ions are illustrated in Schemes S1–S3.† Thus, without significant sample preparation and prior chromatographic separation, this work takes advantage of the multi-dimensional nature of mass spectrometers to enable separation of oligosaccharides based on molecular weight (via full MS) and structure (via CID MS/MS) through the adduction to the saccharide of Cl− ions, which are abundant in raw urine, all enabling the conventional low-energy CID method to be effective when coupled with direct-infusion nESI for small-volume analysis.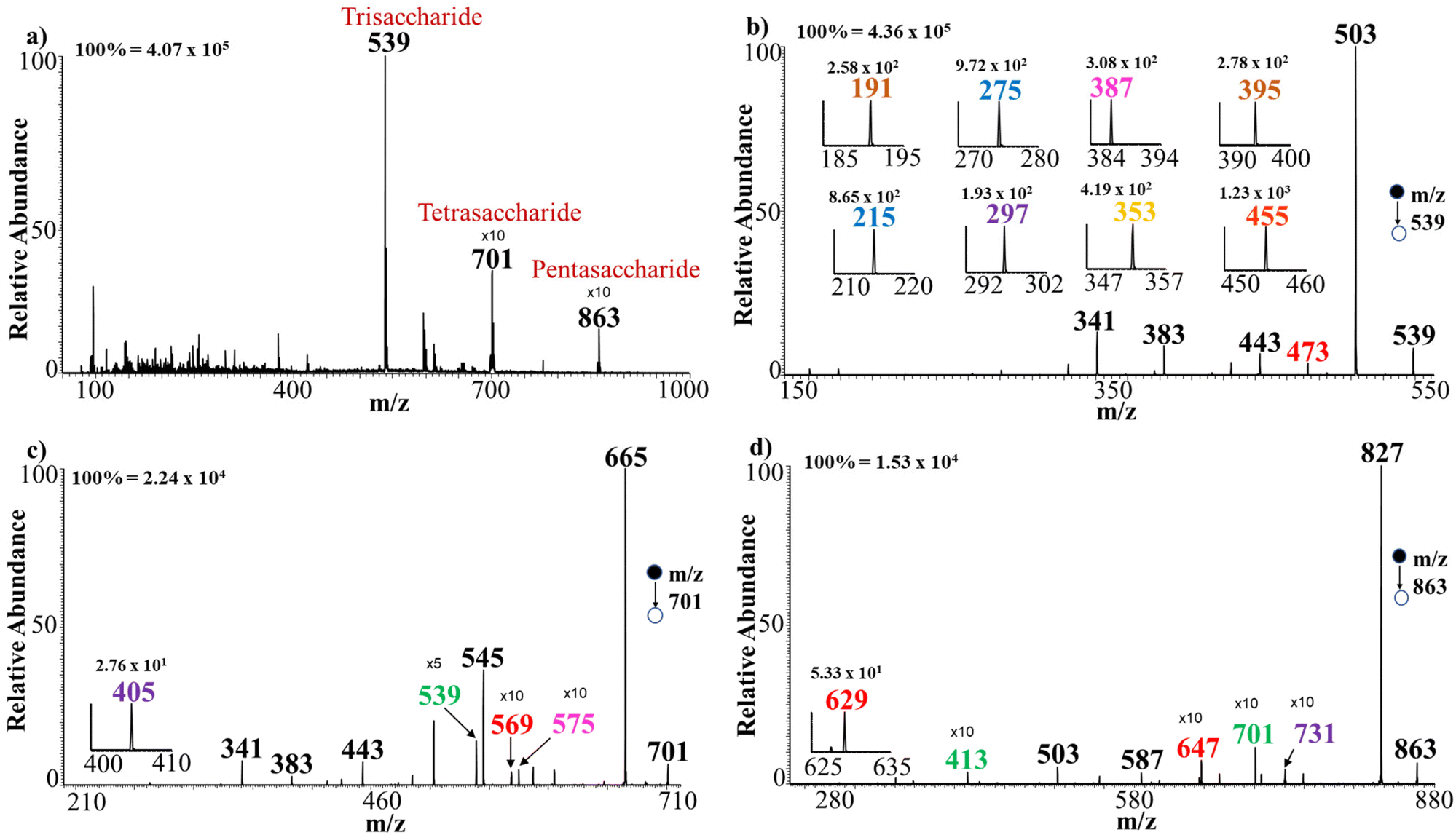 | ||
| Fig. 6 (a) Negative-ion mode mass spectrum showing chloride adducts formed from spraying a mixture of 14 oligosaccharides in urine and ammonium chloride from the theta capillary. Negative-ion mode tandem MS analysis of (b) m/z 539 for trisaccharide adducts, (c) m/z 701 for tetrasaccharide adducts, and (d) m/z 863 for pentasaccharide adducts. Insets show signals for diagnostic ions. | ||

|
Effect of relative concentrations of saccharide isomers
Finally, we evaluated the effect of relative isomer concentrations on our ability to detect them via chloride adduction. This experiment was necessary to investigate ion suppression effects. For this, we first analyzed the mixture of all fourteen oligosaccharides in relative concentrations of 5 μM for trisaccharides, 25 μM for tetrasaccharides, and 50 μM for pentasaccharides (5:25:50). In this case, the seven trisaccharides will result in the mixture having a total concentration of 35 μM. The corresponding total concentrations for the four tetrasaccharides and three pentasaccharides in the mixture will be 100 and 150 μM, respectively. Despite having the lowest relative concentration in the mixture, the trisaccharides showed higher ion abundance compared with the higher-order oligosaccharides (Fig. S19a†). This result reveals higher ionization/adduction efficiency for trisaccharides, as already discussed above. Compared with the results for the equimolar (50 μM) mixture discussed in Fig. 6a (total concentrations: 350 μM triasaccharides, 200 μM tetrasaccharides, and 150 μM pentasaccharides), the reduction of the total concentration of trisaccharides provided opportunities to detect the high-order oligosaccharides. That is, we observed an increase in ion intensity for the tetrasaccharides and pentasaccharides when the constituents in the mixture had different initial concentrations (Fig. S19a†), signifying reduced ion suppression effects. As expected, by changing the relative concentrations from 5:25:50 to 50:5:25 (trisaccharide:tetrasaccharide:pentasaccharide), the ion intensities of both the tetrasaccharide and pentasaccharide adducts decreased accordingly (Fig. S19b†), but clear peaks were observed at m/z 701 and 863, which are sufficient for MS/MS analysis and subsequent identification of the respective isomers packed under each signal. Similar observations were made when a relative concentration of 25:50:5 was used (Fig. S19c†). Here, significant suppression was observed for the pentasaccharides at 5 μM concentration but a discernible signal was detected that allowed MS/MS analysis for isomer differentiation. Therefore, at the relative oligosaccharide concentrations analyzed, prior separation is not necessary. For lower concentrations, however, a front-end separation will be needed to eliminate the ion suppression effects observed here. The fact that the main oligosaccharides have distinct molecular weights is advantageous, for the current direct-infusion experiment, although matrix effects cannot be avoided. Additionally, our halide-adduct method will provide a means to detect the presence of isomers that might coelute during liquid chromatography. This aspect of the work is currently being pursued in our laboratory where online droplet-phase adduct formation between halides and saccharides is accomplished on an LC-MS platform using a contained-ESI source.
Conclusions
In summary, we have shown that negative-ion mode analysis of saccharides through halide adduction can serve as an effective approach for differentiation of saccharide isomers when subjected to tandem mass spectrometry. The present study focused on direct differentiation of oligosaccharide isomers using minute volumes of samples. Herein, we have demonstrated that the oligosaccharides readily form adducts with halides such as Cl− and Br− using a nano-electrospray ionization method. When the adducts were subjected to collision-induced dissociation MS analyses, the various isomers yielded unique diagnostic ions that enabled complete differentiation of homo- and hetero-linked isomers without prior separation or instrument modification. Stereoisomers of trisaccharides were differentiated with ease, both via the use of distinct diagnostic fragment ions and by principal component analysis of the entire MS/MS fragment ion profiles. The ability to differentiate stereoisomers was reproduced for tetrasaccharides and pentasaccharides. In addition, structural, linkage, and positional isomers were differentiated based on the MS/MS diagnostic fragment ions of the halide adduct in negative-ion mode. We also found that the ammonium halide salts provide a two-fold benefit where it generates halide ions for effective negative-ion mode analysis and ammonium adducts in positive-ion mode analysis, both of which afforded diagnostic fragment ions. We extended the applicability of the method to the differentiation of oligosaccharide isomers in raw urine, where we successfully differentiated a mixture of seven trisaccharide isomers, four tetrasaccharide isomers, and three pentasaccharide isomers in a two-dimensional analysis (MS for molecular weight information and MS/MS for structural/isomer differentiation). We believe that these results are important because the method presented showcases a straightforward, widely available platform for direct analysis of saccharide isomers in complex mixtures for saccharide biomarker detection and characterization of rare synthetic sugars.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Data availability
The data that support the findings of this study are available in the ESI† of this article.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by funding from the National Institute of General Medical Sciences (Award Number R01GM149080).References
- S. I. Mussatto and I. M. Mancilha, Non-Digestible Oligosaccharides: A Review, Carbohydr. Polym., 2007, 68(3), 587–597 CrossRef.
- S. A. Belorkar and A. K. Gupta, Oligosaccharides: A Boon from Nature's Desk, AMB Express, 2016, 6(1), 82 CrossRef PubMed.
- S. Patel and A. Goyal, Functional Oligosaccharides: Production, Properties and Applications, World J. Microbiol. Biotechnol., 2011, 27(5), 1119–1128 CrossRef.
- O. O. Ibrahim, Functional Oligosaccharides: Chemicals Structure, Manufacturing, Health Benefits, Applications and Regulations, J. Food Chem. Nanotechnol., 2018, 4(4), 65–76 Search PubMed.
- N. M. Delzenne, Oligosaccharides: State of the Art, Proc. Nutr. Soc., 2003, 62(1), 177–182 CrossRef CAS PubMed.
- X. Qiang, C. YongLie and W. QianBing, Health Benefit Application of Functional Oligosaccharides, Carbohydr. Polym., 2009, 77(3), 435–441 CrossRef.
- S. Hughes and R. A. Rastall, Health-Functional Carbohydrates: Properties and Enzymatic Manufacture, in Novel Enzyme Technology for Food Applications, 2007, pp. 215–242 Search PubMed.
- R. Crittenden and M. Playne, Production, Properties and Applications of Food-Grade Oligosaccharides.Pdf, Trends Food Sci. Technol., 1996, 7(11), 353–361 CrossRef CAS.
- W. Cheng, J. Lu, B. Li, W. Lin, Z. Zhang, X. Wei, C. Sun, M. Chi, W. Bi, B. Yang, A. Jiang and J. Yuan, Effect of Functional Oligosaccharides and Ordinary Dietary Fiber on Intestinal Microbiota Diversity, Front. Microbiol., 2017, 8(9), 1750 CrossRef PubMed.
- R. A. Rastall, Functional Oligosaccharides: Application and Manufacture, Annu. Rev. Food Sci. Technol., 2010, 1(1), 305–339 CrossRef CAS PubMed.
- S. Chockchaisawasdee and C. E. Stathopoulos, Functional Oligosaccharides Derived from Fruit-and-Vegetable By-Products and Wastes, Horticulturae, 2022, 8(10), 911 CrossRef.
- M. Sabater-Molina, E. Larqué, F. Torrella and S. Zamora, Dietary Fructooligosaccharides and Potential Benefits on Health, J. Physiol. Biochem., 2009, 65(3), 315–328 CrossRef CAS PubMed.
- S. Nishat and P. R. Andreana, Entirely Carbohydrate-Based Vaccines: An Emerging Field for Specific and Selective Immune Responses, Vaccines, 2016, 4(2), 19 CrossRef PubMed.
- H. M. I. Osborn, P. G. Evans, N. Gemmell and S. D. Osborne, Carbohydrate–based Therapeutics, J. Pharm. Pharmacol., 2004, 56(6), 691–702 CrossRef CAS PubMed.
- E. Lattová, H. Perreault and M. Poláková, Glycans as Potential Diagnostic Biomarkers and the Importance of Developing Methods for Glycan Analysis, Glycans: Biochem., Charact., Appl, 2012, 1–26 Search PubMed.
- A. Varki, Biological Roles of Glycans, Glycobiology, 2017, 27(1), 3–49 CrossRef CAS.
- C. B. Lebrilla and H. J. An, The Prospects of Glycan Biomarkers for the Diagnosis of Diseases, Mol. Biosyst., 2009, 5(1), 17–20 RSC.
- S. E. Stefan and J. R. Eyler, Differentiation of Glucose-Containing Disaccharides by Infrared Multiple Photon Dissociation with a Tunable CO2 Laser and Fourier Transform Ion Cyclotron Resonance Mass Spectrometry, Int. J. Mass Spectrom., 2010, 297(1–3), 96–101 CrossRef CAS.
- N. C. Polfer, J. J. Valle, D. T. Moore, J. Oomens, J. R. Eyler and B. Bendiak, Differentiation of Isomers by Wavelength-Tunable Infrared Multiple-Photon Dissociation-Mass Spectrometry: Application to Glucose-Containing Disaccharides, Anal. Chem., 2006, 78(3), 670–679 CrossRef CAS PubMed.
- J. T. Adamson and K. Hakansson, Electron Capture Dissociation of Oligosaccharides Ionized with Alkali, Alkaline Earth, and Transition Metals, Anal. Chem., 2007, 79(7), 2901–2910 CrossRef CAS PubMed.
- J. J. Wolff, F. E. L. Iii, T. N. Laremore, D. A. Kaplan, M. L. Easterling, R. J. Linhardt and I. J. Amster, Glycosaminoglycans, 2010, 82(9), 3460–3466 CrossRef CAS PubMed.
- J. J. Wilson and J. S. Brodbelt, Ultraviolet Photodissociation at 355 Nm of Fluorescently Labeled Oligosaccharides, Anal. Chem., 2008, 80(13), 5186–5196 CrossRef CAS PubMed.
- A. Devakumar, Y. Mechref, P. Kang, M. V. Novotny and J. P. Reilly, Identification of Isomeric N-Glycan Structures by Mass Spectrometry with 157 Nm Laser-Induced Photofragmentation, J. Am. Soc. Mass Spectrom., 2008, 19(7), 1027–1040 CrossRef CAS PubMed.
- L. Han and C. E. Costello, Electron Transfer Dissociation of Milk Oligosaccharides, J. Am. Soc. Mass Spectrom., 2011, 22(6), 997–1013 CrossRef CAS PubMed.
- M. J. Kailemia, L. R. Ruhaak, C. B. Lebrilla and I. J. Amster, Oligosaccharide Analysis by Mass Spectrometry: A Review of Recent Developments, Anal. Chem., 2014, 86(1), 196–212 CrossRef CAS PubMed.
- S. G. Penn, M. T. Cancilla and C. B. Lebrilla, Collision-Induced Dissociation of Branched Oligosaccharide Ions with Analysis and Calculation of Relative Dissociation Thresholds, Anal. Chem., 1996, 68(14), 2331–2339 CrossRef CAS PubMed.
- M. Salema-Oom, V. V. Pinto, P. Gonçalves and I. Spencer-Martins, Maltotriose Utilization by Industrial Saccharomyces Strains: Characterization of a New Member of the α-Glucoside Transporter Family, Appl. Environ. Microbiol., 2005, 71(9), 5044–5049 CrossRef PubMed.
- V. C. Seeburger, B. Shaaban, K. Schweikert, G. Lohaus, A. Schroeder and M. Hasselmann, Environmental Factors Affect Melezitose Production in Honeydew from Aphids and Scale Insects of the Order Hemiptera, J. Apic. Res., 2022, 61(1), 127–137 CrossRef.
- A. Watanabe, Y. Kadota, R. Kamio, T. Tochio, A. Endo, Y. Shimomura and Y. Kitaura, 1-Kestose Supplementation Mitigates the Progressive Deterioration of Glucose Metabolism in Type 2 Diabetes OLETF Rats, Sci. Rep., 2020, 10(1), 1–11 CrossRef PubMed.
- R. M. Schaller-Duke, M. R. Bogala and C. J. Cassady, Electron Transfer Dissociation and Collision-Induced Dissociation of Underivatized Metallated Oligosaccharides, J. Am. Soc. Mass Spectrom., 2018, 29(5), 1021–1035 CrossRef PubMed.
- R. Daniel, L. Chevolot, M. Carrascal, B. Tissot, P. A. S. Mourão and J. Abian, Electrospray Ionization Mass Spectrometry of Oligosaccharides Derived from Fucoidan of Ascophyllum Nodosum, Carbohydr. Res., 2007, 342(6), 826–834 CrossRef PubMed.
- B. Spengler, J. W. Dolce and R. J. Cotter, Infrared Laser Desorption Mass Spectrometry of Oligosaccharides: Fragmentation Mechanisms and Isomer Analysis, Anal. Chem., 1990, 62(17), 1731–1737 CrossRef.
- B. Quéméner, J. Vigouroux, E. Rathahao, J. C. Tabet, A. Dimitrijevic and M. Lahaye, Negative Electrospray Ionization Mass Spectrometry: A Method for Sequencing and Determining Linkage Position in Oligosaccharides from Branched Hemicelluloses, J. Mass Spectrom., 2015, 50(1), 247–264 CrossRef PubMed.
- N. P. J. Price, Oligosaccharide Structures Studied by Hydrogen-Deuterium Exchange and MALDI-TOF Mass Spectrometry, Anal. Chem., 2006, 78(15), 5302–5308 CrossRef CAS PubMed.
- Y. Kostyukevich, A. Kononikhin, I. Popov and E. Nikolaev, Conformations of Cationized Linear Oligosaccharides Revealed by FTMS Combined with In-ESI H/D Exchange, J. Mass Spectrom., 2015, 50(10), 1150–1156 CrossRef CAS PubMed.
- T. Tian, N. Rumachik, A. J. G. Sinrod, D. Barile and Y. Liu, Coupling an Ion Chromatography to High Resolution Mass Spectrometry (IC-MS) for the Discovery of Potentially Prebiotic Oligosaccharides in Chardonnay Grape Marc, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2023, 40(1), 1214–123540 Search PubMed.
- M. C. Hagemeijer, J. C. van den Bosch, M. Bongaerts, E. H. Jacobs, J. M. P. van den Hout, E. Oussoren and G. J. G. Ruijter, Analysis of Urinary Oligosaccharide Excretion Patterns by UHPLC/HRAM Mass Spectrometry for Screening of Lysosomal Storage Disorders, J. Inherited Metab. Dis., 2023, 46(2), 206–219 CrossRef CAS PubMed.
- B. Fong, K. Ma and P. McJarrow, Quantification of Bovine Milk Oligosaccharides Using Liquid Chromatography-Selected Reaction Monitoring-Mass Spectrometry, J. Agric. Food Chem., 2011, 59(18), 9788–9795 CrossRef CAS PubMed.
- L. R. Ruhaak, A. M. Deelder and M. Wuhrer, Oligosaccharide Analysis by Graphitized Carbon Liquid Chromatography-Mass Spectrometry, Anal. Bioanal. Chem., 2009, 394(1), 163–174 CrossRef CAS PubMed.
- M. Wuhrer, C. A. M. Koeleman, A. M. Deelder and C. H. Hokke, Normal-Phase Nanoscale Liquid Chromatography-Mass Spectrometry of Underivatized Oligosaccharides at Low-Femtomole Sensitivity, Anal. Chem., 2004, 76(3), 833–838 CrossRef CAS PubMed.
- J. Charlwood, H. Birrell, E. S. P. Bouvier, J. Langridge and P. Camilleri, Analysis of Oligosaccharides by Microbore High-Performance Liquid Chromatography, Anal. Chem., 2000, 72(7), 1469–1474 CrossRef CAS PubMed.
- A. G. Galermo, E. Nandita, M. Barboza, M. J. Amicucci, T. T. T. Vo and C. B. Lebrilla, Liquid Chromatography-Tandem Mass Spectrometry Approach for Determining Glycosidic Linkages, Anal. Chem., 2018, 90(21), 13073–13080 CrossRef CAS PubMed.
- D. Ropartz, M. Fanuel, J. Ujma, M. Palmer, K. Giles and H. Rogniaux, Structure Determination of Large Isomeric Oligosaccharides of Natural Origin through Multipass and Multistage Cyclic Traveling-Wave Ion Mobility Mass Spectrometry, Anal. Chem., 2019, 91(18), 12030–12037 CrossRef CAS PubMed.
- Q. Yang, Y. Guo, Y. Jiang and B. Yang, Structure Identification of the Oligosaccharides by UPLC-MS/MS, Food Hydrocolloids, 2023, 139(1), 108558 CrossRef CAS.
- S. P. Huang, H. C. Hsu, C. Y. Liew, S. T. Tsai and C. K. Ni, Logically Derived Sequence Tandem Mass Spectrometry for Structural Determination of Galactose Oligosaccharides, Glycoconjugate J., 2021, 38(2), 177–189 CrossRef CAS PubMed.
- J. Yan, J. Ding, G. Jin, D. Yu, L. Yu, Z. Long, Z. Guo, W. Chai and X. Liang, Profiling of Sialylated Oligosaccharides in Mammalian Milk Using Online Solid Phase Extraction-Hydrophilic Interaction Chromatography Coupled with Negative-Ion Electrospray Mass Spectrometry, Anal. Chem., 2018, 90(5), 3174–3182 CrossRef CAS PubMed.
- H. Buck-Wiese, M. Fanuel, M. Liebeke, K. Le Mai Hoang, A. Pardo-Vargas, P. H. Seeberger, J. H. Hehemann, H. Rogniaux, G. P. Jackson and D. Ropartz, Discrimination of β-1,4- And β-1,3-Linkages in Native Oligosaccharides via Charge Transfer Dissociation Mass Spectrometry, J. Am. Soc. Mass Spectrom., 2020, 31(6), 1249–1259 CrossRef CAS PubMed.
- J. Song, H. Lee, I. Park and H. Lee, Analysis of Oligosaccharides in Korean Fermented Soybean Products by the Combination of Mass Spectrometry and Gas Chromatography, J. Agric. Food Chem., 2023, 71(1), 760–769 CrossRef CAS PubMed.
- N. Tao, E. J. DePeters, J. B. German, R. Grimm and C. B. Lebrilla, Variations in Bovine Milk Oligosaccharides during Early and Middle Lactation Stages Analyzed by High-Performance Liquid Chromatography-Chip/Mass Spectrometry, J. Dairy Sci., 2009, 92(7), 2991–3001 CrossRef PubMed.
- Y. Liu, S. Urgaonkar, J. G. Verkade and D. W. Armstrong, Separation and Characterization of Underivatized Oligosaccharides Using Liquid Chromatography and Liquid Chromatography-Electrospray Ionization Mass Spectrometry, J. Chromatogr A, 2005, 1079(1–2), 146–152 CrossRef PubMed.
- D. T. Gass, A. V. Quintero, J. B. Hatvany and E. S. Gallagher, Metal Adduction in Mass Spectrometric Analyses of Carbohydrates and Glycoconjuagtes, Mass Spectrom. Rev., 2022, e21801 Search PubMed.
- H. C. Hsu, C. Y. Liew, S. P. Huang, S. T. Tsai and C. K. Ni, Simple Method for de Novo Structural Determination of Underivatised Glucose Oligosaccharides, Sci. Rep., 2018, 8(1), 1–12 Search PubMed.
- M. T. Abutokaikah, J. W. Frye, J. Tschampel, J. M. Rabus and B. J. Bythell, Fragmentation Pathways of Lithiated Hexose Monosaccharides, J. Am. Soc. Mass Spectrom., 2018, 29(8), 1627–1637 CrossRef PubMed.
- T. Yamagaki and Y. Makino, Fragmentation of Oligosaccharides from Sodium Adduct Molecules Depends on the Position of N-Acetyl Hexosamine Residue in Their Sequences in Mass Spectrometry, Mass Spectrom., 2017, 6(2), S0073 CrossRef PubMed.
- E. Amoah, D. S. Kulyk, C. S. Callam, C. M. Hadad and A. K. Badu-Tawiah, Mass Spectrometry Approach for Differentiation of Positional Isomers of Saccharides: Toward Direct Analysis of Rare Sugars, Anal. Chem., 2022, 95(13), 5635–5642 CrossRef PubMed.
- Y. Jiang and R. B. Cole, Oligosaccharide Analysis Using Anion Attachment in Negative Mode Electrospray Mass Spectrometry, J. Am. Soc. Mass Spectrom., 2005, 16(1), 60–70 CrossRef CAS PubMed.
- E. C. H. Wan and Z. Y. Jian, Analysis of Sugars and Sugar Polyols in Atmospheric Aerosols by Chloride Attachment in Liquid Chromatography/Negative Ion Electrospray Mass Spectrometry, Environ. Sci. Technol., 2007, 41(7), 2459–2466 CrossRef CAS PubMed.
- B. Guan and R. B. Cole, MALDI Linear-Field Reflectron TOF Post-Source Decay Analysis of Underivatized Oligosaccharides: Determination of Glycosidic Linkages and Anomeric Configurations Using Anion Attachment, J. Am. Soc. Mass Spectrom., 2008, 19(8), 1119–1131 CrossRef CAS PubMed.
- N. R. Vinueza, V. A. Gallardo, J. F. Klimek, N. C. Carpita and H. I. Kenttämaa, Analysis of Carbohydrates by Atmospheric Pressure Chloride Anion Attachment Tandem Mass Spectrometry, Fuel, 2013, 105, 235–246 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1: Full MS analysis of isomers via chloride adduction. Fig. S2: Analysis of positional isomers via bromide adduction. Fig. S3: Mixture of trisaccharide positional isomers. Fig. S4: Tandem MS analysis of other trisaccharide isomers via chloride adduction. Fig. S5: Analysis of other trisaccharides via bromide adduction. Fig. S6: Mixture of seven isomers via chloride adduction. Fig. S7: Positive-ion mode adduction of oligosaccharide isomers. Fig. S8: Tandem MS analysis of oligosaccharide isomers in positive-ion mode via NH4+ adduction. Fig. S9: Tandem MS analysis of oligosaccharide isomers in positive-ion mode via Na+ adduction. Fig. S10: PCA results from hetero-linked trisaccharide isomers. Fig. S11: Heatmap of hetero-linked trisaccharide isomers (PDF). See DOI: https://doi.org/10.1039/d4an01142b |
| This journal is © The Royal Society of Chemistry 2024 |