
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA candidate reference measurement procedure for the quantification of α-synuclein in cerebrospinal fluid using an SI traceable primary calibrator and multiple reaction monitoring†
Leran
Zhang
 *a,
Eva
Illes-Toth
a,
Adam
Cryar
a,
Giles
Drinkwater
a,
Lucia
Di Vagno
a,
Marie-Laure
Pons
b,
Julia
Mateyka
a,
Bryan
McCullough
a,
Eli
Achtar
a,
Cailean
Clarkson
a,
Laura
Göschel
cd,
Peter
Körtvélyessy
ce,
Chris
Mussell
a,
Christopher J.
Hopley
a,
Agnes
Flöel
f,
Christophe
Hirtz
b,
Sylvain
Lehmann
b and
Milena
Quaglia
a
*a,
Eva
Illes-Toth
a,
Adam
Cryar
a,
Giles
Drinkwater
a,
Lucia
Di Vagno
a,
Marie-Laure
Pons
b,
Julia
Mateyka
a,
Bryan
McCullough
a,
Eli
Achtar
a,
Cailean
Clarkson
a,
Laura
Göschel
cd,
Peter
Körtvélyessy
ce,
Chris
Mussell
a,
Christopher J.
Hopley
a,
Agnes
Flöel
f,
Christophe
Hirtz
b,
Sylvain
Lehmann
b and
Milena
Quaglia
a
aLGC Group, Queens Road, TW11 0LY Teddington, UK. E-mail: l.zhang16@alumni.imperial.ac.uk
bLBPC-PPC, Univ Montpellier, IRMB CHU Montpellier, INM INSERM, 34295 Montpellier, France
cCharité – Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Department of Neurology, 10117 Berlin, Germany
dCharité – Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Neuroscience Clinical Research Center, 10117 Berlin, Germany
eLabor Berlin, Innovations, Sylter Strasse 2, 13353 Berlin, Germany
fDepartment of Neurology, Universitätsmedizin Greifswald, 17475 Greifswald, Germany
First published on 23rd July 2024
Abstract
α-synuclein aggregation is an important hallmark of neurodegenerative diseases such as Parkinson's disease (PD) and Lewy body dementia. α-synuclein has been increasingly used as a diagnostic biomarker in PD and other synucleinopathies. Current clinical assays rely on antibody-based immunoassays to detect α-synuclein, which possess high sensitivity, afford high throughput and require small sample volumes. The utility of these assays, however, may be compounded by the specificity, selectivity and batch-to-batch heterogeneity of the antibody used, which can lead to deviations in the total amount of the protein measured when comparing results among different laboratories. Similarly, current mass spectrometry-based quantification methods for α-synuclein lack well-defined, value assigned calibrators to ensure comparability of measurements. Therefore, there is still an unmet need for the standardisation of clinical measurements for α-synuclein that can be achieved by the development of reference measurement procedures (RMPs) utilising calibrators traceable to the SI (International System of Units). Here, we report a candidate RMP for α-synuclein, using an SI traceable primary calibrator and an isotope dilution mass spectrometry (IDMS) approach to address this need. The gravimetrically prepared primary calibrator was traceably quantified utilising a combination of amino acid analysis (AAA) and quantitative nuclear magnetic resonance (qNMR) for value assignment. An optimised targeted sample clean-up procedure involving a non-denaturing Lys-C digestion and solid-phase extraction strategy was devised, followed by the development of a targeted multiple reaction monitoring (MRM) method for the quantification of α-synuclein in cerebrospinal fluid (CSF). This candidate RMP was then deployed for the sensitive detection and accurate quantification of multiple proteotypic α-synuclein peptides in patient derived CSF samples. The LC-MS based results were subsequently compared to immunoassay data to assess the overall performance of our approach. The development and adoption of this candidate RMP, along with the availability of the SI traceable primary calibrator will allow for reliable quantifications of α-synuclein in CSF by an LC-MS based assay. The RMP will potentially contribute towards the standardisation of this important biomarker and may lead to future interlaboratory comparisons.
Introduction
α-synuclein is an intrinsically disordered presynaptic protein that is expressed predominantly in the central nervous system.1 α-synuclein is the main constituent of Lewy bodies and has been implicated in both familial and sporadic forms of Parkinson's Disease (PD) and Lewy body dementia. α-synuclein gained significant interest as a potential therapeutic target and has been used as a clinical biomarker in the diagnosis of a range of neurodegenerative diseases (NDD).2 In addition, α-synuclein's potential not only as a diagnostic- but also as a prognostic biomarker in PD and Lewy body dementia has been emerging.3–6 Due to the localised production of CSF in the brain, it represents one of the most tractable biofluids for directly monitoring molecular changes occurring due to NDD in laboratory medicine. Publications have demonstrated lower total α-synuclein concentrations in the CSF of PD patients vs. healthy controls7–10 and more recently, data from real-time quaking-induced conversion (RT-QuIC) assays of α-synuclein showed the potential to discriminate between disease and healthy states.11,12 Current challenges for reliable and comparable clinical diagnostic outcomes include variations amongst the antibody-based quantification methods and the lack of established analytical reference measurement procedures (RMP) which can ensure spatiotemporal traceability of results through the use of SI. Variability in clinical measurements was reported to be especially apparent when the absolute concentrations of α-synuclein in CSF were compared.13,14 Previously, two mass spectrometry-based methods were presented as alternatives to more reliably measure the absolute concentration of α-synuclein in CSF.13,14 Interestingly, despite similar methodologies, the two publications reported an average α-synuclein concentration of 0.14 ng mL−1–0.46 ng mL−1 (ref. 13) and 1.46 ng mL−1–2.04 ng mL−1 (ref. 14) in CSF samples, indicating that the use of mass spectrometry alone is not inherently a solution to reducing measurement variability. Both immunoassay-based methods and mass spectrometry methods measure the total amount of synuclein, but mass spectrometry has the unique the capability to differentiate between different isoforms and proteoforms present in clinical samples.15 More recently, the feasibility of measuring misfolded states of α-synuclein showed great potential in clinical practice through the application of RT-QuIC measurements and seed amplification assays.12,16 Nevertheless, challenges in the reproducibility of calibration curves were commonly described across these different measurement techniques and the need for characterisation requirements of synuclein calibrators was highlighted. Therefore, there is still an unmet need to date to standardise clinical measurements of α-synuclein to provide accurate results for differential diagnosis. Here, an SI traceable candidate RMP was developed for the detection of α-synuclein in patient samples to bridge this gap.Analytically validated RMPs are recognised ways to the standardisation of clinical measurements17,18 and will be crucial in minimising measurement variability observed for α-synuclein in CSF. Metrological traceability, whereby quantitative measurements are directly anchored to the SI, is an established route to reliable measurements and interlaboratory comparability. The current conventional metrological methods for the development of primary calibrators mainly focus on the determination of the total amount of protein via AAA and purity assessment. It is, however, critical that for α-synuclein that the higher order structure of the protein is also considered, particularly if methods such as RT-QuIC are to be further developed into more widespread clinical applications and uptake.
Herein, we report the production and application of an SI traceable primary calibrator for α-synuclein that was used for the development of a candidate RMP. Using this primary calibrator, we have developed the first liquid chromatography-mass spectrometry candidate RMP based on MRM for the quantification of α-synuclein in CSF. The method was applied to quantify fifteen patient derived CSF samples, and the total values of the respective α-synuclein peptides are given with associated uncertainty values. To evaluate and compare the performance of this candidate RMP, immunoassay-based data of LC-MS-based methods were compared, and the results presented.
Experimental
Reagents
The α-synuclein primary calibrator and isotopically labelled α-synuclein (13C and 15N) were purchased from Promise Advanced Proteomics (Grenoble, France). Other materials were rLys-C (Promega, WI, USA), ultrapure water (18.2 MΩ cm−1), formic acid (Fisher Scientific, MA, USA), methanol, acetonitrile (Honeywell, NC, USA), tris base and ammonium formate (Merck, NJ, USA). Reagents for artificial CSF (aCSF) were sodium chloride, sodium bicarbonate, potassium chloride, sodium phosphate monobasic, magnesium chloride, D-(+)-glucose and calcium chloride (Merck, NJ, USA).Cerebrospinal fluid
A pool of 100 CSF samples from the biological resource centre (CRB) of CHU-Montpellier was used for validation experiments. Samples for generating pools were retrospectively selected from the NeuroCognition Biobank of Montpellier's University Hospital from patients recruited in the Resource and Research Memory Centre. Patients gave informed and written consent to have their samples stored in an officially registered and ethically approved biological collection (#DC-2008-417) and later used for scientific research. For the CSF panel, all participants gave written informed consent before participation in the NeuroMET and NeuroMET2 studies. The study was approved by the ethics committee of the Charité University Hospital (EA1/197/16 and EA2/121/19).Amino acid analysis (AAA)
Amino acid quantification was performed by microwave assisted hydrolysis and exact matching isotope dilution mass spectrometry (IDMS) with calibration against SI traceable amino acid standards by using a previously described method.19 SI traceability was achieved by using amino acid certified reference materials from the National Metrology Institute of Japan (NMIJ). Briefly three sample blends containing the isotopically labelled amino acids and the sample to be quantified (α-synuclein or peptide internal standards) were gravimetrically prepared together with three calibration blends containing the natural and the isotopically labelled standard amino acids in an equimolar amount to the sample blend. Reagent blanks, natural amino acid standard solution without addition of labelled standard and labelled amino acid standard solution without addition of natural standard were prepared. All sample solutions were freeze-dried in a Genevac EZ-2 plus vacuum centrifuge (SP Scientific, PA, USA) and hydrolysed by acid hydrolysis in an Ethos EZ Microwave Digestion System (Analytix, Tyne and Wear, UK) with 6 M high purity hydrochloric acid. Samples were derivatized for 1 hour at 85 °C using N-tert-butyldimethylsilyl-N-methyl-trifluroacetamide with 1% tert-butyldimethylchlorosilane (Merck, NJ, USA) and immediately analysed by GC-MS. Analysis of the sample solutions was performed on an Agilent 5975C Mass Spectrometer (Agilent Technologies, CA, USA) using a Zebron ZB-5HT Inferno GC column (30 m × 0.25 mm i.d., 0.25 mm film thickness) with a Z-guard column (2 m, 0.53 mm i.d.) (Phenomenex, CA, USA) coupled to an Agilent 5890A gas chromatography system with CTC CombiPAL autosampler. Sample blends were bracketed with calibration blends and injected five times. Amino acid level data was then corrected for peptide impurities, according to the abundance of amino acid residues in those impurities.SI-traceable quantification of α-synuclein
Aliquots of α-synuclein (Promise Proteomics, Grenoble, France) at a nominal concentration of 0.1 μM were prepared and stored at −80 °C. Quantification was performed in triplicates by tryptic digestion and exact matching IDMS using peptides as internal standards. SI traceability was achieved through the utilisation of in-house SI traceable quantified synthetic peptides. For practical considerations, including availability of labelled peptides, purity, suitability for derivatisation, tryptic digestion and kinetics of digested peptide release, four peptides (MDVFMK_T1, QGVAEAAGK_T6, EGVLYVGSK_T8, TVEGAGSIAAATGFVK_T13) and four isotopically labelled analogues (all K(13C6, 15N2) labelled) were purchased from GL Biochem (Shanghai, China) with a stated purity ≥95%. In-house assessment of the purity of the peptides was carried out as previously described.20 Traceability was ensured by AAA and qNMR. The calculated purity (as a percentage) of peptides was: MDVFMK: 95.1 ± 0.6; QGVAEAAGK: 78.7 ± 0.8; EGVLYVGSK: 99.0 ± 0.2; TVEGAGSIAAATGFVK: 26.1 ± 0.4. Additional purification of T13 was conducted, but the peptide was not used for quantification of the primary calibrator due to its different kinetic release when compared with that of T1, T6 and T8. For the quantification of α-synuclein sample blends, three calibration blends and reagent blanks were gravimetrically prepared.21,22 Each sample and calibration blend was digested in duplicate for calculating the contribution of digestion variability to the measurement uncertainty. 2.4 μg of Lys-C and 4.8 μg of trypsin were added to solutions containing a nominal concentration of α-synuclein corresponding to 0.3 μg g−1. Digestion was performed for 4.5 h at 37 °C with gentle agitation. Following tryptic digestion, formic acid was added to each blend to a final concentration of 1% to stop the digest. Each blend was analysed (1 μL injection) on a Waters H-Class UPLC system (Waters, Wilmslow, UK) coupled to a Waters Xevo G2-XS QToF operated in sensitivity mode with a typical resolving power of 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. Peptides were separated by reverse phase chromatography using a CORTECS 90 Å C18 3.0 × 150 mm column (Waters, Wilmslow, UK). Sample blends were bracketed with calibration blends and were injected five times.
000. Peptides were separated by reverse phase chromatography using a CORTECS 90 Å C18 3.0 × 150 mm column (Waters, Wilmslow, UK). Sample blends were bracketed with calibration blends and were injected five times.
Candidate reference measurement procedure (RMP)
CSF (0.5 mL), the α-synuclein primary calibrator (33 nM) and labelled α-synuclein (3.5 nM) were thawed at room temperature for 30 min. Nine calibration samples in 500 μL of aCSF with 0.175 mg mL−1 BSA were gravimetrically prepared from the primary calibrator at 0.1, 0.5, 1, 1.5, 2, 4, 6, 8 and 10 ng g−1, along with the addition of labelled α-synuclein (100 μL). Tris buffer (1 M, pH 8.2) was added to a final concentration of 50 mM. Samples were digested overnight at 27 °C using Lys-C (1.17μg). Solid phase extraction (SPE) cartridges (Phenomenex StrataX 33 μm, 30 mg mL−1) were conditioned with MeOH (1 mL), MeCN (1 mL) and ammonium formate (10 mM, pH 10, 2 mL). Samples were loaded and washed with ammonium formate (10 mM, pH 10, 2 mL). Elution was performed at pH 10, using ammonium formate buffer (10 mM) with different percentages of acetonitrile (MeCN). The T6 peptide was eluted using 3% MeCN (1.8 mL) and T8 was eluted using 10% MeCN (3 mL). The T12 and T13 peptides were eluted using 15% MeCN (1.8 mL) and combined with the T6 fraction. Alternatively, during the later stages of method development, the T6 and T8 elution steps were replaced with a 10% MeCN wash, followed by the focussed elution and further processing of T12/T13 peptides. Samples were dried to completion overnight under vacuum at 37 °C. Samples were resuspended in 0.1% FA, 3% MeCN (70 μL), vortexed, sonicated (5 min) and centrifuged (3000g, 5 min). Final peptide samples were transferred into total recovery vials (Waters, Wilmslow, UK) and centrifuged (3000g, 5 min).Measurement uncertainty
The standard and combined uncertainties were calculated in accordance with the EURACHEM/CITAC Guide CG4 and with the ISO Guide to the Expression of Uncertainty in Measurements.22,23The uncertainty (u) associated with the double exact matching IDMS values from the AAA or tryptic digestion experiments for quantification of the α-syn primary calibrator were calculated as previously reported.19,21 Particularly the combined uncertainty associated to the α-synuclein primary calibrator was calculated as by eqn (1):
 | (1) |
 | (2) |
 | (3) |
The uncertainty associated with the calibration curve quantification was estimated for each of the three peptides quantified using eqn (4):
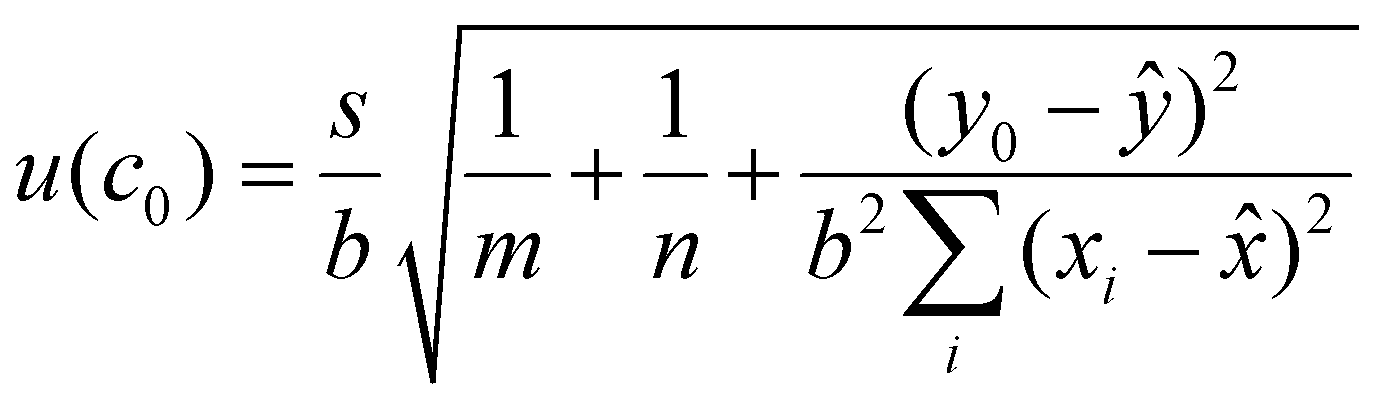 | (4) |
![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) is the average concentration of all the calibration standards, y0 is the observed peak area ratio between the natural and labelled peptides and ȳ is the average natural to labelled ratio of the calibration curve.
is the average concentration of all the calibration standards, y0 is the observed peak area ratio between the natural and labelled peptides and ȳ is the average natural to labelled ratio of the calibration curve.
Results and discussion
Primary calibrator characterisation and quantification
A fundamental requirement for developing SI traceable reference methods is access to traceable value assigned standards. A commercial stock of α-synuclein (Promise Proteomics, Grenoble, France) was sourced. This stock was quantified using a tryptic digestion IDMS approach previously reported.21 Purity was confirmed to be >95% by LC-MS (Fig. S1†). Three peptides (MDVFMK: T1, QGVAEAAGK: T6 and EGVLYVGSK: T8) were selected for α-synuclein quantification based on measured digestion kinetics, with rapid enzymatic release from the standard. These peptide stocks were traceably quantified using a combination of AAA, IDMS and qNMR (Fig. S2†). Note that peptide T1 contained oxidised methionines (singly and doubly oxidised methionines, at 0.7% in abundance relative to the unmodified main peak) as determined by mass spectrometry, for which during value assignment an impurity correction was considered to account for those (Table S1a†). AAA and qNMR quantifications showed good agreement with the T1 and T8 peptides. The AAA results of these two peptides were used for quantifying the primary calibrator. However, the T6 peptide quantifications did not agree between AAA and qNMR, due to the conversion of the N-terminal glutamine into pyroglutamate. Following tryptic digestion optimisation and confirmation of complete enzymatic release from the protein, quantification of the purified α-synuclein stock was achieved (Fig. 1).24 Results gave a final α-synuclein stock concentration of 477 ng g−1 with a combined uncertainty (k = 2) of 9.1% (Table S1b†). The combined uncertainty calculation included uncertainties from AAA/qNMR quantification, proteolytic digestion and homogeneity of the sample. | ||
| Fig. 1 Plot showing the agreement between individual peptide level values for T1, T6 and T8 tryptic peptides liberated from the α-synuclein primary calibrator stock (n = 9). Combining the peptide level data permits value assignment of the primary calibrator with its expanded uncertainty (k = 2), representing the 95% confidence interval. | ||
Structural characterisation of the primary calibrator
For an RMP, the aggregation properties and structure of the primary calibrator must be characterised. Bovine serum albumin (BSA) was used as a carrier protein to reduce sample loss of the primary calibrator in the calibration curve of the candidate RMP. Therefore, structural characterisation was performed with and without BSA using ion mobility mass spectrometry under native-like conditions (Fig. S3†). A previous report suggested that human serum albumin targets α-synuclein and redirects its aggregation into different intermediates.25 No apparent changes were detected in the charge state distribution of α-synuclein encompassing ions from 6+ to 16+ suggesting that BSA could be used under our conditions. Analyses of the corresponding drift times revealed that BSA caused only a minor compaction of ions ranging from 8+ to 11+. The full series from charge states 6+ to 16+ can be found in Fig. S4.† The shifts in drift times in the presence of BSA did not result in significant changes in the calculated mean collision cross sections (TWCCSN2→He) (Table S2†). To probe the stability of α-synuclein with and without BSA, collision induced unfolding (CIU) was employed under native-like conditions. The samples did not show major differences in their CIU fingerprints (Fig. S5†). α-Synuclein is known to self-assemble into oligomers which are often observed in recombinant sources.26 On closer inspection of the mass spectra, we detected various preformed oligomeric populations (Tables S3–S4 and Fig. S6†) of α-synuclein with very low intensities. Importantly, no differences were found in the distributions of these different multimeric α-synuclein complexes ranging from dimers to hexamers in the absence or presence of BSA (Fig. S7–S9†). In order to address the effect of freeze and thaw cycles during sample preparation of the primary calibrator, 1 μM α-synuclein with 0.175 mg mL−1 BSA (2.6 μM) was subjected to one or two cycles of freeze and thaw. It was observed that up to two freeze thaw cycles did not lead to discernible differences in the charge state distributions (Fig. S10†). Lastly, brief vortexing steps were included during the preparation of the primary calibrator. To investigate whether any aggregation occurred and to study the effects of shaking on α-synuclein, 45 μL of 15 μM protein was incubated at pH 7.0 at 21–22 °C in the presence of 0.175 mg mL−1 BSA (2.6 μM) with or without shaking at 350 rpm on an orbital benchtop shaker. Between the two conditions monitored, neither significant changes, nor any indications of aggregation were observed, which often exhibits itself in the appearance of bimodal distributions in the data of intact hydrogen deuterium exchange mass spectrometry (Fig. S11†).Quantification of α-synuclein in CSF
Based on the results obtained from the quantification of the primary calibrator, the T6, T8, T12 and T13 peptides were chosen as measurands for the RMP due to their sensitivity, selectivity, and equimolar release between the natural and labelled proteins (Fig. 2A). Previous literature reports CSF α-synuclein concentrations to be in the region of low ng mL−1, placing the required method limit of quantification (LOQ) at the sub ng mL−1.14 Mass spectrometry is a highly sensitive analytical technique, where attomole concentrations can be detected in solvent standards. At low concentrations in complex biological samples however, an optimised sample preparation protocol is required to reduce ion suppression and the presence of closely eluting isobaric compounds which may introduce bias. To meet these requirements, a bespoke sample clean up method was developed (Fig. 2B). Initial experiments confirmed the previously reported use of a non-denaturing and non-reducing overnight tryptic digestion protocol for increased method sensitivity towards α-synuclein in CSF. A high pH SPE protocol was optimised to allow the concurrent removal of intact proteins, salts and other small molecules. The monitored peptides (T6, T8, T12 and T13) were fractionated during SPE elution using increasing concentrations of MeCN. The T6, T12 and T13 peptides were collected together in the same fraction because T6 eluted early in the chromatographic gradient, while T12 and T13 co-eluted near the end of the gradient. This allowed for the concurrent analysis of these three peptides in the same chromatographic run. Samples were concentrated and injected in triplicate into an ultra-performance liquid chromatography (UPLC) system with a forty-minute acidic reverse phase gradient prior to MRM MS analysis. Both standard flow and capillary flow chromatographic methods were developed and compared. The capillary method produced higher signal to noise (S/N) and was taken forward for sample analysis. MRM transitions with the best signal intensity along with their collision energies were optimised using Skyline (Table S5†).27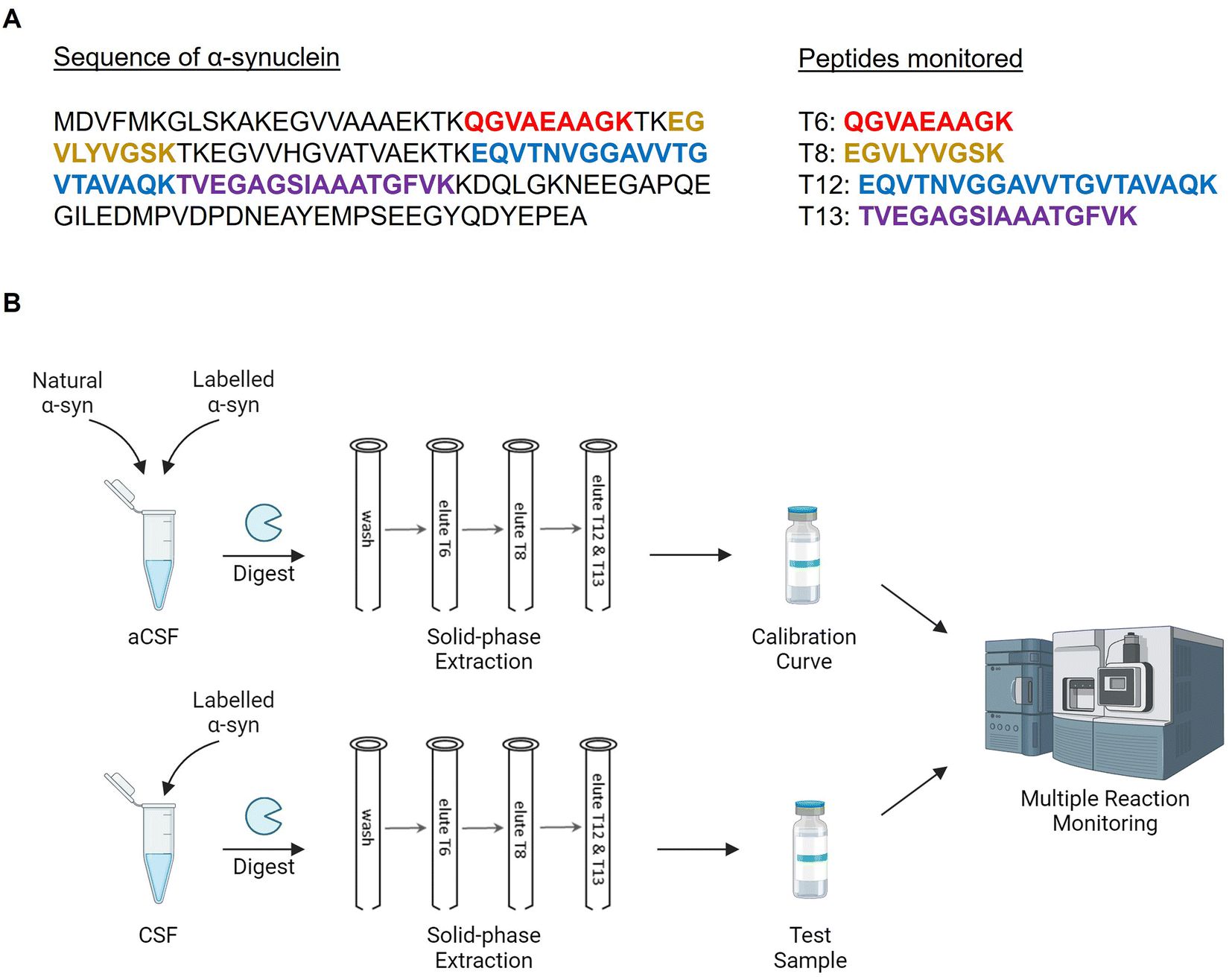 | ||
| Fig. 2 Protocol for the candidate reference method procedure. (A) Sequence of α-synuclein and the monitored peptides. (B) Sample preparation and analysis workflow for artificial CSF (aCSF) or real CSF. | ||
Initial trials applying the above method showed no quantifiable signal with the T6, T12 and T13 peptides in CSF. It was hypothesised that despite the non-reducing tryptic digestion, matrix suppression was still an issue with these peptides. To address this, a more specific protease was required for the digestion of α-synuclein to reduce the complexity of the sample. α-synuclein is unique in the fact that it does not contain any arginine residues. Therefore, the use of Lys-C allowed for the full digestion of α-synuclein while background proteins were only partially digested compared to a tryptic digestion. Furthermore, Lys-C has a greater digestion efficiency for lysine, which permitted a more complete digestion of the measurand.
The switch from trypsin to Lys-C resulted in a four to thirty-fold increase in sensitivity compared to trypsin across the four peptides in spiked samples, with T12 and T13 showing the best improvements. In real CSF samples, matrix effects of T12 and T13 were reduced significantly to a point where the peak intensities were comparable to aCSF. Unfortunately, the T6 peptide was still poorly detectable in CSF. In addition, T8 has a shared sequence with β-synuclein, so that the protocol was simplified to only isolate and analyse the T12/T13 fraction at the final stages of quantification of clinical samples.
The optimised method, nevertheless, showed excellent linearity across three orders of magnitude (0.1 ng g−1–10 ng g−1) of α-synuclein, with r2 = 0.999 for all four peptides (Fig. 3A). The LOQ was assigned to be the lowest point on the calibration curve (0.1 ng g−1), which fulfils the requirement of having a sub ng mL−1 method. Precision between aCSF injections was between 1.1%–5.8% (Fig. 3B). Intra-assay precision between six replicates of the same CSF samples was satisfactory for the T8, T12 and T13 peptides (2.8%–6.4%). CSF samples that have been over spiked with primary calibrator to 5 ng g−1 showed even higher precision (2.1%–2.2%). However, T6 peptides displayed poor sensitivity and was excluded in follow up experiments.
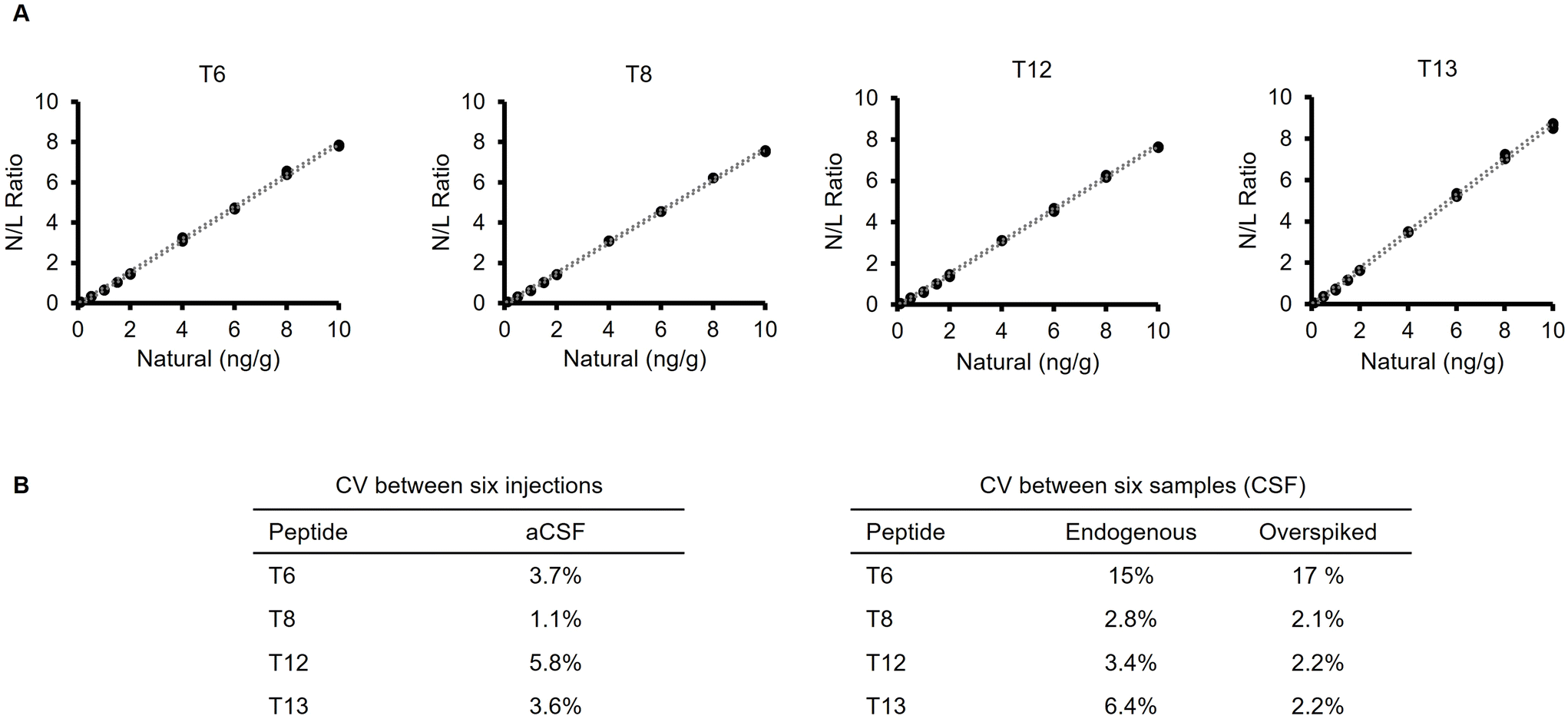 | ||
| Fig. 3 Initial validation experiments for the candidate RMP. (A) Calibration curves for the four monitored peptides (T6, T8, T12 and T13), plotting the natural/labelled peak area ratio against the volumetrically prepared natural concentration. Upper and lower limits of uncertainty at 95% confidence are represented by dotted lines. r2 = 0.999 for all peptides. (B) Method precision between injections in aCSF and between samples in real CSF. | ||
Quantification of α-synuclein in fifteen CSF samples
Fifteen CSF samples from Charité University Hospital were analysed using the candidate RMP (Fig. 4A). These samples were first analysed via immunoassay (Meso Scale, MD, USA). 500 μL of each sample was used for MS analysis. Calibration curves showed good linearity for all four peptides (r2 = 0.996–0.999). The full calibration curve covered three orders of magnitude (0.1 ng g−1–10 ng g−1), but only the lower half was used for the quantification of α-synuclein in the CSF samples. The curves were plotted using the gravimetrically calculated concentration of primary calibrator against the ratio of peak areas between the labelled and natural MRM transitions. The amount of α-synuclein in the fifteen samples were calculated using the same ratios. The method monitored three peptides, T8 (which is a sequence that is shared with β-synuclein) and the T12 and T13 peptides which were the main targets (Fig. 4B). Although, the T6 calibration curve was in line with the other peptides, the T6 peptide was not detected in the CSF samples. This may have been due to the CSF matrix suppressing the peptide signal. | ||
| Fig. 4 Analysis of a panel of CSF samples from Charité University Hospital. (A) Levels of α-synuclein in the CSF samples analysed from an immunoassay and the mass spectrometry methods (MS). * = omitted result due to isobaric interferences. (B) Comparison of concentration values obtained from the T8, T12 and T13 peptides. Error bars represent the expanded uncertainty at 95% confidence. (C) T12 and T13 values were plotted to show linearity. (D) Scatter plot comparing the quantification results from the immunoassay and the MS method for the T8, T12 and T13 peptides. | ||
The calculated concentration values between T12 and T13 peptides showed good linearity (r2 = 0.97), with Sample 1 being the only outlier (Fig. 4C). A thorough inspection of the peak areas of this sample showed that the natural channel had a slightly higher peak area compared to the other two MRMs of the peptide, suggesting the presence of an isobaric interference. Further analysis of the ratios between the main and qualifier MRMs of the other CSF samples have shown that only the aforementioned sample had significant isobaric interferences (Table S6†).
A degree of linearity was observed for all three peptides when plotting the immunoassay results with each of the individual peptides (Fig. 4D). However, the values associated with the immunoassay were consistently above the values obtained from the T12 and T13 peptides, while being below the T8 values obtained by the MS method. No data is available about homology of the epitope sequences between α-synuclein and other synucleins such as β- and γ-synuclein. Other proteoforms may have been also present and specifically recognised, which may have been a contributing factor for the discrepancies observed in total levels of α-synuclein. The mass spectrometry-based method however uses unique peptides and therefore can rely on a single protein isoform of interest.
Conclusion
An SI traceable candidate RMP for the quantification of α-synuclein from CSF samples is reported here. The method covers the physiological concentrations of α-synuclein over three orders of magnitude (0.1 ng g−1–10 ng g−1). The candidate RMP showed correlation with immunoassay results based on the preliminary analysis of CSF from a small patient cohort, including fifteen patient samples. Furthermore, the structurally characterised and value assigned SI traceable primary calibrator of α-synuclein may allow better comparability of results in the future upon wider availability and utilisation. It can be potentially harnessed in phenotypic assays or deployed for the advancement of clinical diagnostics. Although the candidate RMP requires further full-scale validation in terms of recovery, linearity and precision, it serves as a proof-of-concept that α-synuclein can be traceably measured in biological fluids.Abbreviations
| AAA | Amino acid analysis |
| aCSF | Artificial cerebrospinal fluid |
| CCS | Collision cross section |
| CIU | Collision induced unfolding |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| EDTA | Ethylenediaminetetraacetic acid |
| FA | Formic acid |
| GC-MS | Gas chromatography mass spectrometry |
| HPLC | High performance liquid chromatography |
| IDMS | Isotope dilution mass spectrometry |
| IE | Ion exchange |
| IPTG | Isopropyl β-D-1-thiogalactopyranoside |
| LC-MS | Liquid chromatography mass spectrometry |
| LOQ | Limit of quantification |
| MeCN | Acetonitrile |
| MRM | Multiple reaction monitoring |
| MS | Mass spectrometry |
| NMIJ | National Metrology Institute of Japan |
| NMR | Nuclear magnetic resonance |
| PD | Parkinson's disease |
| RT-QuIC | Real-time quaking-induced conversion |
| SEC | Size exclusion chromatography |
| SI | International System of Units |
| SPE | Solid phase extraction |
| UPLC | Ultra-performance liquid chromatography |
Author contributions
L. Z., E. I.-T., A. C., L. D. V. and M. Q. contributed to conceptualisation, investigation, methodology and formal analysis. C. C., C. M., C. J. H., A. F., C. H., S. L. and M. Q. contributed to supervision. G. D., M.-L. P., J. M., B. M. and E. A. contributed to investigation. M.-L. P., L. G. and P. K. provided clinical CSF samples. L. Z. and E. I.-T. contributed to validation, visualisation and writing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding was provided by the EMPIR programme co-financed by the Participating States and from the European Union's Horizon 2020 research and innovation programme (15HLT09-NeuroMET and 18HLT09-NeuroMET2). Biorender.com was used in the generation of Fig. 2B and the table of contents image. We thank Dr Francesca Robertson and Dr Christopher McElroy for critical review of the manuscript.References
- R. Jakes, M. G. Spillantini and M. Goedert, FEBS Lett., 1994, 345, 27–32 CrossRef CAS.
- M. Fayyad, S. Salim, N. Majbour, D. Erskine, E. Stoops, B. Mollenhauer and O. M. A. El-Agnaf, J. Neurochem., 2019, 150, 626–636 CrossRef CAS PubMed.
- L. Stefanis, Cold Spring Harbor Perspect. Med., 2012, 2, a009399–a009399 Search PubMed.
- D. J. Irwin, V. M.-Y. Lee and J. Q. Trojanowski, Nat. Rev. Neurosci., 2013, 14, 626–636 CrossRef CAS PubMed.
- D. Twohig and H. M. Nielsen, Mol. Neurodegener., 2019, 14, 23 CrossRef.
- M. G. Spillantini, R. A. Crowther, R. Jakes, M. Hasegawa and M. Goedert, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6469–6473 CrossRef CAS PubMed.
- B. Mollenhauer, J. J. Locascio, W. Schulz-Schaeffer, F. Sixel-Döring, C. Trenkwalder and M. G. Schlossmacher, Lancet Neurol., 2011, 10, 230–240 CrossRef CAS PubMed.
- B. Mollenhauer, O. M. El-Agnaf, K. Marcus, C. Trenkwalder and M. G. Schlossmacher, Biomarkers Med., 2010, 4, 683–699 CrossRef CAS PubMed.
- W. Sako, N. Murakami, Y. Izumi and R. Kaji, Mov. Disord., 2014, 29, 1599–1605 CrossRef CAS PubMed.
- T. Tokuda, S. A. Salem, D. Allsop, T. Mizuno, M. Nakagawa, M. M. Qureshi, J. J. Locascio, M. G. Schlossmacher and O. M. A. El-Agnaf, Biochem. Biophys. Res. Commun., 2006, 349, 162–166 CrossRef CAS PubMed.
- C. Bargar, W. Wang, S. A. Gunzler, A. LeFevre, Z. Wang, A. J. Lerner, N. Singh, C. Tatsuoka, B. Appleby, X. Zhu, R. Xu, V. Haroutunian, W.-Q. Zou, J. Ma and S. G. Chen, Acta Neuropathol. Commun., 2021, 9, 62 CrossRef CAS PubMed.
- A. Iranzo, G. Fairfoul, A. C. N. Ayudhaya, M. Serradell, E. Gelpi, I. Vilaseca, R. Sanchez-Valle, C. Gaig, J. Santamaria, E. Tolosa, R. L. Riha and A. J. E. Green, Lancet Neurol., 2021, 20, 203–212 CrossRef CAS.
- L. Yang, T. Stewart, M. Shi, G. Pottiez, R. Dator, R. Wu, P. Aro, R. J. Schuster, C. Ginghina, C. Pan, Y. Gao, W. Qian, C. P. Zabetian, S.-C. Hu, J. F. Quinn and J. Zhang, Proteomics: Clin. Appl., 2017, 11, 1700045 Search PubMed.
- P. Oeckl, F. Metzger, M. Nagl, C. A. F. von Arnim, S. Halbgebauer, P. Steinacker, A. C. Ludolph and M. Otto, Mol. Cell. Proteomics, 2016, 15, 3126–3138 CrossRef CAS PubMed.
- M.-L. Pons, N. Loftus, J. Vialaret, S. Moreau, S. Lehmann and C. Hirtz, Front. Aging Neurosci., 2022, 14, 818606 CrossRef CAS PubMed.
- L. Concha-Marambio, S. Pritzkow, M. Shahnawaz, C. M. Farris and C. Soto, Nat. Protoc., 2023, 18, 1179–1196 CrossRef CAS.
- H. Liu, L. Wong, S. Yong, Q. Liu and T. K. Lee, Anal. Bioanal. Chem., 2015, 407, 7579–7587 CrossRef CAS PubMed.
- J.-O. Jeppsson, U. Kobold, J. Barr, A. Finke, W. Hoelzel, T. Hoshino, K. Miedema, A. Mosca, P. Mauri, R. Paroni, L. Thienpont, M. Umemoto and C. Weykamp, Clin. Chem. Lab. Med., 2002, 40, 78–89 CAS.
- A. F. Torma, K. Groves, S. Biesenbruch, C. Mussell, A. Reid, S. Ellison, R. Cramer and M. Quaglia, Clin. Chem. Lab. Med., 2017, 55, 1397–1406 CAS.
- R. D. Josephs, G. Martos, M. Li, L. Wu, J. E. Melanson, M. Quaglia, P. J. Beltrão, D. Prevoo-Franzsen, A. Boeuf, V. Delatour, M. Öztug, A. Henrion, J.-S. Jeong and S.-R. Park, Metrologia, 2019, 56, 044006 CrossRef CAS.
- C. Pritchard, M. Quaglia, C. Mussell, W. I. Burkitt, H. Parkes and G. O'Connor, Clin. Chem., 2009, 55, 1984–1990 CrossRef CAS PubMed.
- S. Ellison and A. Williams, Quantifying uncertainty in analytical measurement, Eurachem, London, 3rd edn, 2012 Search PubMed.
- ISO, Guide to the expression of uncertainty in measurement, International Organisation for Standardisation, Geneva, 1st edn, 1995 Search PubMed.
- R. D. Josephs, N. Stoppacher, S. Westwood, R. I. Wielgosz, M. Li, M. Quaglia, J. Melanson, G. Martos, D. Prevoo, L. Wu, S. Scapin, M. Öztug Senal, L. Wong, J.-S. Jeong, K. W. Y. Chan, C. G. Arsene and S.-R. Park, J. Chem. Metrol., 2017, 11, 1–8 CrossRef.
- R. Ahmed, J. Huang, D. K. Weber, T. Gopinath, G. Veglia, M. Akimoto, A. Khondker, M. C. Rheinstädter, V. Huynh, R. G. Wylie, J. C. Bozelli, R. M. Epand and G. Melacini, J. Am. Chem. Soc., 2020, 142, 9686–9699 CrossRef CAS PubMed.
- A. S. Phillips, A. F. Gomes, J. M. D. Kalapothakis, J. E. Gillam, J. Gasparavicius, F. C. Gozzo, T. Kunath, C. MacPhee and P. E. Barran, Analyst, 2015, 140, 3070–3081 RSC.
- B. MacLean, D. M. Tomazela, N. Shulman, M. Chambers, G. L. Finney, B. Frewen, R. Kern, D. L. Tabb, D. C. Liebler and M. J. MacCoss, Bioinformatics, 2010, 26, 966–968 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4an00634h |
| This journal is © The Royal Society of Chemistry 2024 |