
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSea urchin nanostructured nickel cobaltite modified carbon cloth integrated wearable patches for the on-site detection of the immunosuppressant drug mycophenolate mofetil†
K.
Niyas
ab,
Bartholomew
Richard
 b,
Menon
Ankitha
b and
P. Abdul
Rasheed
*ab
b,
Menon
Ankitha
b and
P. Abdul
Rasheed
*ab
aDepartment of Biological Sciences and Engineering, Indian Institute of Technology Palakkad, Palakkad, Kerala 678557, India. E-mail: abdulrasheed@iitpkd.ac.in
bDepartment of Chemistry, Indian Institute of Technology Palakkad, Palakkad, Kerala 678557, India
First published on 8th May 2024
Abstract
Mycophenolate mofetil (MpM) is a medication used to prevent the rejection of transplanted organs, particularly in kidney, heart, and liver transplant surgeries. It is extremely important to be conscious that MpM can raise the risk of severe infections and some cancers if it exceeds the recommended dose while lower doses will result in organ rejections. So, it is essential to monitor the dosage of MpM in real time in the micromolar range. In this work, we have synthesized 3-aminopropyltriethoxysilane (APTES) functionalized nickel cobaltite (NiCo2O4) and this amino functionalization was chosen to enhance the stability and electrochemical activity of NiCo2O4. The enhanced activity of NiCo2O4 was used for developing an electrochemical sensor for the detection of MpM. APTES functionalized NiCo2O4 was coated on carbon cloth and used as the working electrode. Surface functionalization with APTES on NiCo2O4 was aimed at augmenting the adsorption/interaction of MpM due to its binding properties. The developed sensor showed a very low detection limit of 1.23 nM with linear ranges of 10–100 nM and 1–100 μM and its practical applicability was examined using artificial samples of blood serum and cerebrospinal fluid, validating its potential application in real-life scenarios.
1. Introduction
Modern-day immunosuppression is linked to a crucial procedure for monitoring organ function after transplantation in medical diagnostics. Evidence-based guidelines recognize that immunosuppressant use increases the rates of transplantation and patient survival. The maintenance phase of treatment typically begins six months to a year after the induction phase, when long-term oral medicine is started.1,2 Two prevalent families of immunosuppressants are commonly employed for maintenance therapy: calcineurin inhibitors, including tacrolimus (TAC) and cyclosporine A (CsA), and purine synthesis inhibitor agents such as azathioprine (AZT) and mycophenolate mofetil (MpM).3 All of these medications have one thing in common: they have narrow therapeutic indices (TIs), which might have harmful side effects at supra-therapeutic concentrations as well as reduce treatment effectiveness at sub-therapeutic levels.4Mycophenolate mofetil (MpM), known as the pro-drug of mycophenolic acid, is a well-tolerated immunosuppressant medication commonly recommended for the prevention of immune-mediated damage in transplanted organs such as the kidneys, liver, heart, and various other organs.5 In addition to its role in transplant medicine, MpM is also utilized in the treatment of conditions like scleroderma and interstitial lung disease. The drug exerts its immunosuppressive effects by inhibiting the action of the enzyme inosine monophosphate dehydrogenase (IMPDH), which controls the conversion of inosine monophosphate into guanosine monophosphate through catalytic oxidation.6 MpM is employed as a therapeutic agent that modulates the immune system's activity, benefiting the treatment of various autoimmune disorders and contributing to anticancer therapies. The dose of MpM exceeds the recommended doses, it causes severe infections and some cancers, while lower doses will result in organ rejections. Furthermore, it can cause potential risks in foetal development and miscarriage when it is used in pregnant women. These negative effects showed that therapeutic drug monitoring (TDM) of MpM is an essential requirement for follow-up post-transplantation and in other treatments. Maintaining a suitable balance between a prescribed therapeutic dose (approximately 18 μM) and the toxicity and possibility of side effects is the primary goal of TDM for immunosuppressive medication. Hence, the need for an effective detection method to quantify MpM in biological fluids has gained significant attention in these contexts.7
The majority of analytical techniques used in the past for the identification of MpM were based on chromatography and immunoassay techniques.8,9 Some disadvantages of immunoassay systems include their high cost, lengthy reaction times, and lack of stability. Furthermore, the cross-reactivity of the antibodies typically affects their sensitivity. Chromatography techniques yield highly sensitive and selective findings for the tests they perform, but they also require expensive equipment and trained personnel to analyze the data.8 To continuously determine the quantity of immunosuppressants in biological fluids, we require simple, quick-to-react analytical techniques that can serve as a platform for on-site point-of-care (POC) detection in complex biological samples like the cerebrospinal fluid, serum, urine, etc. Electrochemical sensing emerges as a promising approach to address this need, providing high sensitivity, rapid response, and compatibility with miniaturized devices suitable for point-of-care applications.10
Ashjari et al. introduced an electrochemical sensor modified with an ionic liquid and MgO/single-walled carbon nanotubes for MpM detection.7 In a separate study, Mahnashi et al. innovatively designed a disposable pencil graphite electrode for the detection of MpM and Tacrolimus (TAC).10 Similarly, Madrakian et al. developed a simultaneous sensor for MpM and its active metabolites utilizing multi-walled carbon nanotube-modified electrodes.5 Sumanth et al. conducted a cyclic voltammetric study, employing copper oxide-modified electrodes to determine MpM concentrations in biological fluids.11 In a very recent study, Vaishag et al. explored a new electrochemical sensing podium for MpM detection in serum samples using APTES-modified Nb2CTx on a carbon cloth yarn electrode.12 The use of carbon cloth electrodes is a better alternative to electrodes like glassy carbon electrodes, pencil graphite electrodes, screen printed electrodes, etc. due to their outstanding features, which include cost-effectiveness, flexibility, portability, and sensitivity.13 Customized carbon cloth-based electrode development and the subsequent fabrication of integrated sensors with appropriate electroactive materials can accomplish innovations for cost-effective wearable POC diagnostic devices.14
Transition metal oxides (TMOs) are gaining prominence as a viable option to enhance and tailor electrodes for the development of flexible sensors capable of detecting biomolecules with high sensitivity. Various TMOs derived from metals like nickel, cobalt, manganese, iron, and zinc have been utilized to modify electrodes, enabling the selective and sensitive detection of biomolecules.15 The morphology of these 2-dimensional (2D) TMOs plays a crucial role in the detection process due to their increased surface-to-volume ratio, adjustable electronic properties, and enhanced interaction with analytes. In electrochemical applications, mixed TMOs, especially nickel cobaltite (NiCo2O4), have garnered widespread attention and have been extensively explored for various electrochemical sensing applications.16,17 In NiCo2O4, one-third of the occupied cations are in the +2 oxidation state, while the remaining two-thirds are trivalent. The Ni species occupy the octahedral sites, while Co is accommodated on both octahedral and tetrahedral sites. Thus, these mixed oxidation states facilitate enhanced electron transfer in addition to easy synthesis, accessibility, and cost-effectiveness that enables their use in electrochemical applications.
The introduction of various functional moieties on the surface of TMOs offers good stability and improved performance as well as expanding the world of applications. (3-Aminopropyl) triethoxy silane (APTES) has become a prominent choice in the field of nanomaterial modification, owing to its capability to introduce amino groups onto various nanomaterial surfaces.18 The modification process entails the covalent attachment of APTES molecules to surface functional groups, resulting in nanomaterials that are amino-functionalized, exhibiting enhanced stability and interaction with target analytes and biomolecules.12,19
In this work, we have used APTES modified NiCo2O4 for the modification of an electrochemical sensing platform for the sensitive and selective detection of MpM with a promising limit of detection (LOD) at the nanomolar level (1.23 nM). The primary objective of this study was to exemplify the successful modification of a NiCo2O4 with APTES to enhance the electrochemical activity and its application in the electrochemical detection of MpM in biological fluids. APTES functionalised NiCo2O4 was coated on carbon cloth (CC) and it is used as the working electrode. The synthesis of APTES modified NiCo2O4 and its application in electrochemical sensing of MpM is given in Scheme 1. By combining the unique properties of CC and the stability of amino functionalized NiCo2O4, we aim for the clinical advancement of electrochemical sensing technology, enabling accurate and efficient monitoring of MpM levels in the current diagnostic scenario especially in TDM. The developed sensor showed a promising LOD of 1.23 nM along with good selectivity, stability, repeatability and reproducibility. In addition, the practical application of the sensor is also evaluated using artificial serum and CSF.
 | ||
| Scheme 1 Synthesis of APTES modified NiCo2O4 and its application in the electrochemical sensing of mycophenolate mofetil (MpM). | ||
2. Experimental section
2.1. Materials
Nickel(II) nitrate hexahydrate (Ni(NO3)2·6H2O, 98%), cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O, 98%), 3-aminopropyltriethoxysilane (APTES), toluene, dimethyl sulphoxide (DMSO), ethanol, ascorbic Acid (AA), glutamic acid (GA), KCl, NaCl, glucose (GLU), glycine (GLA) and alanine (ALN) were purchased from SRL Pvt Ltd, India, and used without any further purification. Urea, dopamine (DA), uric acid (UA) and artificial human serum were purchased from Sigma Aldrich, India. Mycophenolate mofetil (MpM) was procured from IPCA Laboratories, India, and carbon cloth (CC) was purchased from Sainergy Fuel Cell India Pvt. Ltd. Phosphate buffer (0.1 M, pH 7.1) solution (PBS) was employed as the assisted electrolyte in all the electrochemical studies. Artificial cerebrospinal fluid (CSF) is prepared by mixing two parent solutions A and B. The solution A was prepared by mixing NaCl (8.66 g), KCl (0.224 g), CaCl2·2H2O (0.206 g) and MgCl2·6H2O (0.163) in 500 mL sterile water. Solution B was prepared by mixing Na2HPO4·7H2O (0.214 g) and NaH2PO4·H2O (0.027 g) in 500 mL sterile water. Combine these solutions, A and B in the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio for preparing CSF since equal volumes are required to end up with a multivalent physiological ion solution of artificial CSF.20
:1 ratio for preparing CSF since equal volumes are required to end up with a multivalent physiological ion solution of artificial CSF.20
2.2. Synthesis nickel cobaltite
NiCo2O4 was prepared by the solvothermal method as reported in the previous literature.21 In this standard procedure, 0.147 g of Ni(NO3)2·6H2O and 0.293 g of Co(NO3)2·6H2O were combined in a 1:2 ratio and added to a solution containing 30 mL of distilled water and 10 mL of ethanol. Subsequently, 0.85 g of urea was introduced into the solution, and the mixture was vigorously stirred for 30 minutes using magnetic stirrers at room temperature to achieve a homogeneous solution. The resulting solution was then transferred to a Teflon-lined stainless steel autoclave with a 50 mL capacity. After sealing the autoclave, the mixture was heated at 100 °C for 6 h, and the autoclave was cooled down to room temperature. The precipitate was centrifuged four times with a 1:1 ratio of ethanol and distilled water and collected and kept for drying at 60 °C for 12 hours. The dried precipitate was further heated at 400 °C for 4 hours at a ramp rate of 2 °C per minute under ambient air conditions.
2.3. Preparation of NiCo2O4-APTES
The APTES modification of NiCo2O4 was carried out by the solution-phase deposition method.22 The reaction was carried out in an anhydrous solution to prevent uncontrollable polymerization in the presence of water.23,24 Initially, 500 mg of NiCo2O4 was transferred into a 1:1 mixture of ethanol and toluene. This solution was further refluxed at 90 °C under constant magnetic stirring, 30 mL ethanol was further added upon subsequent reduction in volume solution. This was repeated four times. To this mixture solution, an adequate amount of APTES was added and stirred for 30 minutes and followed by sonication for 4 hours and the product was washed several times with ethanol to remove the organic residue and dried in a vacuum oven for 12 hours.
2.4. Fabrication of a modified electrode
Carbon cloth (CC) having a geometrical surface area of 1 cm2 served as the working electrode. Onto the CC, 60 μL of the NiCo2O4-APTES dispersion in DMSO (1 mg mL−1) was drop cast. Following this, the CC was subjected to a 1 hour drying period in an oven and then utilized as the working electrode for electrochemical analysis. Platinum wire and the saturated calomel electrode were used as the counter electrode and reference electrode, respectively. All electrochemical experiments such as cyclic voltammetry (CV), current versus time plot and electrochemical impedance spectroscopy (EIS) were performed in a conventional three-electrode system on a CHI6038 electrochemical workstation.2.5. Material characterization
The surface morphology of the prepared materials was examined by scanning electron microscopy (SEM) using a Gemini SEM 300, Carl Zeiss, equipped with an energy dispersive spectrometer (EDS). The structural properties of the products were examined using Fourier-transform infrared (FTIR) spectroscopy using an IR Tracer-100, Shimadzu, in the wavenumber range of 400–4000 cm−1 on thin pellets of the sample made using the KBr method. Raman spectroscopy was performed using a Horiba LabRAM HR Evolution confocal Raman spectrometer equipped with a 532 nm laser. The crystalline nature and phase of the product were confirmed using powder X-ray diffraction (PXRD) studies with a Rigaku XRD SmartLab (source: CuKα). The presence of different elements and their chemical states were studied using X-ray electron photoelectron spectroscopy (XPS) with a PHI 5000 VersaProbe II, ULVAC-PHI Inc., equipped with a micro-focused (200 μm, 15 kV) monochromatic Al Kα X-ray source (hυ = 1486.6 eV).3. Results and discussion
3.1 Material characterization
The structure and morphology of NiCo2O4 were examined using SEM analysis and the corresponding images at different magnifications are given in Fig. 1. The sea urchin nanostructured shape of NiCo2O4 can be attributed to the presence of urea in the hydrothermal process, which adjust the pH to a mildly acidic range of 5.5 or 6.25 This particular pH can initiate the formation of both individual nanoneedles (Fig. 1(a and b)) and featuring small nanorods protruding from their centers (Fig. 1(c)). Upon meticulous examination of the structure, the aggregation of nanospheres played a pivotal role in the formation of nanoneedles as seen in Fig. 1(d). The continuous evolution of these nanostructured sphere bundles terminated in the captivating formation of sea urchin nanostructured spheres. The amino modification of NiCo2O4 did not cause a considerable change in the overall morphology of the system as shown in Fig. S1.† The distribution of NiCo2O4-APTES on CC after the modification was analysed by SEM images and it confirmed the successful distribution of NiCo2O4-APTES on CC (Fig. S2†). | ||
| Fig. 1 SEM images of (a) sea urchin nanostructured NiCo2O4. (b) Magnified image of the urchin model of NiCo2O4. (c) Needles of the sea urchin model of NiCo2O4. (d) Magnified image of nanoneedles showing the assembled nanospheres. | ||
The crystalline nature and phase properties were analysed by XRD analysis. The XRD spectrum of synthesized NiCo2O4 and surface functionalized NiCo2O4-APTES is given in Fig. 2(a), which indicates the fine crystalline nature of the prepared nanomaterials. The XRD pattern of NiCo2O4 matches well with previous reports of NiCo2O4.21,26 The high intense peak that appeared at 36.7° corresponds to the (311) diffraction plane for both spinel NiCo2O4 and NiCo2O4-APTES. Also, the fine diffraction peaks were observed at 2θ values of 19.1°, 31.1°, 44.6°, 59.1° and 65.0° in NiCo2O4 and NiCo2O4-APTES as well, which correspond to the (111), (220), (400), (511) and (440) diffraction planes of spinel NiCo2O4. This confirms the modification of APTES did not affect the structural features of NiCo2O4. Scherrer's equation (eqn (1) in the ESI†) was used to calculate the average crystallite size of both NiCo2O4 and NiCo2O4-APTES.27 The average crystallite size of NiCo2O4 and its APTES functionalized NiCo2O4 was calculated to be 10.32 nm and 9.82 nm, respectively, across all diffraction peaks (please refer to the ESI†). This observation again confirms that the modification had no discernible impact on the crystallinity of the material by APTES modification.
 | ||
| Fig. 2 (a) XRD diffraction peaks of NiCo2O4 and NiCo2O4-APTES. (b) FT-IR spectra of NiCo2O4 and NiCo2O4-APTES. (c) XPS survey spectrum of NiCo2O4 and NiCo2O4-APTES. | ||
The FTIR spectra shown in Fig. 2(b) confirm the formation of bonds between NiCo2O4 and APTES by the presence of characteristic absorption bands relevant to the bonding of APTES. The typical NiCo2O4 peaks arise at approximately 559 and 645 cm−1, corresponding to metal–oxygen (M–O) vibrations of NiCo2O4.28 The absorption bands at around 1625 cm−1 and 3422 cm−1 could be attributed to the O–H bending and stretching modes of the vibration of physisorbed H2O molecules. The adsorption peak at 450 cm−1 suggests the bending vibration of the siloxane (Si–O–Si) bond and the peaks at 1116 cm−1 and 1047 cm−1 correspond to the stretching vibrations of Si–O–Si for NiCo2O4-APTES.29,30 The IR bands for the N–H stretching and bending –NH2 groups are not apparent in the spectra because they overlap with the IR band of the –OH stretching and bending modes of vibrations. These IR absorption bands demonstrate that the silane coupling agent APTES was covalently bound to the NiCo2O4 surface.
The elemental composition and oxidation state elements in NiCo2O4 and NiCo2O4-APTES were tested and evaluated using XPS. The characteristic peaks of Ni, Co, O and C elements can be observed in the survey spectrum of the porous NiCo2O4 as well as the modified NiCo2O4-APTES. The surface functionalization with APTES can be clearly seen by the appearance of N 1s, Si 2s and Si 2p peaks in the survey spectra as seen in Fig. 2(c).31 The high resolution (HR) scans of Ni 2p, Co 2p, Si 2p and N 1s are shown in Fig. 3. Both the Ni 2p and Co 2p can be fitted into two spin–orbit doublets and two shakeup satellites by using a Gaussian fitting method. The HR scan of Ni 2p (Fig. 3(a)) shows two peaks at 853.78 eV and 871.37 eV corresponding to Ni3+ 2p3/2 and Ni3+ 2p1/2, respectively. Other strong peaks at 855.43 eV and 872.96 eV are observed which are characteristic of Ni2+ 2p3/2 and Ni2+ 2p1/2, respectively. Peaks at 861.64 eV and 880.04 eV correspond to the satellite peaks of Ni 2p3/2 and Ni 2p1/2 which are consistent with the reported articles.32,33 Similarly, in the HR scan of Co 2p spectrum (Fig. 3(b)), it can be seen that peaks at 779.46 eV and 794.42 eV correspond to the presence of Co3+ 2p3/2 and Co3+ 2p1/2 respectively, while the peaks at 780.95 eV and 795.94 eV can be assigned to the presence of Co2+ 2p3/2 and Co2+ 2p1/2 respectively. The spin–orbital splitting of Co 2p3/2 and 2p1/2 was found to be 15.01 eV with two weakly accompanied satellite peaks located at 790.07 eV and 804.16 eV, respectively which are well in agreement with the reported literature.32,34 The HR scan of Si 2p in NiCo2O4-APTES is shown in Fig. 3(c). After functionalization with APTES, the Si 2p spectrum exhibits a peak at 102.80 eV to Si–C from APTES bonded to O of the NiCo2O4 surface.35 Additionally, the HR spectrum of N 1s shows two peaks at 399.72 eV and 401.46 eV which could be attributed to amine groups and protonated amines, respectively.31,36,37
 | ||
| Fig. 3 HR XPS spectra of (a) Ni 2p and (b) Co 2p of NiCo2O4 and NiCo2O4-APTES. HR spectra of (c) Si 2p and (d) N 1s of NiCo2O4-APTES. | ||
3.2. Electrochemical analysis
The electrochemical analysis of bare CC, NiCo2O4 modified CC (NiCo2O4/CC), and NiCo2O4-APTES modified CC (NiCo2O4-APTES/CC) was analysed initially by CV analysis. The CV analysis was performed in the potential range of −0.2 to +0.6 V in a solution containing 0.1 M KCl and 10 mM [Fe (CN)6]3−/4− solutions at the scan rate of 100 mV s−1. As illustrated in Fig. 4(a), well-defined redox peaks were observed; however, the bare CC exhibited a subpar current response in contrast to the NiCo2O4/CC and NiCo2O4-APTES/CC. The current response of bare CC was increased after the deposition of the electroactive material NiCo2O4 over CC due to its conducting nature and the electrochemical activity was further enhanced after the APTES functionalization owing to the introduction of amino groups on the surface. The protonation of amino groups in the aqueous solution increases the easy electronic transport at the electrode–solution interface.38 | ||
| Fig. 4 (a) CV of bare CC, NiCo2O4/CC and NiCo2O4-APTES/CC in 0.1 M KCl and 10 mM [Fe (CN)6]3−/4− at 100 mV s−1. (b) Nyquist plots of bare CC, NiCo2O4/CC and NiCo2O4-APTES/CC in 0.1 M KCl and 10 mM [Fe (CN)6]3−/4− (inset: a simple Randles electrical circuit). | ||
Additionally, the amino functionalization of through silane coupling exhibited good electrochemical cycling stability in the potential window of −0.2 to +0.8 V in PBS and PBS containing MpM as depicted in Fig. S3(a) and (b)†, respectively. There is no significant change in the peak current and peak potential even after ten consecutive scans, and this signifies the reliability and durability of NiCo2O4-APTES for the electrochemical oxidation of MpM.
To examine the impedance variations and charge transfer characteristics at the electrode–electrolyte interface for differently modified electrodes, EIS analysis was performed in a 0.1 M KCl and 10 mM [Fe (CN)6]3−/4− redox system. As shown in Fig. 4(b), the Nyquist plot of bare CC, NiCo2O4/CC and NiCo2O4-APTES/CC which was fitted by a simple Randles electrical circuit. The diameter of the semicircular region of the Nyquist plot represents the electron transfer limited process, which is equivalent to the charge transfer resistance (Rct). It comes out that the Rct value of the CC electrode was decreased from 26.9 Ω to 18.8 Ω upon the modification of NiCo2O4 and further decreased to 10.7 Ω after the NiCo2O4-APTES modification. This suggests that the charger transfer resistance decreased about 3 times with the introduction of NiCo2O4-APTES over CC. The lowest Rct value of NiCo2O4-APTES/CC over NiCo2O4/CC depicts that the amine functionalization on NiCo2O4 improved the electron charge transfer and hence increased the conductivity which is complementary to the CV analysis.
In the same way, the capability of bare CC, NiCo2O4/CC and NiCo2O4-APTES/CC to oxidize MpM was also analyzed by using CV in the potential range of +0.2 to +0.8 V at a scan rate of 100 mV s−1. Fig. 5(a) shows the CV data of bare CC, NiCo2O4/CC and NiCo2O4-APTES/CC in 0.1 M PBS containing 100 μM MpM. It is shown that the anodic oxidation peak current for MpM was observed at 0.61 V and the peak current is higher for NiCo2O4-APTES/CC than for NiCo2O4/CC and bare CC. Hence, we have selected NiCo2O4-APTES/CC as a remarkable catalyst for the electrochemical detection of MpM. The electrochemical sensing capability of NiCo2O4-APTES/CC was further investigated by CV analysis in 0.1 M PBS containing various concentrations of MpM from 10 nM to 100 μM and the data are given in Fig. 5(b). The observed current responses exhibit a noticeable upward trend as the MpM concentration increases from 10 nM to 100 μM, thereby validating the electrocatalytic sensing efficacy of the NiCo2O4-APTES/CC electrode towards MpM oxidation.
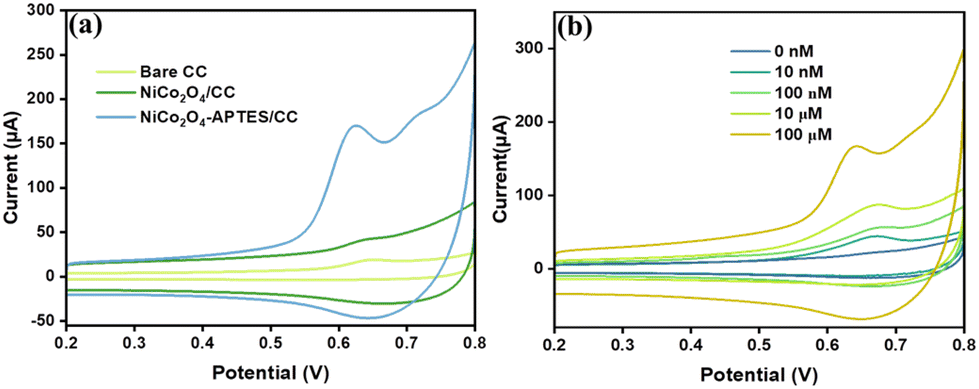 | ||
| Fig. 5 (a) CV of bare CC, NiCo2O4/CC and NiCo2O4-APTES/CC in 100 μM MpM solution and 0.1 M PBS at 100 mV s−1. (b) CV of NiCo2O4-APTES/CC at different concentrations of MpM in 0.1 M PBS at 100 mV s−1. | ||
A comprehensive analysis of the CV plot (Fig. 5(a)) provides detailed insights into the electrochemical oxidation of MpM. The oxidation peak of MpM at 0.61 V can be attributed to the presence of the enolic –OH group in the MpM molecule. Throughout the electrochemical oxidation process, the terminal keto group forms a hydrogen bond with the amino group on the NiCo2O4-APTES surface.12Scheme 2 illustrates the covalent bonding of the silanol group in APTES within the sea urchin shaped NiCo2O4, and the amine groups of APTES extending as a tail interacts with the incoming oxygen terminals of MpM, establishing hydrogen bonding. This hydrogen bonding significantly influences electron transfer between the NiCo2O4-APTES surface and MpM. Upon oxidation, the MpM molecule releases 2e− to the electrolyte and is converted to mycophenolic acid,11 and there is a gradual rise in the current response when the MpM concentration increases.
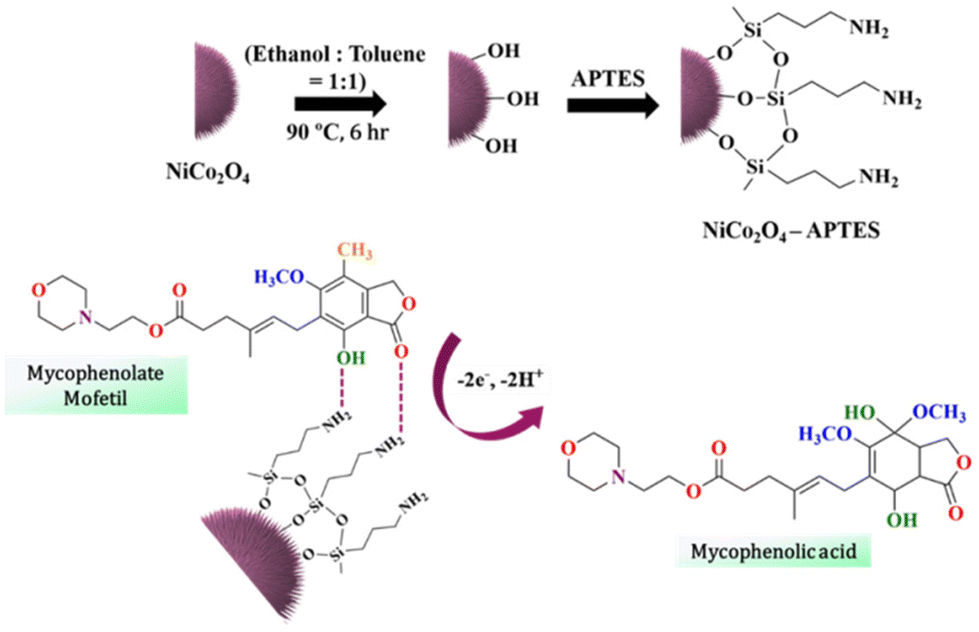 | ||
| Scheme 2 The schematics of the covalent bonding of the silanol group in APTES within the sea urchin shaped NiCo2O4 and the mechanism of electrochemical oxidation of MpM resulting in the formation of mycophenolic acid. | ||
To assess the pH dependent behavior of the electrochemical oxidation of MpM on the NiCo2O4-APTES/CC electrode, CV curves were obtained by varying the pH of electrolyte solution from 5 to 9 in the presence of 100 μM MpM at a scan rate of 100 mV s−1. The CV data of the NiCo2O4-APTES/CC electrode at different pH values is given in Fig. S4(a)† and it shows that the peak current was initially increased up to 7.1 and then decreased gradually by increasing the pH. A plot between the oxidation peak current (Ipa) and pH was made and it is given in Fig. S4(b).† From the data, we have selected the optimum pH of the electrolyte solution as 7.1, and it was used for all further electrochemical experiments. The optimum pH of 7.1 is near to the normal human body pH and hence the proposed sensor can be used for the detection of MpM from the human samples. Moreover, as the pH increased, the NiCo2O4-APTES/CC electrode's anodic potential changed to lesser positive potential values (Fig. S4(a)†), which suggests that protons (H+) are directly participating in the electro-oxidation reaction of MpM. Fig. S4(c)† shows the linear correlation between the anodic peak potential (Epa) and pH of the analyte solution with linear fitting equation of Epa(V) = −49.96pH + 95.81 (R2 = 0.9986). However, the slope value of ∼50 mV per pH is almost exactly equivalent to the theoretical Nernstian value of 59 mV per pH. It implies that the electrooxidation of MpM involves the transfer of the same number of electrons and protons.
The reaction kinetics of NiCo2O4-APTES/CC towards the electrochemical oxidation of MpM at different scan rates were studied from 20 mV s−1 to 200 mV s−1. The corresponding CV data are given in Fig. S5(a).† As observed in the figure, the anodic peak current was gradually increased with an increase in the scan rate. Fig. S5(b)† shows the relationship between the anodic peak current vs. square root of scan rate which shows a precise linear relationship with a high correlation coefficient (R2) value of 0.98237. In addition, a calibration curve was plotted between logcurrent vs. log scan rate (Fig. S5(c)†). The slope value of 0.49 of the calibration curve further confirms the diffusion-controlled electron transfer mechanism. This behavior suggests that the electrocatalytic oxidation of MpM on NiCo2O4-APTES/CC adheres to a standard diffusion-controlled electron transfer mechanism.
For quantitative detection of MpM using a NiCo2O4-APTES/CC electrode, the current vs. time analysis at a constant potential was conducted at a potential of 0.61 V, while varying the concentration of MpM from 0 nM to 100 μM. The response is given in Fig. 6(a) and it shows that the current value increases as the concentration of MpM increases within the range of 10 nM to 100 μM. From the current vs. time curve, a calibration curve was plotted, correlating the current value (at t = 8 s) with the concentration of MpM. This calibration plot displayed two distinct linear ranges spanning from 10 nM to 100 nM and from 1 μM to 100 μM with an impressive regression coefficient of 0.9902 and 0.9988, respectively, as shown in Fig. 6(b). The limit of detection (LOD) was determined from the calibration plot using the three-sigma method, and the LOD was found to be 1.23 nM (please refer to the ESI† for calculation). The heightened sensor response with a promising LOD of 1.23 nM was achieved with the developed sensor which can be attributed to the increased number of active sites, structural stability, and outstanding electrochemical properties arising from the NiCo2O4-APTES modified CC. A comparison of various electrochemical sensors developed for the detection of MpM is given in Table 1. Notably, the LOD achieved with the developed NiCo2O4-APTES modified CC sensor is exceptionally promising when contrasted with other electrochemical sensors developed for the electrochemical detection of MpM.
 | ||
| Fig. 6 (a) Current vs. time response of NiCo2O4-APTES/CC in 0.1 M PBS solution containing different concentrations of MpM ranging from 0 nM to 100 μM. The inset shows the response at concentrations of MpM ranging from 0 nM to 100 nM. (b) Calibration plot of the anodic peak current at t = 8 s vs. concentration of MpM. The inset graph contains a calibration plot of anodic peak current vs. concentration of MpM in the 10 nM to 100 nM range. | ||
| Electrode | Technique | Linear range | LOD (nM) | Ref. |
|---|---|---|---|---|
| DPV: differential pulse voltammetry; SWV: square wave voltammetry; and AASDPV: adsorptive anodic stripping differential pulse voltammetry. | ||||
| GCE/MWCNTs | DPV | 5–160 μM | 900 | 39 |
| CPE/MIP/MWCNTs | SWV | 9.9 nM–87 μM | 7.0 | 40 |
| GCE/Fe3O4/MWCNTs | AASDPV | 50 nM–200 μM | 9.0 | 41 |
| CuO/FCPE | CV | 10–80 μM | 150 | 11 |
| GCE | DPV | 500 nM–750 μM | 148 | 42 |
| Nb2CTx-APTES/CCY | DPV | 10–100 μM | 1000 | 12 |
| NiCo2O4-APTES/CC | CA | 10 nM–100 nM And 1–100 μM | 1.23 | This work |
The selectivity of the NiCo2O4-APTES/CC sensor for the detection of MpM was assessed in the presence of potential interference compounds typically found in real samples using current vs. time analysis. Fig. 7(a) illustrates the impact of these interference compounds on the electrochemical sensing capability of the NiCo2O4-APTES/CC even in the presence of a surplus of potential interfering analytes, including AA, DA, GA, KCl, NaCl, GLU, UA, GLA and ALN at a concentration of 10 times more than that of MpM. The same experiment was also carried out in the presence of some drugs which are in the same category as immunosuppressant drugs such as Tacrolimus and cyclosporine. As shown in Fig. 7(b), the selectivity plots illustrate that the NiCo2O4-APTES/CC exhibits the most significant current response to MpM compared to other immunosuppressant drugs. The corresponding current vs. time data are given in Fig. S6.† These results explicitly affirm the sensor's exceptional selectivity for detecting MpM in biological samples in the presence of interfering molecules.
 | ||
| Fig. 7 (a) Selectivity of MpM in the presence of different interfering analytes GA, GLA, ALA, AA, DA, GLU, KCl, UA, and the mixture. (b) selectivity of MpM in the presence of tacrolimus (Tac) and cyclosporine at a 10 times higher concentration than MpM. | ||
The reproducibility and long-term storage stability of the electrodes were systematically evaluated at regular intervals, extending up to three weeks. Three different NiCo2O4-APTES/CC electrodes were independently prepared and stored at 4 °C and the current vs. time analysis was performed after 7, 14 and 21 days. As observed in Fig. 8(a), the plots depict the storage stability assessments conducted at two distinct concentrations of MpM (1 μM and 100 μM) over the course of 21 days. The corresponding current vs. time plots are given in Fig. S7(a).† The repeatability of the NiCo2O4-APTES/CC electrodes was examined through consecutive runs of current vs. time plot with a 5-minute interval between measurements on the same electrode in a 100 μM MpM solution. As illustrated in Fig. 8(b), negligible changes in current responses were observed, with the standard deviation from 2.6 to 3.9% across three different electrodes. The corresponding current vs. time plots are given in Fig. S7(b).† The reproducibility of the sensor was evaluated by using three different NiCo2O4-APTES/CC electrodes and the corresponding current vs. time data are given in Fig. S7(c),† which shows the promising repeatability of the sensor with a standard deviation of 1.54%. These results underscore the proposed NiCo2O4-APTES/CC electrode sensor's excellent storage stability, reproducibility and repeatability, which further supports the reliability and promising performance of the sensor.
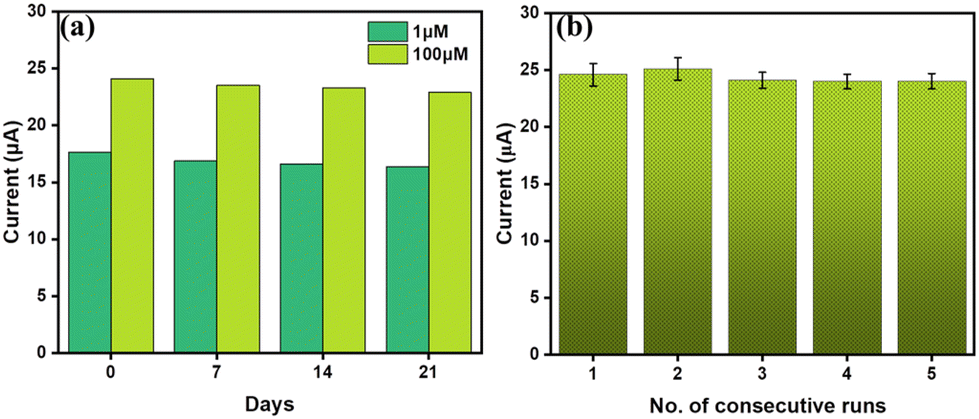 | ||
| Fig. 8 (a) Bar plot for the current vs. time curves showing the stability of different NiCo2O4-APTES/CC electrodes from 0 to 21 days in 1 μM and 100 μM MpM in 0.1 M PBS solution. (b) Bar plot of the CA curves for 5 consecutive runs. | ||
3.3. Real sample analysis
The practical applications of the NiCo2O4-APTES/CC electrochemical sensor were evaluated by investigating its performance in detecting MpM within artificial blood serum samples and cerebrospinal fluid (CSF). The experiments were conducted under optimal conditions, involving a tenfold dilution of the serum using 0.1 M PBS solution without any additional treatments. The current vs. time responses of the NiCo2O4-APTES/CC in spiked serum and CSF samples of known amounts were thoroughly examined and compared to responses obtained in 0.1 M PBS solution. The obtained data are presented in Fig. 9(a) and (c), providing a clear representation of the sensor's efficacy in real-time MpM detection in blood serum samples and CSF. Similar to the experiments in PBS, the serum experiment shows two linear ranges of 10 nM to 100 nM and 1 μM to 100 μM as displayed in Fig. 9(b) with regression coefficients (R2) of 0.9956 and 0.9894, respectively. Similarly, the real sample experiment with CSF also shows a similar linear range with R2 values of 0.9855 and 0.9960, respectively (Fig. 9(d)). The LODs of the sensor in serum and CSF experiments were calculated and found to be 2.47 nM and 1.47 nM, respectively. This compelling evidence underscores the NiCo2O4-APTES/CC sensor's outstanding capability for accurate and real-time detection of MpM in complex biological samples. | ||
| Fig. 9 Analytical performance of the NiCo2O4-APTES/CC sensor in CSF and serum. (a) Current vs. time analysis in the concentration range of 0 to 100 μM in MpM containing CSF. The inset shows the response at concentrations of MpM ranging from 0 nM to 100 nM. (b) calibration plot of the concentration of MpM in CSF vs. anodic peak current at t = 8 s (inset graph contains a calibration plot of the concentration in the lower (nM) range of MpM vs. current). (c) Current vs. time analysis of MpM in blood serum diluted in 0.1 M PBS solution varying in the range of 0 to 100 μM. The inset shows the response at concentrations of MpM ranging from 0 nM to 100 nM. (d) calibration plot of the concentration of MpM in blood serum vs. anodic peak current at t = 8 s (inset graph contains a calibration plot of the concentration in the lower (nM) range of MpM vs. current). | ||
3.4. Evaluation of the sensor prototype
We developed a prototype device with the proposed NiCo2O4-APTES/CC electrode sensor for the on-site detection of MpM. NiCo2O4-APTES/CC was taken as the working electrode, Ag/AgCl wire was taken as the reference electrode, and a bare carbon cloth thread was taken as the counter electrode. These electrodes were evenly spaced on a locally available band aid patch and firmly placed using adhesive tape to ensure that the patch is leak-proof (Fig. 10(a)). The sensing capability of the prototype was evaluated by current vs. time analysis by connecting this prepared patch prototype sensor to a portable potentiostat (Palmsense EmStat4S.LR). The current vs. time analysis responses of the prototype towards the oxidation of MpM are depicted in Fig. 10(b) and it shows that the current value increases with increasing concentration of MpM from 10 nM to 100 nM. A calibration plot between the current (at t = 8 s) and the concentration of MpM is demonstrated in Fig. 10(c), which shows a good linear range with an R2 value of 0.99541. The LOD of the prototype was found to be 6.8 nM which shows its potential application in POC devices for the onsite detection of MpM. | ||
| Fig. 10 (a) Schematics of the fabrication of the band-aid based prototype sensor patch and its connection to the portable potentiostat. (b) Current vs. time analysis conducted in the range of 0 to 100 nM MpM. (c) Calibration plot between the current (at t = 8 s) and the concentration of MpM. | ||
4. Conclusion
In summary, a low cost material comprising NiCo2O4 and APTES was synthesized and employed as a flexible electrochemical platform for the sensitive and selective detection of MpM from biological samples. The surface tailoring of NiCo2O4 with APTES significantly enhanced the charge transfer and stability, resulting in an outstanding electrocatalytic performance for MpM oxidation, covering a broad linear concentration range (0–100 μM) and achieving an impressively low detection limit of 1.23 nM. Furthermore, the modified NiCo2O4-APTES/CC electrode displayed significant selectivity, stability, repeatability, and reproducibility in MpM analysis. The sensor was successful in detecting MpM in artificial serum samples as well as CSF medium, which suggests the real-time application of the proposed sensor. A carbon cloth-based flexible device prototype was also successfully built and tested, displaying exceptional sensing capabilities and offering great promise for application in POC devices for biological and clinical diagnostics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
PAR acknowledges the Ramalingaswami re-entry fellowship (BT/RLF/Re-entry/75/2020) from the Department of Biotechnology (DBT), Govt. of India. MA acknowledges the PMRF fellowship (31202903) from the Govt. of India. The authors acknowledge the Central Instrumentation Facility, Indian Institute of Technology Palakkad for material characterization.References
- M. Naesens, D. R. Kuypers and M. Sarwal, Clin. J. Am. Soc. Nephrol., 2009, 4, 481–508 CrossRef CAS PubMed.
- L. C. Martial, R. E. Aarnoutse, M. F. Schreuder, S. S. Henriet, R. J. Brüggemann and M. A. Joore, PLoS One, 2016, 11, e0167433 CrossRef.
- M. Sahman, S. Mugosa and N. Rancic, Front. Public Health, 2021, 9, 671316 CrossRef.
- C. Ponticelli and R. J. Glassock, J. Nephrol., 2019, 32, 851–870 CrossRef PubMed.
- T. Madrakian, M. Soleimani and A. Afkhami, Mater. Sci. Eng., C, 2014, 42, 38–45 CrossRef CAS PubMed.
- M. M. Tielemans, G. A. J. van Boekel, T. van Gelder, E. T. Tjwa and L. B. Hilbrands, Transplant. Rev., 2019, 33, 55–63 CrossRef PubMed.
- M. Ashjari, H. Karimi-Maleh, F. Ahmadpour, M. Shabani-Nooshabadi, A. Sadrnia and M. A. Khalilzadeh, J. Taiwan Inst. Chem. Eng., 2017, 80, 989–996 CrossRef CAS.
- M. Plätzer, K. Jahn, J. Wohlrab and R. H. Neubert, J. Chromatogr. B: Biomed. Sci. Appl., 2001, 755, 355–359 CrossRef.
- U. D. Renner, C. Thiede, M. Bornhäuser, G. Ehninger and H.-M. Thiede, Anal. Chem., 2001, 73, 41–46 CrossRef CAS.
- M. H. Mahnashi, A. M. Mahmoud, S. A. Alkahtani, R. Ali and M. M. El-Wekil, Anal. Bioanal. Chem., 2020, 412, 355–364 CrossRef CAS PubMed.
- G. S. Sumanth, B. E. K. Swamy and K. Chetankumar, Mater. Chem. Phys., 2023, 307, 128118 CrossRef CAS.
- P. V. Vaishag, M. Ankitha and P. A. Rasheed, ACS Appl. Nano Mater., 2023, 6, 23324–23331 CrossRef CAS.
- M. Ankitha, P. V. Vaishag, P. Muhsin and P. A. Rasheed, J. Ind. Eng. Chem., 2024, 131, 449–458 CrossRef CAS.
- M. Ankitha, F. Shamsheera and P. A. Rasheed, ACS Appl. Electron. Mater., 2024, 6, 599–610 CrossRef CAS.
- A. S. Agnihotri, A. Varghese and M. Nidhin, Appl. Surf. Sci. Adv., 2021, 4, 100072 CrossRef.
- X. B. Joseph, S. Kogularasu, S.-F. Wang and J.-K. Sheu, ACS Appl. Nano Mater., 2021, 4, 12788–12797 CrossRef CAS.
- R. R. Poolakkandy, A. R. Neelakandan, M. F. Puthiyaparambath, R. G. Krishnamurthy, R. Chatanathodi and M. M. Menamparambath, J. Mater. Chem. C, 2022, 10, 3048–3060 RSC.
- H. Liu and G. Jin, Thin Solid Films, 2023, 769, 139738 CrossRef CAS.
- M. Khan, X. Liu, J. Zhu, F. Ma, W. Hu and X. Liu, Biosens. Bioelectron., 2018, 108, 76–81 CrossRef CAS PubMed.
- W. H. Zheng, C. Yan, T. Chen and D. Z. Kang, J. Physiol. Pharmacol., 2020, 71, 919–925 CAS.
- R. Zou, K. Xu, T. Wang, G. He, Q. Liu, X. Liu, Z. Zhang and J. Hu, J. Mater. Chem. A, 2013, 1, 8560–8566 RSC.
- A. S. Agnihotri, C. G. Ann Maria, A. Varghese, P. Mane, B. Chakraborty and M. Nidhin, Surf. Interfaces, 2022, 35, 102406 CrossRef CAS.
- N. S. K. Gunda, M. Singh, L. Norman, K. Kaur and S. K. Mitra, Appl. Surf. Sci., 2014, 305, 522–530 CrossRef CAS.
- M. Sypabekova, A. Hagemann, D. Rho and S. Kim, Biosensors, 2023, 13, 36 CrossRef CAS PubMed.
- D. Guragain, C. Zequine, T. Poudel, D. Neupane, R. Gupta and S. Mishra, J. Nanosci. Nanotechnol., 2020, 20, 2526–2537 CrossRef CAS PubMed.
- S. Khalid, C. Cao, L. Wang and Y. Zhu, Sci. Rep., 2016, 6, 22699 CrossRef CAS PubMed.
- M. M. Rahman, T. A. Sheikh, A. M. Asiri and M. R. Awual, New J. Chem., 2019, 43, 4620–4632 RSC.
- M. Kaur, P. Chand and H. Anand, Chem. Phys. Lett., 2022, 786, 139181 CrossRef CAS.
- J. Liu, J. Wang, Y. Zhang, W. Zheng, Y. Yao, Q. Liu, X. Zhang, Y. Yang and X. Wang, ACS Catal., 2023, 13, 14737–14745 CrossRef CAS.
- B. K. Sodipo and A. A. Aziz, Beilstein J. Nanotechnol., 2014, 5, 1472–1476 CrossRef PubMed.
- H. H. Kyaw, S. H. Al-Harthi, A. Sellai and J. Dutta, Beilstein J. Nanotechnol., 2015, 6, 2345–2353 CrossRef CAS PubMed.
- X. Xiao, X. Zhang, Z. Zhang, J. You, S. Liu and Y. Wang, Microchim. Acta, 2020, 187, 1–9 CrossRef PubMed.
- C. Pan, Z. Liu, W. Li, Y. Zhuang, Q. Wang and S. Chen, J. Phys. Chem. C, 2019, 123, 25549–25558 CrossRef CAS.
- Y. Pan, W. Zeng, L. Li, Y. Zhang, Y. Dong, D. Cao, G. Wang, B. L. Lucht, K. Ye and K. Cheng, Nano-Micro Lett., 2017, 9, 1–9 CrossRef CAS.
- G. G. Lara, G. F. Andrade, M. F. Cipreste, W. M. da Silva, P. L. Gastelois, D. A. Gomes, M. C. de Miranda, W. A. de Almeida Macedo, M. J. Neves and E. M. B. de Sousa, J. Mater. Sci.: Mater. Med., 2018, 29, 130 CrossRef PubMed.
- R. G. Acres, A. V. Ellis, J. Alvino, C. E. Lenahan, D. A. Khodakov, G. F. Metha and G. G. Andersson, J. Phys. Chem. C, 2012, 116, 6289–6297 CrossRef CAS.
- M. Šetka, R. Calavia, L. Vojkůvka, E. Llobet, J. Drbohlavová and S. Vallejos, Sci. Rep., 2019, 9, 8465 CrossRef PubMed.
- M. Z. H. Khan, X. Liu, J. Zhu, F. Ma, W. Hu and X. Liu, Biosens. Bioelectron., 2018, 108, 76–81 CrossRef CAS PubMed.
- T. Madrakian, M. Soleimani and A. Afkhami, Mater. Sci. Eng., C, 2014, 42, 38–45 CrossRef CAS PubMed.
- H. Momeneh and M. B. Gholivand, Anal. Biochem., 2018, 557, 97–103 CrossRef CAS PubMed.
- M. B. Gholivand and M. Solgi, Anal. Biochem., 2017, 520, 1–8 CrossRef CAS PubMed.
- S. N. Prashanth, K. C. Ramesh and J. Seetharamappa, Int. J. Electrochem., 2011, 2011, 193041 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4an00592a |
| This journal is © The Royal Society of Chemistry 2024 |