
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWireless rotating bipolar electrochemiluminescence for enzymatic detection†
Chunguang
Li
a,
Minghui
Feng
a,
Dalibor
Stanković
 b,
Laurent
Bouffier
c,
Feifei
Zhang
*a,
Zonghua
Wang
a and
Neso
Sojic
*c
b,
Laurent
Bouffier
c,
Feifei
Zhang
*a,
Zonghua
Wang
a and
Neso
Sojic
*c
aCollege of Chemistry and Chemical Engineering, Shandong Sino-Japanese Center for Collaborative Research of Carbon Nanomaterials, Instrumental Analysis Center of Qingdao University, Qingdao University, Qingdao 266071, China. E-mail: zhangfeifei@qdu.edu.cn
bUniversity of Belgrade – Faculty of Chemistry, Studentski trg 12-16, 11000 Belgrade, Serbia
cUniv. Bordeaux, Bordeaux INP, CNRS, UMR 5255, 33607 Pessac, France. E-mail: sojic@u-bordeaux.fr
First published on 27th March 2024
Abstract
New dynamic, wireless and cost-effective analytical devices are developing rapidly in biochemical analysis. Here, we report on a remotely-controlled rotating electrochemiluminescence (ECL) sensing system for enzymatic detection of a model analyte, glucose, on both polarized sides of an iron wire acting as a bipolar electrode. The iron wire is controlled by double contactless mode, involving remote electric field polarization, and magnetic field-induced rotational motion. The former triggers the interfacial polarization of both extremities of the wire by bipolar electrochemistry, which generates ECL emission of the luminol derivative (L-012) with the enzymatically produced hydrogen peroxide in presence of glucose, at both anodic and cathodic poles, simultaneously. The latter generates a convective flow, leading to an increase in mass transfer and amplifying the corresponding ECL signals. Quantitative glucose detection in human serum samples is achieved. The ECL signals were found to be a linear function of the glucose concentration within the range of 10–1000 μM and with a limit of detection of 10 μM. The dynamic bipolar ECL system simultaneously generates light emissions at both anodic and cathodic poles for glucose detection, which can be further applied to biosensing and imaging in autonomous devices.
Introduction
Bipolar electrochemistry (BPE) is a wireless technique that induces remote polarization of conducting objects named bipolar electrodes (BEs) through the application of an external voltage between two feeder electrodes.1–3 If the electric field imposed across an electrolyte is high enough, oxidation reactions occur at the anodic pole of the BE simultaneously with reduction reactions at the cathodic pole. BPE has been exploited to initiate other functionalities such as light emission, as for example electrochemiluminescence (ECL) and LED (light-emitting diode) readouts.4–8 In particular, bipolar ECL (BP-ECL) is a phenomenon where luminescence is generated through redox reactions involving a luminophore and a co-reactant and producing light at the surface of BEs.9–14 Thus, ECL generates an optical readout opening various bioanalytical and microscopy opportunities.15–21 In the analytical applications of ECL, two main strategies were employed in the open bipolar configurations: (i) the analytical target is involved directly or indirectly in the ECL mechanism and the ECL intensity generated at one pole of the BE is proportional to its concentration; (ii) the redox reaction of the analyte occurs at a given pole named sensing pole and is coupled with the counter redox reaction generating the ECL signal at the opposite pole named reporting pole.11,22 This latter strategy was introduced by Crooks and co-workers.23,24In dynamic BP-ECL systems, the polarization of the BE is regulated by the electric field, while the wireless motion of these electrodes is controlled by two primary ways.25–29 The first one involves the production of gas bubbles (H2 or O2) at the electrode surface, which is commonly employed to activate the translation, rotation, and vertical motion of the BE.27,28 These systems have been reported for the selective and sensitive detection of different analytes.26–28 The second mechanism, which is the focus of this study, relies on the utilization of an external magnetic field.25 The manipulation of objects by magnetic fields has been proposed for chemical and biochemical sensing due to its contactless, versatile, and cost-effective nature.30,31
Imaging the motion of BP-ECL systems can be easily achieved with commercial cameras or smartphones due to the high intensity of the emitted light and negligible background light interference for rapid, portable, and user-friendly detection.32–35 For instance, a BP-ECL array aptasensor was fabricated for detecting aflatoxin M1 concentrations using a smartphone as a photodetector.36 In addition to static BEs, the motion of the electrode can also be monitored with a digital camera. For example, a wireless rotating BP-ECL device was developed with a gold-coated iron wire BE system that employs both magnetic and electric fields for a double contactless approach.25
Luminol ECL possesses high luminescent quantum yield and low oxidation potential, making it an excellent luminophore in analytical chemistry.37–43 The sensitive ECL-based detection of hydrogen peroxide (H2O2) through its reaction with luminol has been extensively used in immunosensing, disease diagnosis, cancer cell imaging, and other bio-related fields.5,44–48 Hydrogen peroxide is an effective co-reactant of luminol ECL as well as a byproduct of numerous enzymatic reactions in biological systems.49–51 However, the use of luminol is limited to alkaline conditions due to poor solubility at neutral pH, impeding its wider application in biological research.43,52 Consequently, analogue molecules with a similar chemical structure and stability in physiological conditions have been reported.52–54 Among them, L-012 (8-amino-5-chloro-7-phenylpyrido[3,4-d]pyridazine-1,4(2H,3H) dione), exhibits a bright ECL emission at physiological pH values, possesses a low oxidation potential together with a long luminescent lifetime.53,55–58 Luminol and L-012 primarily exhibit ECL at the anode, initiated by oxidation reactions. However, in recent years, several authors reported the cathodic ECL emission in various conditions,55,59,60 such as with catalytic nanomaterials, e.g. zinc oxide nanoparticle-modified Ni-foam,61 carbonized polydopamine nanotubes,62 catalase-like CoO nanorods,63 acetylene black oxide,64 two-dimensional NiM metal–organic framework (MOF, with M = Fe, Co, and Zn).65 By adjusting iron single atom structures, the enhancement mechanism of luminol cathodic ECL was investigated and found to involve the production of massive reactive oxygen species for the generation of luminol radicals.66–68 In a recent study, bright L-012 emission from a simple iron wire was observed in a wireless bipolar ECL configuration, which displayed both cathodic and anodic ECL emission with the naked eye.55 This appealing finding of ECL emission generated both in oxidation and reduction provides the basis for potential applications in biosensing, remote detection, and bipolar ECL analysis.
In this study, we present an original biosensing approach based on a wireless rotating open BP-ECL enzymatic system for glucose detection on both cathodic and anodic poles of a BE made of an iron wire (Scheme 1). External magnetic and electric fields were combined with ECL readout to induce rotation of the BE and ensure regular convection within the solution. It accelerates the mass transfer process and results in an increase of the ECL intensity emitted by the rotating iron wire electrode. Under physiological pH conditions, glucose oxidase (GOD) selectively catalyzes the oxidation of glucose and the reduction of O2 into H2O2. In the meantime, H2O2 acts as a coreactant of L-012 ECL, promoting its blue light emission. The quantitative detection of glucose is achieved by monitoring the ECL intensity.
 | ||
| Scheme 1 The principle of BP-ECL biosensing system for glucose detection with two sensing poles. Schematic view of the experimental device with the reaction principle of L-012/GOD/glucose system. The rotating bipolar electrode generates ECL emission on both anodic and cathodic poles. The inset shows the molecular structure of L-012. | ||
Experimental
All chemicals were of analytical grade and used as received. Enzymes were stored at −20 °C, did not require purification, and could be used directly. Solutions were prepared using a Milli-Q ultrapure water instrument (resistivity of 18.2 MΩ cm). Hydrogen peroxide (H2O2), glucose, sodium phosphate monobasic (Na2HPO4·2H2O), and disodium hydrogen phosphate dodecahydrate (NaH2PO4·12H2O) were purchased from Sinopharm Chemical Reagent Co., Ltd. GOD was purchased from Aladdin Reagent Co., Ltd. L-012 sodium salt (inset in Scheme 1) was purchased from Sigma-Aldrich. Stainless steel plates were used as the feeder electrode material (Magnetism Materials Co., Ltd, 20 mm × 20 mm × 1 mm), and iron wire was used as the BE (with a diameter of 200 μm and a length of 20 mm). A power supply (GWINSTEK PSR 60–6, Programmable Power Supply, 60 V, 6 A, 150 W) was used to apply a DC voltage between the two feeder electrodes. A rotational movement was generated using a magnetic stirrer (ThermoFisher Scientific). BP-ECL images and videos were recorded using a commercial digital camera (Canon EOS 6D Mark II, Objective Canon Macro Lens, 100 mm, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.8). Data analysis was processed using Image J software (AVI encoding).
:2.8). Data analysis was processed using Image J software (AVI encoding).
The enzymatic reaction was conducted prior to mixing the enzyme system with the ECL luminophore. For the L-012/GOD system, 20 U mL−1 GOD was mixed with different concentrations of glucose in 0.1 M phosphate buffer (pH 7.4) and incubated at 37 °C for 30 min. After the enzymatic reaction, the enzymatic system was supplemented with the luminescent reagent (L-012), and subsequently, the pH was adjusted using a NaOH solution.
A wireless rotating open BP-ECL biosensing system was constructed with an iron wire serving as the BE. The images and videos were processed using Image J software (AVI encoding) and ultimately converted to WMV format. The average intensity of the ECL signal generation area was calculated using Image J.
The blue ECL signal is visible with the naked eye and ECL emission occurs at both the cathode and anode of the wire. The camera is set to capture 24 frames per second in these imaging experiments, where each frame represents a 37.5° angular displacement of the wire. ImageJ software is utilized for image processing. To show the stability of the emitted light signal in the wireless rotating BP-ECL sensing configuration, the average ECL intensity of each video snapshot is documented. We recorded ECL signals for 10 seconds, which corresponds to capturing 240 frames as the wire completes 25 revolutions (i.e. 150 rpm). To demonstrate the reproducibility of the light emission signal variations, the average value of light intensity is calculated for each video snapshot.
Results and discussion
The principle of the dynamic sensing approach based on BP-ECL is presented in Scheme 1. To serve as the BE, a 2 cm-long iron wire with a diameter of 200 μm was placed between two feeder electrodes (schematized as black rectangles in Fig. 1a–d), and a uniform electric field was generated by applying a DC voltage between them. The surface of the iron wire and the surrounding solution would produce a potential difference gradient, leading to a polarization difference with a maximum amplitude at the opposite poles of the iron wire. Redox reactions occur at the ends of these BEs when the polarization voltage reaches a threshold value.4 A magnetic stirrer is placed under the bipolar cell to induce the rotation of the iron wire in the horizontal plane located at the bottom of the cell, leading to a regular stirring of the electrolyte.25 The ECL signal is ultimately detected using a commercial digital camera. | ||
| Fig. 1 (a and d) Top-view optical images of the iron wire used as a bipolar electrode and placed between two feeder electrodes before the application of the electric field. (b and e) Top-view of static and rotating (150 rpm) iron wires, respectively, under an electric field of 10 V cm−1 in the dark. (c and f) Horizontal ECL intensity profiles for static and rotating iron wires, respectively. The longitudinal axis of the wire is initially oriented parallel to the applied electric field (i.e. θ = 0°). The solution is composed of 1 mM glucose, 20 U mL−1 GOD and 1 mM L-012. | ||
The L-012/GOD system, depicted in Scheme 1, is utilized to convert enzymatically dioxygen (O2) into H2O2 during the enzymatic reaction. The concentration of H2O2 is directly proportional to the glucose substrate and the produced H2O2 acts as a co-reactant of L-012. Under neutral or alkaline conditions, L-012 is electrochemically oxidized to form an intramolecular diaza-quinone, which is then oxidized by H2O2 (HOO˙ or superoxide radical O2˙−) to generate the excited state. Upon de-excitation, it emits light with a typical blue color.
The longitudinal axis of the BE was initially oriented parallel to the electric field lines (i.e. θ = 0°). Application of an electric field of 10 V cm−1 resulted in a blue ECL emission at both cathodic and anodic regions of the BE (Fig. 1b and e, before and during rotation), which was visible to the naked eye. As reported previously,25 maximum ECL intensity on both poles is obtained when θ = 0. Indeed, the polarization difference is maximal in this position. The ECL intensity appears to be more intense at the anodic pole compared to the cathodic one. This means that under these experimental conditions, the anodic ECL mechanism is indeed more efficient. Upon applying a rotation speed of 150 rpm, the ECL emission from the anode of the BE was significantly enhanced whereas the cathodic emission is less intense (Fig. 1f). The ECL emission from the cathode of the BE was less intense probably due to the involvement of surface species55 in this ECL mode contrarily to the anodic pathway which purely involves diffusive species whose transport is enhanced by the stirring. Fig. 1c and f display the intensity distribution of ECL alongside the electrodes (i.e. axial profile) for stationary and rotating configurations, respectively. Under a voltage of 60 V (i.e. 10 V cm−1), the polarization threshold required for ECL emission is reached at both poles of the BE and with a maximum intensity peaking at an axial position of 5 mm and 18 mm for the cathodic and anodic pathways, respectively (static electrode). However, during rotation at 150 rpm, the polarization threshold required to trigger ECL emission is observed in oxidation with an almost constant intensity from 13 mm and 19 mm and in reduction at 2 mm. A better understanding of the L-012 mechanism in reduction is required and may guide alternative analytical approaches to enhance the cathodic ECL emission.
The magnetic field induces the continuous stirring of the wire, and the application of the electric field leads to the blue ECL signal as recorded by a commercial digital camera (Video S1†). Fig. S1† depicts the time-dependent variations in ECL intensity at the anode end of the wire during rotation for a glucose concentration of 0.5 and 1 mM. The ECL emission is initiated upon application of the electric field at t = 6 s and for 10 s. A series of ECL transients are observed as soon and as long as the electric field is applied (Fig. S1a and b†). The intensity of the transients appears to be quite uniform with a peak height of 37 ± 3 a.u. and 62 ± 32 a.u. for 0.5 mM (Fig. S1c†) and 1 mM of glucose (Fig. S1d†), and the relative standard deviations for 1 mM of glucose was 3.3%. The advantage of the method is the generation of multiple measurement points that can be used to perform a statistical analysis. Also, the ECL signal is found to be very stable and reliable, and the amplitude of the signal is clearly correlated to the analyte concentration.
The experimental conditions were optimized to investigate the variations of the ECL signal. Optimization of the applied voltage and rotation speed of the L-012/GOD system was initially performed. As previously observed, L-012 produces blue light at both cathodic and anodic poles of the BE, with the ECL intensity at the anode being significantly stronger than that at the cathode.55 Thus, the influence of the applied voltage and rotation speed of the system on the ECL intensity at the cathodic end of BE was primarily examined, as shown in Fig. S2.† Fig. S2a† reports the correlation between the applied voltage and the maximal ECL intensity on the BE. The observed trend shows a progressive rise in ECL intensity with an increasing electric field. A first local maximum is observed at about 40 V followed by a main maximum at 60 V before a rapid decrease of the signal. This trend probably reflects two sequential mechanistic pathways that do not involve the same electrogenerated species depending on the driving force. Fig. S2b† reports the variation in cathodic ECL peak values with a rotation speed varying from 0 to 250 rpm. The ECL intensity first increases to reach a maximum at 150 rpm before a progressive decrease. Furthermore, we studied the influence of the GOD concentration since it should directly impact the ECL intensity. This was performed with 1 mM of glucose and the ECL intensity displayed regular calibration curves obtained both at the anodic and cathodic ends (Fig. 2). As already observed, ECL on the anodic side is much stronger than on the cathodic one. An increase of the signal up to 15 U mL−1 is observed first before a plateau reflecting a steady response at larger GOD concentration (20 or 25 U mL−1). Such a plateau indicates that a maximum amount of H2O2 is reached for the glucose-GOD reaction. Consequently, the quantitative determination of glucose was carried out under these optimized conditions, which included an applied voltage of 60 V, a rotating speed of 150 rpm, and a GOD concentration of 20 U mL−1.
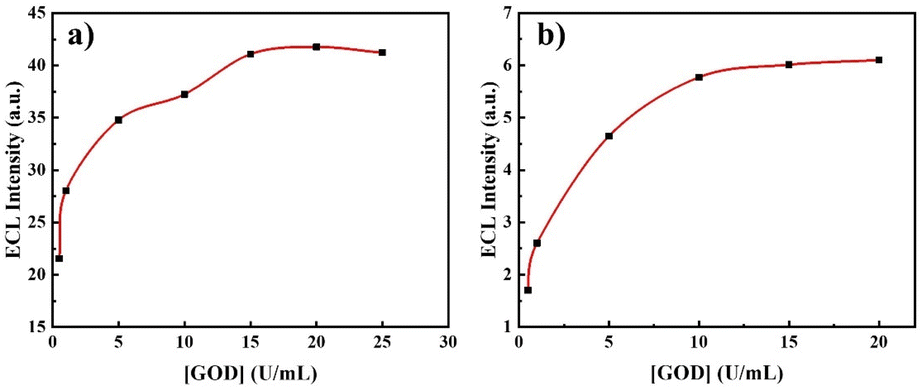 | ||
| Fig. 2 Variations of the ECL peak intensity with the concentration of GOD at the (a) anodic and (b) cathodic ends of the BE. The solution consists of 1 mM L-012, 1 mM glucose, and different contents of GOD (0.5, 1, 5, 10, 15, 20 and 25 U mL−1). The rotation is set at 150 rpm. | ||
Under the optimal experimental conditions, we performed the glucose sensing and the quantitative results obtained for the cathode and the anode are reported in Fig. 3. Once again, the ECL signals were recorded on the two poles of the wire and by varying the glucose concentration in the range from 0 to 5000 μM. However, the ECL response was linear only up to 1000 μM. The integrated values of ECL intensity were found to have a linear relationship with glucose concentration within the range of 10–1000 μM. The limits of detection were 10 μM for both cathodic and anodic ECL modes. At the anode of the bipolar electrode, the linear equation between glucose concentration (x/μM) and the integrated ECL intensity (y) was y = 0.0909x + 15.625, R2 = 0.985, whereas at the cathode, the linear equation was determined as y = 0.0318x + 4.268, R2 = 0.994.
 | ||
| Fig. 3 Quantitative determination of glucose. The linear relationship between glucose concentration and ECL intensity at the (a) anodic and (b) cathodic ends of the BE, with a rotation speed of 150 rpm. The composition of the solution is 1 mM L-012, 20 U mL−1 GOD, and different concentrations of glucose (0, 0.01, 1, 10, 50, 250, 500, 1000, 2500, and 5000 μM). | ||
To evaluate the reliability of the method to detect glucose in real samples, a recovery experiment was performed. Human serum samples were diluted 100-fold and were spiked with different concentrations of glucose (100, 200, and 500 μM) for ECL detection (Tables 1 and 2). The recovery rate was calculated as shown in the ESI.† These findings demonstrate the high accuracy and feasibility of the method for detecting glucose in pharmaco-relevant samples.
| Sample number | Original sample glucose content (μM) | Addition of glucose (μM) | Estimated value (μM) | Recovery rate (%) |
|---|---|---|---|---|
| 1 | 34.1 | 100.0 | 117.0 | 81.9 ± 1.0 |
| 2 | 200.0 | 227.4 | 96.6 ± 0.9 | |
| 3 | 500.0 | 580.6 | 108.9 ± 0.6 |
| Sample number | Original sample glucose content (μM) | Addition of glucose (μM) | Estimated value (μM) | Recovery rate (%) |
|---|---|---|---|---|
| 1 | 31.2 | 100.0 | 124.7 | 105.7 ± 0.4 |
| 2 | 200.0 | 225.5 | 102.4 ± 0.1 | |
| 3 | 500.0 | 434.4 | 82.8 ± 0.1 |
Conclusions
In this work, we report the rapid detection of glucose in serum based on the anodic and cathodic BP-ECL emission originating from L-012 dye. In this assay, GOD converts glucose substrate while molecular oxygen is reduced to H2O2. The latter acts as a sacrificial coreactant for L-012 ECL at physiological pH. The main originality involves a double remote addressing of the electrode by applying a constant electric field and also a continuous homogenization of the solution by using magnetic stirring. A low-cost iron wire was used as the electrode material for ECL sensing. The external magnetic field was introduced during the experiment to induce rotation of the wire, which enhanced the ECL signal by ensuring a forced convection. Furthermore, the rotation resulted in more uniform reaction solution, thus avoiding experimental variability. The same underlying principle can be potentially applied to smaller conductive objects and extended for designing multifunctional systems. There is potential for utilizing this method in dynamic enzyme detection or immunoassays, generating numerous opportunities for future development.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the support from the Natural Science Foundation of Shandong Province (ZR2021MB069, ZR2020MB063), the Taishan Scholar Project of Shandong Province (ts201511027) and the Sino-French International Research Network ELECTROSENS (CNRS).References
- S. E. Fosdick, K. N. Knust, K. Scida and R. M. Crooks, Angew. Chem., Int. Ed., 2013, 52, 10438–10456 Search PubMed.
- K. L. Rahn and R. K. Anand, Anal. Chem., 2021, 93, 103–123 CAS.
- L. Bouffier, D. Zigah, N. Sojic and A. Kuhn, Annu. Rev. Anal. Chem., 2021, 14, 65–86 CAS.
- E. Villani and S. Inagi, Anal. Chem., 2021, 93, 8152–8160 CAS.
- E. Villani, N. Shida and S. Inagi, Electrochim. Acta, 2021, 138718 Search PubMed.
- W. Gao, K. Muzyka, X. Ma, B. Lou and G. Xu, Chem. Sci., 2018, 9, 3911–3916 Search PubMed.
- F. Du, Z. Dong, Y. Guan, A. M. Zeid, D. Ma, J. Feng, D. Yang and G. Xu, Anal. Chem., 2022, 94, 2189–2194 CAS.
- M. Liu, G. Salinas, J. Yu, A. Cornet, H. Li, A. Kuhn and N. Sojic, Chem. Sci., 2023, 14, 10664–10670 CAS.
- Z. Liu, W. Qi and G. Xu, Chem. Soc. Rev., 2015, 44, 3117–3142 Search PubMed.
- H. Qi and C. Zhang, Anal. Chem., 2020, 92, 524–534 CAS.
- L. Bouffier, S. Arbault, A. Kuhn and N. Sojic, Anal. Bioanal. Chem., 2016, 408, 7003–7011 CAS.
- X. Ma, W. Gao, F. Du, F. Yuan, J. Yu, Y. Guan, N. Sojic and G. Xu, Acc. Chem. Res., 2021, 54, 2936–2945 Search PubMed.
- S. F. Douman, D. Collins, L. R. Cumba, S. Beirne, G. G. Wallace, Z. Yue, E. I. Iwuoha, F. Melinato, Y. Pellegrin and R. J. Forster, Chem. Commun., 2021, 57, 4642–4645 CAS.
- S. F. Douman, E. Brennan, E. I. Iwuoha and R. J. Forster, Anal. Chem., 2017, 89, 11614–11619 CAS.
- C. Ma, Y. Cao, X. Gou and J.-J. Zhu, Anal. Chem., 2020, 92, 431–454 CAS.
- J. Zhang, S. Arbault, N. Sojic and D. Jiang, Annu. Rev. Anal. Chem., 2019, 12, 275–295 CAS.
- F. Ben Trad, J. Delacotte, M. Guille-Collignon, F. Lemaître, S. Arbault, N. Sojic, F. Burlina, E. Labbé and O. Buriez, Chem. Biomed. Imaging, 2023, 1, 58–65 CAS.
- Y. Feng, W. Zhou, X. Wang, J. Zhang, M. Zou, C. Zhang and H. Qi, Chem. Biomed. Imaging, 2023, 1, 648–658 CAS.
- L. Ding, P. Zhou, Y. Yan and B. Su, Chem. Biomed. Imaging, 2023, 1, 558–565 CAS.
- Y. Lu, Y. Ning, B. Li and B. Liu, Anal. Chem., 2024, 96, 463–470 Search PubMed.
- D. Han, B. Goudeau, D. Manojlovic, D. Jiang, D. Fang and N. Sojic, Angew. Chem., Int. Ed., 2021, 60, 7686–7690 Search PubMed.
- M.-S. Wu, D.-J. Yuan, J.-J. Xu and H.-Y. Chen, Chem. Sci., 2013, 4, 1182–1188 CAS.
- W. Zhan, J. Alvarez and R. M. Crooks, J. Am. Chem. Soc., 2002, 124, 13265–13270 CAS.
- W. Zhan, J. Alvarez, L. Sun and R. M. Crooks, Anal. Chem., 2003, 75, 1233–1238 CAS.
- A. L. Dauphin, A. Akchach, S. Voci, A. Kuhn, G. Xu, L. Bouffier and N. Sojic, J. Phys. Chem. Lett., 2019, 10, 5318–5324 CAS.
- L. Bouffier, D. Zigah, C. Adam, M. Sentic, Z. Fattah, D. Manojlovic, A. Kuhn and N. Sojic, ChemElectroChem, 2014, 1, 95–98 Search PubMed.
- M. Sentic, S. Arbault, B. Goudeau, D. Manojlovic, A. Kuhn, L. Bouffier and N. Sojic, Chem. Commun., 2014, 50, 10202–10205 Search PubMed.
- M. Sentic, G. Loget, D. Manojlovic, A. Kuhn and N. Sojic, Angew. Chem., Int. Ed., 2012, 51, 11284–11288 Search PubMed.
- V. Eßmann, J. Clausmeyer and W. Schuhmann, Electrochem. Commun., 2017, 75, 82–85 Search PubMed.
- J. D. Kraus and D. A. Fleisch, Electromagnetics with Applications, McGraw-Hill, Singapore, 1999 Search PubMed.
- K. M. Grant, J. W. Hemmert and H. S. White, J. Am. Chem. Soc., 2002, 124, 462–467 Search PubMed.
- J. L. Delaney, C. F. Hogan, J. Tian and W. Shen, Anal. Chem., 2011, 83, 1300–1306 Search PubMed.
- J. Totoricaguena-Gorriño, M. Dei, A. F. Alba, N. Peřinka, L.-R. Rubio, J. L. Vilas-Vilela and F. J. del Campo, ACS Sens., 2022, 7, 1544–1554 Search PubMed.
- L. Zhu, S. Li, W. Liu, J. Chen, Q. Yu, Z. Zhang, Y. Li, J. Liu and X. Chen, Biosens. Bioelectron., 2021, 187, 113284 Search PubMed.
- E. Kerr, R. Farr, E. H. Doeven, Y. H. Nai, R. Alexander, R. M. Guijt, B. Prieto-Simon, P. S. Francis, M. Dearnley, D. J. Hayne, L. C. Henderson and N. H. Voelcker, Sens. Actuators, B, 2021, 330, 129261 Search PubMed.
- S. M. Khoshfetrat, H. Bagheri and M. A. Mehrgardi, Biosens. Bioelectron., 2018, 100, 382–388 Search PubMed.
- M. Xi, Z. Wu, Z. Luo, L. Ling, W. Xu, R. Xiao, H. Wang, Q. Fang, L. Hu, W. Gu and C. Zhu, Angew. Chem., Int. Ed., 2023, 62, e202302166 Search PubMed.
- M. Guo, D. Du, J. Wang, Y. Ma, D. Yang, M. A. Haghighatbin, J. Shu, W. Nie, R. Zhang, Z. Bian, L. Wang, Z. J. Smith and H. Cui, Chem. Biomed. Imaging, 2023, 1, 179–185 CAS.
- H. Cui, G.-Z. Zou and X.-Q. Lin, Anal. Chem., 2003, 75, 324–331 CAS.
- H. Cui, W. Wang, C.-F. Duan, Y.-P. Dong and J.-Z. Guo, Chem. – Eur. J., 2007, 13, 6975–6984 CAS.
- J. Shu, Z. Han, T. Zheng, D. Du, G. Zou and H. Cui, Anal. Chem., 2017, 89, 12636–12640 CAS.
- W. Xu, Y. Wu, X. Wang, Y. Qin, H. Wang, Z. Luo, J. Wen, L. Hu, W. Gu and C. Zhu, Angew. Chem., Int. Ed., 2023, 62, e202304625 CAS.
- P. Zhou, S. Hu, W. Guo and B. Su, Fundam. Res., 2022, 2, 682–687 CAS.
- Y. Wang, R. Jin, N. Sojic, D. Jiang and H.-Y. Chen, Angew. Chem., Int. Ed., 2020, 59, 10416–10420 CAS.
- C. Cui, R. Jin, D. Jiang, J. Zhang and J.-J. Zhu, Anal. Chem., 2020, 92, 578–582 CAS.
- G. Ma, J. Zhou, C. Tian, D. Jiang, D. Fang and H. Chen, Anal. Chem., 2013, 85, 3912–3917 CAS.
- J. Zhou, G. Ma, Y. Chen, D. Fang, D. Jiang and H.-y. Chen, Anal. Chem., 2015, 87, 8138–8143 CAS.
- J. Xu, P. Huang, Y. Qin, D. Jiang and H.-y. Chen, Anal. Chem., 2016, 88, 4609–4612 CAS.
- B. Leca-Bouvier, B. Doumèche and L. J. Blum, in Analytical Electrogenerated Chemiluminescence: From Fundamentals to Bioassays, The Royal Society of Chemistry, 2020, pp. 331–385, 10.1039/9781788015776-00331.
- A. de Poulpiquet, B. Diez-Buitrago, M. Dumont Milutinovic, M. Sentic, S. Arbault, L. Bouffier, A. Kuhn and N. Sojic, Anal. Chem., 2016, 88, 6585–6592 Search PubMed.
- E. F. Laespada, J. L. P. Pavon and B. M. Cordero, Anal. Chim. Acta, 1996, 327, 253–260 Search PubMed.
- S. Rink, A. Duerkop and A. J. Baeumner, Analysis Sensing, 2023, 3, e202200111 CAS.
- J. Zielonka, J. D. Lambeth and B. Kalyanaraman, Free Radical Biol. Med., 2013, 65, 1310–1314 CAS.
- M. Mayer, S. Takegami, M. Neumeier, S. Rink, A. Jacobi von Wangelin, S. Schulte, M. Vollmer, A. G. Griesbeck, A. Duerkop and A. J. Baeumner, Angew. Chem., Int. Ed., 2018, 57, 408–411 CAS.
- M. Feng, A. L. Dauphin, L. Bouffier, F. Zhang, Z. Wang and N. Sojic, Anal. Chem., 2021, 93, 16425–16431 Search PubMed.
- K. Hiramoto, K. Ino, K. Komatsu, Y. Nashimoto and H. Shiku, Biosens. Bioelectron., 2021, 181, 113123 CAS.
- J. Yu, H. Saada, N. Sojic and G. Loget, Electrochim. Acta, 2021, 381, 138238 CAS.
- H. Zhu, J.-L. Zhou, C. Ma, D. Jiang, Y. Cao and J.-J. Zhu, Anal. Chem., 2023, 95, 11526–11534 CAS.
- M. Tan, Y. Wang, Z. Hong, P. Zhou, J. Jiang and B. Su, Analyst, 2024, 149, 1496–1501 CAS.
- H. Zhang, Y. Liu, M. Yao, W. Han and S. Zhang, Anal. Chem., 2023, 95, 570–574 CAS.
- M. A. Kamyabi, Z. Alipour and M. Moharramnezhad, J. Solid State Electrochem., 2021, 25, 445–456 CAS.
- H. Li, H. Zhou, T. Zhang, Z. Zhang, G. Zhao and C. Wang, ACS Sustainable Chem. Eng., 2022, 10, 10361–10368 CAS.
- Z. Xu, Y. Zhou, M. Li, Z. Guo and X. Zheng, Anal. Chem., 2023, 95, 10457–10463 CAS.
- C. Zhao, C. Ma, F. Zhang, W. Lai, C. Hong and Y. Qi, J. Colloid Interface Sci., 2023, 645, 997–1004 CAS.
- C. Zhao, C. Ma, F. Zhang, W. Li, C. Hong and Y. Qi, Chem. Eng. J., 2023, 466, 143156 CAS.
- W. Gu, X. Wang, M. Xi, X. Wei, L. Jiao, Y. Qin, J. Huang, X. Cui, L. Zheng, L. Hu and C. Zhu, Anal. Chem., 2022, 94, 9459–9465 CAS.
- H. Xia, X. Zheng, J. Li, L. Wang, Y. Xue, C. Peng, Y. Han, Y. Wang, S. Guo, J. Wang and E. Wang, J. Am. Chem. Soc., 2022, 144, 7741–7749 CAS.
- W. Gu, H. Wang, L. Jiao, Y. Wu, Y. Chen, L. Hu, J. Gong, D. Du and C. Zhu, Angew. Chem., Int. Ed., 2020, 59, 3534–3538 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4an00365a |
| This journal is © The Royal Society of Chemistry 2024 |