
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimultaneous quantitative LC-MS/MS analysis of 13 apolipoproteins and lipoprotein (a) in human plasma†
Yuxuan
Zhang
ab,
Xuanru
Ren
ab,
Zhitong
Zhou
ab,
Dao Wen
Wang
ab,
Xiaoquan
Rao
ab,
Hu
Ding
ab and
Junfang
Wu
 *ab
*ab
aDivision of Cardiology, Department of Internal Medicine, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China. E-mail: junfang.wu@tjh.tjmu.edu.cn
bHubei Key Laboratory of Genetics and Molecular Mechanisms of Cardiological Disorders, Wuhan, China
First published on 13th May 2024
Abstract
Numerous studies have revealed a close correlation between the levels of apolipoproteins (Apos) (including lipoprotein(a) [Lp(a)]) and an increased risk of cardiovascular disease in recent decades. However, clinically, lipid profiling remains limited to the conventional plasma levels of cholesterol, triglyceride, ApoA1, and ApoB, which brings the necessity to quantify more apolipoproteins in human plasma. In this study, we simultaneously quantified 13 apolipoproteins and Lp(a) in 5 μL of human plasma using the LC-MS/MS platform. A method was developed for the precise detection of Lp(a), ApoA1, A2, A5, B, C1, C2, C3, D, E, H, L1, M, and J. Suitable peptides were selected and optimized to achieve clear separation of each peak. Method validation consisting of linearity, sensitivity, accuracy and precision, recovery, and matrix effects was evaluated. The intra-day CV ranged from 0.58% to 14.2% and the inter-day CV ranged from 0.51% to 13.3%. The recovery rates ranged from 89.8% to 113.7%, while matrix effects ranged from 85.4% to 113.9% for all apolipoproteins and Lp(a). Stability tests demonstrated that these apolipoproteins remained stable for 3 days at 4 °C and 7 days at −20 °C. This validated method was successfully applied to human plasma samples obtained from 45 volunteers. The quantitative results of ApoA1, ApoB, and Lp(a) exhibited a close correlation with the results from the immunity transmission turbidity assay. Collectively, we developed a robust assay that can be used for high-throughput quantification of apolipoproteins and Lp(a) simultaneously for investigating related risk factors in patients with dyslipidemia.
1. Introduction
Apolipoproteins are essential structural constituents of plasma, facilitating the transport of cholesterol and other lipids.1 An increasing number of works over the past decade have substantiated the causal relationship between the concentrations of apolipoproteins (including lipoprotein(a) [Lp(a)]) and an elevated risk of atherosclerotic disease.2–5 Clinically, lipid profiling continues to concentrate on conventional markers, including total cholesterol, very low-density lipoprotein cholesterol (VLDL-c), low-density lipoprotein cholesterol (LDL-c), high-density lipoprotein cholesterol (HDL-c), and triglyceride, for assessing the status of hyperlipidemia patients. Recently, there has been increased focus on the levels of apolipoprotein subtypes (such as apolipoprotein A1 (ApoA1), ApoB) because the ratio (ApoB/ApoA1) is reported to be independently associated with disease severity in stroke6 or in critical illnesses.7 In a standardized case-control study of acute myocardial infarction in 52 countries, Yusuf et al. revealed that the ratio of ApoB/ApoA1 is the most important risk factor with the highest population-attributable risk among myocardial infarction patients.8 Additionally, the other subtypes of apolipoprotein, including ApoC1, ApoC2, and ApoC3,9–11 were also reported to play important roles in coronary heart diseases. Hamsten et al. discovered that the ApoC1 content of triglyceride-rich lipoproteins independently predicts early atherosclerosis in healthy middle-aged men.12 The specific measurement of ApoB, C3, and E of VLDL takes a greater risk than plasma triglycerides for assessing recurrent coronary events in a prospective Cholesterol and Recurrent Events (CARE) trial.13 Moreover, ApoE serves as a determinant of cardiovascular disease risk and atherosclerosis, with apoE4 playing an important role in the pathogenesis of Alzheimer's disease.14,15Apart from the apolipoprotein, the lipoprotein (a) [Lp(a)] comprised of apolipoprotein (a) covalently bound to apolipoprotein B-100 via a single disulfide bridge poses challenges for accurate detection due to its distinctive structural attributes and size heterogeneity of apolipoprotein (a).16,17 Typically, the concentration of apo(a) exhibits an inverse relationship with its size. This is attributed to the kringle IV (KIV) domain of apo(a) consisting of 1 to 10 distinct KIV types, of which the KIV-2 repeats range from 3 to 40 copies.18,19 Consequently, the mass of the measured particle does not reflect the number of Lp(a) particles, which makes it difficult for accurate quantification of this apolipoprotein subtype. Therefore, the quantification of Lp(a) may be under-estimated or over-estimated when employing the immunochemical method.5
The quantitation of multiple apolipoproteins and Lp(a) is the prerequisite to understanding their physiological and pathophysiological functions. Furthermore, high-throughput methods are required to simultaneously quantify multiple apolipoproteins in a single measurement, particularly when dealing with large sample numbers with limited amounts. Previously, triglyceride-rich lipoproteins, such as ApoC1 and ApoB, were usually sub-fractionated using cumulative density gradient ultracentrifugation.12 In the current clinical laboratory, the quantification of apolipoproteins mostly relies on the immunity transmission turbidity method, which is susceptible to endogenous interference or the hook effect.20 On the other hand, the accuracy of the immunoassay was reduced obviously for those samples with an extreme concentration (too low or too high).21 With the state-of-the-art mass spectrometry, Li et al. simultaneously quantified three apolipoproteins (Apo A1, E and J) in human plasma using solid phase extraction.22 Additionally, Roddy et al. developed a method for the simultaneous quantification of apoB48 and A5, employing a combination of immune-enrichment and LC-MS measurement.23 Furthermore, Marcovina et al. proposed the LC-MS/MS method as a candidate reference for standardization of analytical methods for Lp(a) measurement.24 Therefore, there is a need for the development of a methodology capable of simultaneously identifying subclasses of apolipoproteins and Lp(a) that are associated with dyslipidemia.
Here, we aim to simultaneously quantify 13 apolipoproteins and Lp(a), including ApoA1, ApoA2, ApoA5, ApoB, ApoC1, ApoC2, ApoC3, ApoD, ApoE, ApoH, ApoL1, ApoM, ApoJ, and Lp(a) in human plasma samples. Our study established a high coverage and precise LC-MS/MS method for quantifying apolipoproteins and Lp(a) in human plasma with a small volume.
2. Materials and methods
2.1. Chemicals and reagents
Ammonium bicarbonate (AmBic), formic acid (FA), sodium deoxycholate (SDC), dithiothreitol (DTT), iodoacetamide (IAA), and dimethyl sulfoxide (DMSO) were purchased from Sigma Aldrich Co. Ltd (St Louis, MO, USA). All reagents used were of the highest available purity. HPLC gradient grade methanol (MeOH, 99.9%)and acetonitrile (ACN, 99.9%) were purchased from Merck Co. Ltd (Darmstadt, Germany). All peptides and isotope internal peptides were purchased from Shanghai Apeptide Co. Ltd (Shanghai, China). Sequencing grade modified trypsin was purchased from Promega Corporation (Madison, USA).2.2. Peptides
The selection of peptides should ideally adhere to the following criteria:25,26 the selected amino acid sequence (1) is specific for the targeted protein; (2) is preferably composed of 8 and 15 amino acid residues in length; (3) should exclude unstable amino acid residues; (4) should not have C-terminal adjacent arginine or lysine amino acid residues; (5) should not contain known human genetic mutations, and (6) should not be located within the KIV-2 domain for Lp(a).Briefly, the NCBI Protein database (https://www.ncbi.nlm.nih.gov/protein/) was initially accessed to acquire the comprehensive sequence and relevant information for the designated protein. Subsequently, the curated feature peptides that meet the specific criteria were selected using the advanced tools embedded in the Skyline software (https://skyline.ms/). Finally, the unequivocal uniqueness of the chosen feature peptides within the genome or protein database was validated by duplicating the selected feature peptide sequences and rigorously examining the search results through the BLAST tool (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Such precautionary steps were undertaken to mitigate the risk of cross-reactivity or ambiguity in subsequent analyses.
All selected peptides for each apolipoprotein and Lp(a) are detailed in ESI Table S1.† Two or three candidate peptides for some apolipoproteins (ApoA1, ApoA2, ApoB, ApoC1, ApoC2, ApoE) and two peptides on unique KIV 5 (TPENYPNAGLTR, Lp(a)-TPEN) and KIV 9 (GISSTTVTGR, Lp(a)-GISS) domain of Lp(a) were selected for quantification according to the standardized reference method.24 The better peptide of each apolipoprotein with higher peak intensity and recovery rate was selected for further method validation (Table 1).
| Peptide | Protein name | Peptide MW | Precursor ion (m/z) | Product ion (m/z) | DP (V) | CE (V) |
|---|---|---|---|---|---|---|
| a Internal standard peptides. | ||||||
| VQPYLDDFQK | ApoA1 | 1252.3718 | 627.1 | 1025.7 | 44 | 23 |
| 1008.4 | 23 | 24 | ||||
| VQPYLDDFQKa | ApoA1-IS | 1260.3859 | 631.1 | 1033.7 | 46 | 21.3 |
| 1016.4 | 40 | 25.6 | ||||
| SPELQAEAK | ApoA2 | 972.0506 | 487.0 | 442.8 | 87.3 | 24 |
| 659.4 | 44 | 26 | ||||
| SPELQAEAKa | ApoA2-IS | 980.0648 | 491.0 | 450.8 | 87.3 | 24 |
| 667.4 | 44 | 26 | ||||
| VQELQEQLR | ApoA5 | 1142.2629 | 571.9 | 673.6 | 38 | 24 |
| 915.4 | 40 | 29.6 | ||||
| VQELQEQLRa | ApoA5-IS | 1152.2711 | 576.9 | 683.6 | 40 | 30 |
| 925.4 | 45 | 28 | ||||
| TEVIPPLIENR | ApoB | 1280.4697 | 641.1 | 838.7 | 67.4 | 25.2 |
| 741.2 | 56 | 33 | ||||
| TEVIPPLIENRa | ApoB-IS | 1290.4780 | 646.1 | 848.7 | 65.2 | 29 |
| 751.2 | 27 | 32 | ||||
| EFGNTLEDK | ApoC1 | 1052.0922 | 527.0 | 391.3 | 41 | 22 |
| 776.4 | 31 | 24 | ||||
| EFGNTLEDKa | ApoC1-IS | 1060.1064 | 531.0 | 399.3 | 41 | 24 |
| 784.4 | 44 | 22 | ||||
| TYLPAVDEK | ApoC2 | 1035.1478 | 518.3 | 658.3 | 60 | 24.2 |
| 771.6 | 68 | 20.8 | ||||
| TYLPAVDEKa | ApoC2-IS | 1043.1619 | 522.3 | 666.3 | 128 | 25 |
| 779.6 | 86 | 23 | ||||
| GWVTDGFSSLK | ApoC3 | 1196.3085 | 598.9 | 854.6 | 60 | 27.1 |
| 343.2 | 40 | 23.6 | ||||
| GWVTDGFSSLKa | ApoC3-IS | 1204.3226 | 598.9 | 854.6 | 60 | 27.1 |
| 343.2 | 40 | 23.6 | ||||
| NILTSNNIDVK | ApoD | 1230.3680 | 616.0 | 890.6 | 131 | 32 |
| 789.4 | 146 | 31 | ||||
| NILTSNNIDVKa | ApoD-IS | 1238.3821 | 620.0 | 898.6 | 144 | 28 |
| 797.4 | 102 | 35 | ||||
| LGPLVEQGR | ApoE | 968.1083 | 484.8 | 588.5 | 60 | 30.6 |
| 701.6 | 50 | 31.1 | ||||
| LGPLVEQGRa | ApoE-IS | 978.1165 | 489.8 | 598.5 | 60 | 30.8 |
| 711.6 | 50 | 31.7 | ||||
| ATVVYQGER | ApoH | 1022.1126 | 512.0 | 652.4 | 66 | 22 |
| 751.4 | 75 | 15 | ||||
| ATVVYQGERa | ApoH-IS | 1032.1209 | 517.0 | 662.4 | 52 | 33 |
| 761.4 | 67 | 15 | ||||
| VTTVASHTSDSDVPSGVTEVVVK | ApoJ | 2314.5021 | 772.4 | 507.9 | 50 | 30.2 |
| 650.9 | 45 | 28.8 | ||||
| ALDNLAR | ApoL1 | 771.8620 | 386.9 | 473.4 | 53 | 27 |
| 588.5 | 60 | 30.6 | ||||
| ALDNLARa | ApoL1-IS | 781.8703 | 391.9 | 483.4 | 51 | 23.8 |
| 598.5 | 93 | 17 | ||||
| FLLYNR | ApoM | 824.9660 | 413.6 | 565.3 | 125 | 20 |
| 452.3 | 100 | 20 | ||||
| FLLYNRa | ApoM-IS | 834.9743 | 418.6 | 575.3 | 119 | 32 |
| 462.3 | 102 | 32 | ||||
| 672.4 | 1015.3 | 65 | 28.2 | |||
![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) ![[I with combining low line]](https://www.rsc.org/images/entities/char_0049_0332.gif) ![[S with combining low line]](https://www.rsc.org/images/entities/char_0053_0332.gif) ![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) ![[V with combining low line]](https://www.rsc.org/images/entities/char_0056_0332.gif) ![[R with combining low line]](https://www.rsc.org/images/entities/char_0052_0332.gif) |
Lp(a) | 978.0585 | 489.9 | 808.3 | 30 | 21.8 |
| 533.3 | 40 | 25.2 | ||||
| GISSTTVTGRa | Lp(a)-GISS-IS | 988.0668 | 495.3 | 543.2 | 30 | 22.5 |
| 818.2 | 31 | 22.8 | ||||
2.3. Standard stock solutions and internal standard preparation
The powder of each standard peptide was individually reconstituted in a solution of 50% acetonitrile (VACN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :VH2O = 1:1) to produce stock solutions with a concentration of 1 mg mL−1. The linearity range based on the physiological concentration and its dilution to the desired concentration by 50% ACN are shown in Table 2. Stable isotope-labeled (SIL) peptides served as internal standards to minimize the variability and improve the precision and accuracy of the measurement. The internal standard solution, consists of ApoA1-IS (20 mg L−1), ApoA2-IS (20 mg L−1), ApoA5-IS (0.025 mg L−1), ApoB-IS (5 mg L−1), ApoC1-IS (2 mg L−1), ApoC2-IS (2 mg L−1), ApoC3-IS (2 mg L−1), ApoD-IS (2 mg L−1), ApoE-IS (5 mg L−1), ApoH-IS (2 mg L−1), ApoL1-IS (0.2 mg L−1), ApoM-IS (0.2 mg L−1), and Lp(a)-GISS-IS (0.4 mg L−1). The solution was then divided into several aliquots, which were stored at −80 °C to avoid repeated freezing and thawing.
:VH2O = 1:1) to produce stock solutions with a concentration of 1 mg mL−1. The linearity range based on the physiological concentration and its dilution to the desired concentration by 50% ACN are shown in Table 2. Stable isotope-labeled (SIL) peptides served as internal standards to minimize the variability and improve the precision and accuracy of the measurement. The internal standard solution, consists of ApoA1-IS (20 mg L−1), ApoA2-IS (20 mg L−1), ApoA5-IS (0.025 mg L−1), ApoB-IS (5 mg L−1), ApoC1-IS (2 mg L−1), ApoC2-IS (2 mg L−1), ApoC3-IS (2 mg L−1), ApoD-IS (2 mg L−1), ApoE-IS (5 mg L−1), ApoH-IS (2 mg L−1), ApoL1-IS (0.2 mg L−1), ApoM-IS (0.2 mg L−1), and Lp(a)-GISS-IS (0.4 mg L−1). The solution was then divided into several aliquots, which were stored at −80 °C to avoid repeated freezing and thawing.
| Peptide | Linear | R 2 | RT (min) | S/N | Rangea (ng ml−1) |
|---|---|---|---|---|---|
| a The concentration here refers to the concentration of the peptide segments. Keys: RT, Retention time; min, minute; S/N, signal to noise ratio. | |||||
| ApoA1 | y = 2.20 × 10−5x + 0.027 | 0.995 | 16.5 | 2786.87 | 2000–400000 |
| ApoA2 | y = 0.013x + 10.26 | 0.997 | 7.3 | 685.25 | 1000–200000 |
| ApoA5 | y = 0.015x + 0.0069 | 0.992 | 12.4 | 80.25 | 0.2–40 |
| ApoB | y = 7.22 × 10−4x + 0.0054 | 0.999 | 18.1 | 309.9 | 100–20000 |
| ApoC1 | y = 6.82 × 10−4x + 0.047 | 0.991 | 11.1 | 1929.59 | 100–20000 |
| ApoC2 | y = 6.09 × 10−4x + 0.045 | 0.999 | 14.3 | 1999.06 | 125–25000 |
| ApoC3 | y = 2.40 × 10−4x − 1.48 × 10−4 | 0.996 | 18.1 | 356.05 | 250–50000 |
| ApoD | y = 5.34 × 10−4x + 0.0037 | 0.999 | 17.1 | 833.49 | 250–50000 |
| ApoE | y = 2.37 × 10−4x + 0.0078 | 0.999 | 12.5 | 1581.9 | 25–5000 |
| ApoH | y = 8.24 × 10−4x + 0.044 | 0.999 | 8.4 | 347.83 | 100–20000 |
| ApoJ | y = 0.0024x − 0.13 | 0.995 | 17.6 | 841.34 | 250–50000 |
| ApoL1 | y = 0.0011x + 0.032 | 0.999 | 10.1 | 258.65 | 50–10000 |
| ApoM | y = 0.0019x + 0.0059 | 0.998 | 16.1 | 2786.87 | 25–5000 |
| Lp(a)-GISS | y = 0.0092x + 0.0075 | 0.999 | 8.7 | 685.25 | 2–400 |
2.4. UPLC-ESI-MS/MS instrumentation and MS method
LC-MS/MS analysis was performed using an AB SCIEX Triple Quad 6500+ mass spectrometer equipped with an electrospray ionization (ESI) source (AB SCIEX, Framingham, MA, USA). Chromatographic separation was achieved using a Waters Acquity UPLC HSS T3 column (2.1 × 50 mm, 1.8 μm) maintained at 45 °C with a flow rate of 0.3 mL min−1. The mobile phases comprised solution A (0.1% FA, 2% DMSO in water) and solution B (0.1% FA, 2% DMSO in MeOH). The gradient elution was optimized for the separation of individual peptides as follows: 0–1 min 3% B, 1–16 min 30% B, 16–21 min 95% B, and 21–21.5 min 3% B. The column was equilibrated for two more minutes, with a run time of 23 minutes in total. The ion source parameters were set in the positive mode under optimal conditions: curtain gas, 35 psi; ion spray voltage, 5500 V; temperature, 500 °C; and ion source gas 1/2, 50 psi. Optimal multiple-reaction monitoring (MRM) transitions were further identified for the analyses of individual peptides as well as their corresponding isotope-labeled internal standards.The [M + 2H] double-charged precursor ions while product ions with a single charge (specifically y-ions) were chosen for quantification (as quantifier ions) and qualification (as qualifier ions) due to their excellent response in mass spectrometry. If possible, product ions with m/z values greater than those of the precursor ion were selected to minimize the possibility of interferences. The simultaneous measurement of transitions in dynamic MRM mode was minimized by using a planned measurement approach for different peptides. Each peptide was allocated a retention time window of 60 seconds. Data acquisition and analysis were conducted using Analyst 1.7.1 software (AB Sciex) and further analyzed using OS 2.1 software (AB Sciex).
2.5. Linearity of calibration curves
Bovine serum albumin (BSA) dissolved in PBS solution was utilized as a substitute for comparing various plasma matrices. The calibration curves were determined by measuring the different concentrations of standard peptides in the BSA solution. For each peptide, the peak area ratio (Y) to the corresponding SIL internal standard versus the nominal concentration (X) of analytes was calculated with weighted (1/X2) least squares linear regression. The lower limit of quantification for different analytes was determined by the minimal concentration with a signal-to-noise ratio (S/N) of 10:1.
2.6. Precision and accuracy
To evaluate the precision and accuracy, the intra-day and inter-day accuracy of quality control (QC) samples (including solution mixture and plasma samples) at different concentrations (low, middle, and high levels) was analyzed in 6 replicates in one day and over three consecutive days, respectively. The precision was determined as the coefficient of variation (CV) of the measured concentration, while the accuracy was evaluated as the relative error (RE) of the mean measured concentration that deviated from the nominal value. The estimation of precision and accuracy was assessed according to the guidelines outlined by the Clinical and Laboratory Standards Institute (CLSI C62-A), which established a maximum permissible RE and CV of 15%.272.7. The matrix effect
The matrix effect of each peptide was evaluated by examining the BSA or mice plasma spiked at the different concentrations (low, medium, and high) of the peptide standard mixture. For each peptide, the spiked concentrations in BSA or mice plasma were calculated based on the calibration curve in the solution. The matrix factor (MF) of each peptide was calculated by dividing the spiked concentration in BSA or mice plasma by the theoretical spiked concentration. Then the matrix effect for the analytes at each concentration was obtained. The matrix factor is considered acceptable if it is below 20%.
2.8. The extraction recovery
The extraction recovery of each peptide was evaluated by examining the QC sample spiked at the different concentrations (low, medium, and high) of the peptide standard mixture. For each peptide, the spiked BSA concentrations were calculated based on the matrix calibration curve. Then the extraction recovery was calculated by subtracting the original concentration from the concentration of the spiked sample and dividing it by the theoretical spiked concentration. The recovery rate is considered acceptable if it is below 20%.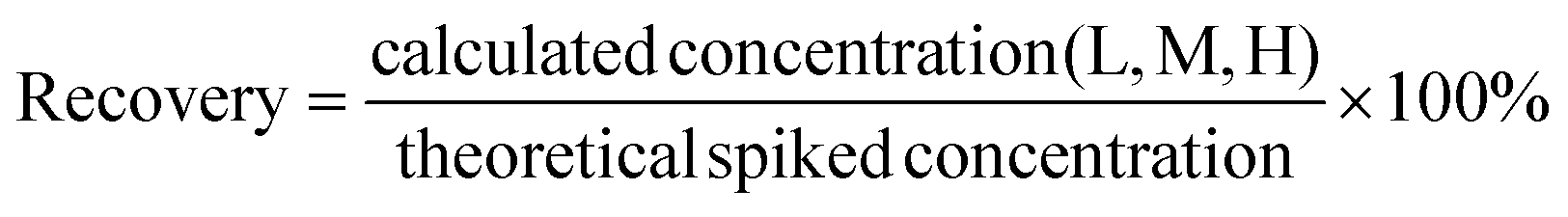
2.9. Stability
The storage conditions chosen for this investigation are practical operations that are usually encountered during routine sample preparation in daily experiments. The stability of peptides in human plasma was tested by measuring the concentrations immediately after sample collection and after storage at various temperatures for different durations (room temperature for 24 hours, 4 °C for 3 days, and −20 °C for 7 days, n = 6). Stability was evaluated by calculating the relative deviation of each peptide concentration (80–120%) compared to its initial concentration.2.10. Sample collection
The newly developed method was applied to 45 plasma samples of Chinese Han volunteers. This study adhered to the principles of the Declaration of Helsinki and received approval from the Ethics Committee of Tongji Medical College. Written informed consent was obtained from all participants. Fasting plasma samples were collected in the morning. The blood samples were centrifuged at 3000g for 8 minutes and the aliquoted plasma was stored at −80 °C immediately. Additionally, ApoA1, ApoB, and Lp(a) in the same sample were measured using the immunity transmission turbidity method following the manufacturer's instructions on a Roche Cobas 8000 (Mannheim, Germany), in the Department of Clinical Laboratory in Tongji Hospital.2.11. Sample extraction
A previously published protocol was optimized for sample preparation.24 Briefly, 10 μL of SIL-internal standard mixture, 5 μL plasma samples, or the calibrators in BSA (prepared as described above) were mixed in a 1.5 mL tube with 75 μL of 1% SDC (w/v) and 50 μL of 100 mmol L−1 AmBic. Then the mixture was reduced with 5 μL of 250 mmol L−1 DTT at 90 °C for 60 minutes at 500 rpm, followed by mixing with 10 μL of 500 mmol L−1 IAA for 30 minutes in the dark at room temperature. Then the 5 μL of 250 mmol L−1 DTT was added to quench the alkylation reaction, and the solution was diluted to 1 mL with 0.5% SDC in 100 mmol L−1 AmBic. After centrifugation at 10000g for 10 minutes at 4 °C, 50 μL of the supernatant was collected and mixed with an equal volume of 0.5% SDC. Then 1 μg of sequencing grade Promega trypsin was used for digestion at 37 °C on a constant temperature oscillator overnight (18 hours). Following digestion, the reaction was terminated by adding 20 μL of 20% formic acid aqueous solution. The mixture was allowed to stand at room temperature for 5 minutes before centrifugation at 13000g for 15 minutes at 4 °C. Finally, 80 μL of the supernatant was transferred to a sample vial and injected into the LC-MS/MS system (Fig. 1).
 | ||
| Fig. 1 The flow chart of sample preparation. Keys: AmBic, ammonium bicarbonate; DTT, dithiothreitol; IAA, iodoacetamide; RT, room temperature; SDC, sodium deoxycholate; SIL, stable isotope-labeled. | ||
Additionally, the usage of solid phase extraction (SPE) or not was compared to optimize the sample extraction. Before SPE, the Oasis Prime HLB cartridge plate was washed three times with 0.6 mL water (0.1% formic acid), followed by elution twice with 0.6 mL of 80% methanol (0.1% formic acid). Then the eluate was subjected to a Termovap sample concentrator to remove the organic solvent. The dried tube was subsequently reconstituted with 100 μL of a 0.1% formic acid solution for further sample acquisition on the LC-MS/MS system.
2.12. Statistical analysis
The peptide levels in plasma were analyzed and expressed as mean ± standard deviation using the GraphPad Prism 9 (San Diego, CA, U.S.A.). The distribution of all variables was measured before statistical analysis. The Welch's test was employed for normally distributed data, while the Mann–Whitney test was used for non-normally distributed data (Prism, GraphPad Software Inc., San Diego, CA, USA). p < 0.05 was considered to be statistically significant.3. Results and discussion
Although LC-MS/MS has emerged as a potent technique for the simultaneous measurement of a series of compounds,28,29 the immunoturbidimetric assay and enzyme-linked immunosorbent assay (ELISA) remain the predominant clinical methods for apolipoprotein and lipoprotein measurement.30,31 For the extreme samples with high antigen or antibody, the formation of soluble complexes may lead to errors or be influenced by hyperlipemia, thereby posing challenges to accurate determination of apolipoprotein concentration in patients.The main goal of the study was to develop a robust and sensitive analytical protocol for quantifying a wide array of apolipoprotein and Lp(a) in human plasma. Challenges in analyzing these peptides include chemical instability, complexity of sample matrixes, and compatibility with a wide range of compounds. Except for the immunity transmission turbidity method, the measurement of apolipoprotein and Lp (a) by LC-MS/MS has been increasing in recent years due to its high sensitivity.32–34 However, to our knowledge, there is currently no reported bioanalytical LC-MS/MS method for the simultaneous analysis of apolipoproteins and Lp(a) in human plasma using a small sample volume.
3.1. Chromatography and MRM-MS separation
The methods, including the separation method by UPLC and the MS/MS fragment transitions of each peptide, were optimized to ensure the resolution among all peptides. The total ion chromatogram and its multiple reaction monitoring (MRM) chromatograms of blank matrix solution containing internal standards are shown in Fig. 2 and ESI Fig. S1,† respectively. | ||
| Fig. 2 The total (A) and dynamic multiple reaction monitoring (B) chromatograms of each peptide were analyzed in a standard solution mixture. | ||
It is better to choose two or more unique peptides of the same apolipoprotein for peptide quantitation. Here two peptides of some apolipoprotein (such as ApoA1, ApoA2, ApoB, ApoC1, ApoC2, ApoE, Lp(a)) were explored for the method validation. Following the comparison of peak intensity and preliminary method validation, the peptides ApoA1-VQPY, ApoA2-SPEL, ApoB-FPEV, ApoC1-EFGN, ApoC2-TYLP, ApoE-LGPL, and Lpa-GISS were selected for subsequent quantification owing to their relatively stable recoveries (Table 1 and ESI Table S1†).
3.2. Method optimization for sample preparation
The sample extraction process, including the usage of solid phase extraction (SPE), the optimization of the trypsin: plasma ratio, and the determination of the optimal concentration of SDC were explored to ensure optimal resolution among all peptides.Firstly, due to the relatively low abundance of endogenous ApoA5 and Lp(a), we found that their peaks were easily interfered with its neighboring peaks originating from the matrix. In addition to attempts to adjust the chromatographic gradient to maximize the separation of the target signal from the interference peak, the SPE or immunoaffinity method was usually used for extracting ApoA5 in previous works.22 The comparison between samples with and without SPE (Oasis Prime HLB cartridge) was presented in ESI Fig. S2.† The usage of SPE does not increase the signals of peptides. In contrast, the peak intensity of ApoA5 and Lp(a) decreased significantly after SPE. It was said that SPE usage is a double-edged sword, offering the advantage of yielding clean samples while mitigating signal suppression through desalting and preventing instrument contamination. Nonetheless, the recovery rate for the low-abundance proteins, such as the aforementioned two peptides, may not be optimal following SPE extraction. Moreover, the peak splitting observed in ApoC1 and ApoL1 may come from the co-elution after SPE extraction. Besides, it should also be taken into account that the usage of SPE would increase the labor intensity and the cost of the method, which can be crucial when dealing with substantial sample numbers.
Secondly, an optimal trypsin protease to plasma protein ratio was evaluated before sample preparation. Although the commonly accepted enzyme-to-substrate ratio typically ranges from 1:20 to 1:100, numerous studies have suggested higher enzyme concentrations positing enhanced digestion efficiency.35,36 Considering the reported plasma protein content per microliter (60–80 g L−1),37 various amounts of trypsin at 1 μg, 2 μg, and 4 μg were investigated to explore the optimal trypsin:plasma ratio during sample preparation. There was no obvious difference for all peptides with three different amounts of trypsin when compared to the digestive enzyme at 1 μg (CV <15%) (ESI Table S2†). Then 1 μg trypsin (trypsin:plasma = 1:14) was chosen for digestion for further validation due to its efficiency and cost-effectiveness. Furthermore, we also compared two types of reducing agents (tris-2-carboxyethyl-phosphine (TCEP) and DTT) to assess their effects on targeted peptides. Although the majority of peptide quantification remained unaffected by using different reducing agents, DTT proved to be more effective in achieving a clear peak for Lp(a) (ESI Fig. S3†). It might be because the DTT is a specific protein-RNA cross-linker and a cell-permeable reducing agent when compared to the cell-impermeable reducing agent TCEP.38,39
Thirdly, the SDC added during the reduction step was used to minimize the inadvertent omission of host cell proteins during digestion.40,41 Here different concentrations of anionic descaling agent-based SDC (0.5%, 1%, 2%) that effectively disrupts protein interactions were explored during sample preparation. Since the SDC's chemical structure easily causes the flocculent precipitates at pH levels below 7.5. It was observed that a high SDC concentration resulted in significant precipitation, which could not be fully resolved even after two rounds of centrifugation for 30 minutes (ESI Fig. S4†). Finally, the 0.5% SDC was chosen to minimize the suspended particles in sample preparation.
3.3. Selection of substitute matrix and the matrix effects
A suitable alternative matrix was necessary to improve the reliability and accuracy of the method. Ideally, human plasma with extremely low levels of apolipoprotein and Lp(a) would have served as an ideal matrix, but it is not easy to meet and its amount for method validation is too much. Given that apolipoproteins and Lp(a) are endogenous in human plasma, the alternative matrices for the calibration curve were explored in bovine serum albumin (BSA) and mice plasma (ESI Fig. S5†). From the representative chromatograms, the SIL-internal standards showed clear peaks without interference in both the BSA and mice plasma matrix. Furthermore, the BSA does not show any interference with the apolipoprotein and Lp(a). But mice plasma possesses the same peak intensity (ApoA2, ApoC1, ApoC3, ApoE, ApoJ, ApoL1, and ApoM) compared to the selected peptides in human plasma. Even though the peak intensity in mice plasma is relatively low compared to human plasma, it easily leads to high background signals, which brings difficulty in quantifying low levels of peptides. Therefore, the 0.5% bovine serum albumin solution was used for future validation purposes with calibration curves. The performance of the separation in terms of peak shapes and retention times was consistent throughout the study.The matrix factor was calculated for all peptides by spiking different levels of standards (low, middle, and high) in BSA and mice plasma, respectively. The consistency spanned from 85.4% to 113.9% in BSA, which also meets the acceptance criteria of a 15% deviation (Fig. 3A). However, the matrix factor of ApoA5, ApoJ, ApoL1, and ApoM was not stable in mice plasma (ESI Table S3†). These results suggested that the matrix effects of BSA have limited effects on accuracy and precision for all peptides.
 | ||
| Fig. 3 Data for matrix factor and recovery of the developed method for all measured apolipoproteins and Lp(a). | ||
3.4. Method validation
The method was validated following the guidelines of the Food and Drug Administration and the Clinical and Laboratory Standards Institute (CLSI C62-A) for bioanalytical method validation. Here the linearity, recovery, accuracy, recovery and stability were determined according to these guidelines. All the spiked concentrations of the standards (low, middle, and high) used in method validation are the same as those listed in ESI Table S3.†:1, with the highest ratio reaching 2786, as detailed in Table 2.
| Peptide | LQC | MQC | HQC | Sample (n = 4) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cona (ng ml−1) | % RE | Intra % CV | Inter % CV | Cona (ng ml−1) | % RE | Intra % CV | Inter % CV | Cona (ng ml−1) | % RE | Intra % CV | Inter % CV | Average cona (ng ml−1) | Intra % CV | Inter % CV | |
| a The concentration here refers to the concentration of peptide segments. | |||||||||||||||
| ApoA1 | 12500 |
100.46 | 0.59 | 2.75 | 25000 |
98.53 | 1.67 | 2.43 | 100000 |
94.99 | 3.52 | 1.99 | 60237.11 |
2.73 | 1.85 |
| ApoA2 | 6250 | 113.93 | 5.34 | 11.43 | 12500 |
94.70 | 4.39 | 3.05 | 50000 |
99.60 | 4.36 | 1.16 | 29763.09 |
6.72 | 0.90 |
| ApoA5 | 1.25 | 93.04 | 2.99 | 4.25 | 2.5 | 82.28 | 9.06 | 13.43 | 10 | 99.27 | 6.30 | 10.93 | 7.36 | 8.82 | 6.19 |
| ApoB | 312.5 | 103.53 | 2.32 | 7.17 | 625 | 113.88 | 7.46 | 5.19 | 2500 | 101.06 | 4.68 | 4.69 | 1951.35 | 4.04 | 4.30 |
| ApoC1 | 625 | 103.52 | 2.03 | 1.97 | 1250 | 104.15 | 3.17 | 0.98 | 5000 | 102.96 | 2.04 | 0.93 | 8489.79 | 2.95 | 2.19 |
| ApoC2 | 781.25 | 97.08 | 1.41 | 1.58 | 1562.5 | 96.91 | 1.96 | 2.01 | 6250 | 98.28 | 2.38 | 1.59 | 8605.01 | 2.67 | 3.25 |
| ApoC3 | 1562.5 | 93.68 | 5.85 | 2.83 | 3125 | 100.53 | 2.52 | 3.63 | 12500 |
87.34 | 6.98 | 8.99 | 13634.86 |
6.93 | 5.17 |
| ApoD | 1562.5 | 99.11 | 2.44 | 0.51 | 3125 | 110.92 | 5.33 | 5.25 | 12500 |
108.28 | 5.79 | 2.62 | 1787.07 | 4.29 | 6.63 |
| ApoE | 156.25 | 102.42 | 1.37 | 1.43 | 312.5 | 101.68 | 1.60 | 1.24 | 1250 | 99.63 | 1.27 | 1.82 | 7721.32 | 5.19 | 2.13 |
| ApoH | 625 | 104.23 | 3.37 | 3.43 | 1250 | 97.53 | 4.14 | 1.01 | 5000 | 98.01 | 5.34 | 3.34 | 2731.68 | 4.05 | 3.15 |
| ApoJ | 1562.5 | 84.38 | 13.80 | 8.06 | 3125 | 90.97 | 14.20 | 12.07 | 12500 |
99.58 | 5.17 | 7.17 | 7194.47 | 14.23 | 13.45 |
| ApoL1 | 312.5 | 99.49 | 2.80 | 1.45 | 625 | 96.39 | 2.57 | 2.09 | 2500 | 97.46 | 3.55 | 2.18 | 812.50 | 5.19 | 7.27 |
| ApoM | 156.25 | 99.95 | 1.22 | 1.96 | 312.5 | 100.06 | 0.58 | 2.36 | 1250 | 100.78 | 7.96 | 4.10 | 741.79 | 7.69 | 4.58 |
| Lp(a) | 31.25 | 110.57 | 3.56 | 5.78 | 62.5 | 104.86 | 0.89 | 5.25 | 250 | 107.55 | 4.15 | 7.60 | 51.16 | 6.16 | 7.04 |
| Fresh analytes | 24 h, room temperature | 3 days, 4 °C | 7 days, −20 °C | ||||
|---|---|---|---|---|---|---|---|
| Cona | % RE | % CV | % RE | % CV | % RE | % CV | |
| a The concentration here refers to the concentration of proteins. | |||||||
| ApoA1 (g L−1) | 1.50 | 0.60 | 3.04 | 0.68 | 2.90 | 0.70 | 6.31 |
| ApoA2 (g L−1) | 0.33 | 2.68 | 5.37 | 2.66 | 2.87 | 2.26 | 3.87 |
| ApoA5 (mg L−1) | 0.25 | 0.72 | 9.35 | 1.88 | 6.83 | 8.86 | 8.87 |
| ApoB (g L−1) | 0.96 | 2.56 | 0.13 | 3.77 | 8.60 | 1.62 | 18.16 |
| ApoC1 (mg L−1) | 73.50 | 3.41 | 0.70 | 4.55 | 9.31 | 4.12 | 15.07 |
| ApoC2 (mg L−1) | 95.84 | 2.56 | 5.29 | 2.65 | 3.78 | 0.83 | 4.31 |
| ApoC3 (mg L−1) | 133.25 | 4.13 | 14.10 | 2.48 | 13.61 | 15.06 | 2.60 |
| ApoD (mg L−1) | 31.80 | 2.28 | 4.45 | 6.49 | 15.18 | 4.92 | 13.67 |
| ApoE (mg L−1) | 286.01 | 1.11 | 1.07 | 1.77 | 3.48 | 2.58 | 1.75 |
| ApoH (mg L−1) | 103.43 | 8.37 | 2.21 | 8.19 | 16.98 | 2.54 | 14.72 |
| ApoJ (mg L−1) | 221.62 | 11.42 | 2.60 | 8.78 | 0.78 | 11.33 | 9.12 |
| ApoL1 (mg L−1) | 59.06 | 4.70 | 0.43 | 4.39 | 5.43 | 3.09 | 1.22 |
| ApoM (mg L−1) | 19.05 | 4.59 | 0.38 | 6.93 | 7.14 | 2.82 | 11.58 |
| Lp(a) (nmol L−1) | 51.20 | 3.36 | 3.75 | 3.82 | 11.39 | 5.67 | 0.24 |
3.5. Immunoassay & LC-MRM/MS
Clinically, only three apolipoproteins including ApoA1, ApoB, and Lp(a) could be quantified by the immunity transmission turbidity method with commercial kits in the Department of Clinical Laboratory. Here the comparative measurement was conducted between the commercial human kits and the mass spectrometry platform. A total of 45 volunteers were recruited with an average age of 57.6 ± 14.7 years. The basic demographic characteristics and distribution of each apolipoprotein and Lp(a) concentration are listed in ESI Table S4.† Among all peptides, ApoA1 exhibited the highest concentration, while Lp(a) showed the lowest. The excellent correlation between the immunity transmission turbidity method and LC-MS/MS quantification was observed from plasma ApoA1, ApoB, and Lp(a) peptides with R2 values ranging from 0.94 to 0.96 (Fig. 4), which implied the robust applicability of our method to human plasma samples in future works. | ||
| Fig. 4 Comparison of the quantitative LC-MRM/MS method and immunity transmission turbidity assays. The protein concentration correlation from 45 volunteers was determined by the LC-MRM/MS method with the immunity transmission turbidity assay for (A) ApoA1, (B) ApoB, and (C) Lp(a) in human plasma. | ||
3.6. Deficiency and improvement
Here we established a method for simultaneous quantification of apolipoproteins and Lp(a). However, this method possesses certain deficiencies or limitations for future study. There are still some apolipoproteins that could not be included in this panel. For example, the subtypes of ApoB, namely ApoB100 and ApoB48, could not be quantified precisely. The ApoB/ApoA-I ratio has been significantly associated with cardiovascular disease risk factors in previous works8 and ApoB100/ApoB48 serves as indicators for understanding the endogenous remnants produced in the liver after fat intake.42 Despite our efforts to quantify ApoB48 (LSQLQTYMI, ESI Table S1†) simultaneously in our panel, we encountered challenges in quantifying it in human plasma, despite the good linearity of ApoB48 in standards. This limitation may come from the fact that the proportion of ApoB48 constitutes only 0.1% of the total B apolipoprotein, necessitating further enrichment processes before measurement.4. Conclusion
In this study, we simultaneously examined a variety of 14 cardiovascular-related lipoproteins in just 5 μL of human plasma. This innovative method not only enables a more individualized assessment of cardiovascular disease risk, but also facilitates the accelerated application of apolipoprotein measurement in clinical laboratories in addition to choosing the Lp(a) and the ApoB/ApoA-I ratio only. Our works provide a novel and robust tool for monitoring lipid profiling including apolipoproteins and Lp(a), particularly in patients with dyslipidemia.Author contributions
Yuxuan Zhang and Junfang Wu performed experiments and contributed to manuscript preparation; Zhitong Zhou and Xuanru Ren collected the samples and clinical information; Xiaoquan Rao and Hu Ding provided useful suggestions in reviewing; Dao Wen Wang, Hu Ding and Junfang Wu: conceptualization, writing and reviewing and editing, and funding acquisition.Data availability
The data that support the findings of this study are available from the corresponding author (Dr Junfang Wu, junfang.wu@tjh.tjmu.edu.cn) upon reasonable request.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors would like to thank Dr Shaochun Zhu from Umea University, Sweden for providing useful suggestions throughout this work. This work was supported by the National Key Research and Development Program of China (2021YFC2500600 and 2022YFC3400700), the National Nature Science Foundation of China (82370397, 31971358), and the Hubei Provincial Key Research and Developmental Program (2022BCA037).References
- I. van den Broek, K. Sobhani and J. E. Van Eyk, Advances in quantifying apolipoproteins using LC-MS/MS technology: implications for the clinic, Expert Rev. Proteomics, 2017, 14, 869–880 CrossRef CAS PubMed.
- P. Willeit, P. M. Ridker, P. J. Nestel, J. Simes, A. M. Tonkin, T. R. Pedersen, G. G. Schwartz, A. G. Olsson, H. M. Colhoun, F. Kronenberg, C. Drechsler, C. Wanner, S. Mora, A. Lesogor and S. Tsimikas, Baseline and on-statin treatment lipoprotein(a) levels for prediction of cardiovascular events: individual patient-data meta-analysis of statin outcome trials, Lancet, 2018, 392, 1311–1320 CrossRef CAS PubMed.
- D. P. Wilson, T. A. Jacobson, P. H. Jones, M. L. Koschinsky, C. J. McNeal, B. G. Nordestgaard and C. E. Orringer, Use of Lipoprotein(a) in clinical practice: A biomarker whose time has come. A scientific statement from the National Lipid Association, J. Clin. Lipidol., 2019, 13, 374–392 CrossRef.
- B. G. Nordestgaard and A. Langsted, Lipoprotein (a) as a cause of cardiovascular disease: Insights from epidemiology, genetics, and biology, J. Lipid Res., 2016, 57, 1953–1975 CrossRef CAS PubMed.
- S. M. Marcovina and J. J. Albers, Lipoprotein (a) measurements for clinical application, J. Lipid Res., 2016, 57, 526–537 CrossRef CAS.
- S. As, S. Sahukar, J. Murthy and K. Kumar, A study of serum apolipoprotein A1, apolipoprotein B and lipid profile in stroke, J. Clin. Diagn. Res., 2013, 7, 1303–1306 Search PubMed.
- J. Wu, Y. Wang, H. Li, W. Tan, X. Chen and S. Ye, Serum apolipoprotein B-to-apolipoprotein A1 ratio is independently associated with disease severity in patients with acute pancreatitis, Sci. Rep., 2019, 9, 7764 CrossRef PubMed.
- S. Yusuf, S. Hawken, S. Ounpuu, T. Dans, A. Avezum, F. Lanas, M. McQueen, A. Budaj, P. Pais, J. Varigos, L. Lisheng and I. S. Investigators, Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study, Lancet, 2004, 364, 937–952 CrossRef.
- D. C. Chan, M. M. Chen, E. M. Ooi and G. F. Watts, An ABC of apolipoprotein C-III: a clinically useful new cardiovascular risk factor?, Int. J. Clin. Pract., 2008, 62, 799–809 CrossRef CAS PubMed.
- N. S. Shachter, Apolipoproteins C-I and C-III as important modulators of lipoprotein metabolism, Curr. Opin. Lipidol., 2001, 12, 297–304 CrossRef CAS PubMed.
- A. A. Kei, T. D. Filippatos, V. Tsimihodimos and M. S. Elisaf, A review of the role of apolipoprotein C-II in lipoprotein metabolism and cardiovascular disease, Metabolism, 2012, 61, 906–921 CrossRef CAS PubMed.
- A. Hamsten, A. Silveira, S. Boquist, R. Tang, M. G. Bond, U. de Faire and J. Bjorkegren, The apolipoprotein CI content of triglyceride-rich lipoproteins independently predicts early atherosclerosis in healthy middle-aged men, J. Am. Coll. Cardiol., 2005, 45, 1013–1017 CrossRef CAS PubMed.
- F. M. Sacks, P. Alaupovic, L. A. Moye, T. G. Cole, B. Sussex, M. J. Stampfer, M. A. Pfeffer and E. Braunwald, VLDL, apolipoproteins B, CIII, and E, and risk of recurrent coronary events in the Cholesterol and Recurrent Events (CARE) trial, Circulation, 2000, 102, 1886–1892 CrossRef CAS.
- Y. Huang, Mechanisms linking apolipoprotein E isoforms with cardiovascular and neurological diseases, Curr. Opin. Lipidol., 2010, 21, 337–345 CrossRef CAS.
- M. H. Dominiczak and M. J. Caslake, Apolipoproteins: metabolic role and clinical biochemistry applications, Ann. Clin. Biochem., 2011, 48, 498–515 CrossRef CAS.
- S. M. Marcovina, J. J. Albers, B. Gabel, M. L. Koschinsky and V. P. Gaur, Effect of the number of apolipoprotein(a) kringle 4 domains on immunochemical measurements of lipoprotein(a), Clin. Chem., 1995, 41, 246–255 CrossRef CAS.
- S. M. Marcovina and J. J. Albers, Lipoprotein (a) measurements for clinical application, J. Lipid Res., 2016, 57, 526–537 CrossRef CAS.
- M. L. Koschinsky and S. M. Marcovina, Structure-function relationships in apolipoprotein(a): Insights into lipoprotein(a) assembly and pathogenicity, Curr. Opin. Lipidol., 2004, 15, 167–174 CrossRef CAS.
- S. M. Marcovina, Z. H. Zhang, V. P. Gaur and J. J. Albers, Identification of 34 apolipoprotein(a) isoforms: Differential expression of apolipoprotein(a) alleles between american blacks and whites, Biochem. Biophys. Res. Commun., 1993, 191, 1192–1196 CrossRef CAS PubMed.
- M. A. Zaydman, J. R. Brestoff, N. Logsdon and A. M. Gronowski, Kinetic Approach Extends the Analytical Measurement Range and Corrects Antigen Excess in Homogeneous Turbidimetric Immunoassays, J. Appl. Lab. Med., 2019, 4, 214–223 CrossRef CAS.
- Y. J. Bae, A. Gaudl, S. Jaeger, S. Stadelmann, A. Hiemisch, W. Kiess, A. Willenberg, M. Schaab, K. von Klitzing, J. Thiery, U. Ceglarek, M. Dohnert and J. Kratzsch, Immunoassay or LC-MS/MS for the measurement of salivary cortisol in children?, Clin. Chem. Lab. Med., 2016, 54, 811–822 CAS.
- P. Li, Y. Cong, W. Zhang, L. Wang, L. Ren, X. Li, S. Yang, Z. Zhang, G. Li and L. Liu, Simultaneous quantification of apolipoproteins A-I, E, and J in human plasma by LC-MS/MS for clinical application to diabetes mellitus complicated with cardiovascular disease, RSC Adv., 2022, 12, 16763–16771 RSC.
- Y. Pan, H. Zhou, A. Mahsut, R. J. Rohm, O. Berejnaia, O. Price, Y. Chen, J. Castro-Perez, M. E. Lassman, D. McLaren, J. Conway, K. K. Jensen, T. Thomas, G. Reyes-Soffer, H. N. Ginsberg, D. E. Gutstein, M. Cleary, S. F. Previs and T. P. Roddy, Static and turnover kinetic measurement of protein biomarkers involved in triglyceride metabolism including apoB48 and apoA5 by LC/MS/MS, J. Lipid Res., 2014, 55, 1179–1187 CrossRef CAS PubMed.
- S. M. Marcovina, N. Clouet-Foraison, M. L. Koschinsky, M. S. Lowenthal, A. Orquillas, M. B. Boffa, A. N. Hoofnagle and T. Vaisar, Development of an LC-MS/MS Proposed Candidate Reference Method for the Standardization of Analytical Methods to Measure Lipoprotein(a), Clin. Chem., 2021, 67, 490–499 CrossRef.
- J. Wu, J. F. Stevens and C. S. Maier, Mass spectrometry-based quantification of myocardial protein adducts with acrolein in an in vivo model of oxidative stress, Mol. Nutr. Food Res., 2011, 55, 1401–1410 CrossRef CAS.
- F. Bonn, S. Maass and D. Becher, Sample Preparation for Mass-Spectrometry Based Absolute Protein Quantification in Antibiotic Stress Research, Methods Mol. Biol., 2017, 1520, 281–289 CrossRef CAS.
- M. J. Satlin, J. S. Lewis, M. P. Weinstein, J. Patel, R. M. Humphries, G. Kahlmeter, C. G. Giske and J. Turnidge, Clinical and Laboratory Standards Institute and European Committee on Antimicrobial Susceptibility Testing Position Statements on Polymyxin B and Colistin Clinical Breakpoints, Clin. Infect. Dis, 2020, 71, e523–e529 CAS.
- L. Anderson and C. L. Hunter, Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins, Mol. Cell. Proteomics, 2006, 5, 573–588 CrossRef CAS.
- A. N. Hoofnagle, Quantitative clinical proteomics by liquid chromatography-tandem mass spectrometry: assessing the platform, Clin. Chem., 2010, 56, 161–164 CrossRef CAS PubMed.
- D. Armbruster and R. R. Miller, The Joint Committee for Traceability in Laboratory Medicine (JCTLM): a global approach to promote the standardisation of clinical laboratory test results, Clin. Biochem. Rev., 2007, 28, 105–113 Search PubMed.
- I. van den Broek, F. P. Romijn, J. Nouta, A. van der Laarse, J. W. Drijfhout, N. P. Smit, Y. E. van der Burgt and C. M. Cobbaert, Automated Multiplex LC-MS/MS Assay for Quantifying Serum Apolipoproteins A-I, B, C-I, C-II, C-III, and E with Qualitative Apolipoprotein E Phenotyping, Clin. Chem., 2016, 62, 188–197 CrossRef CAS.
- C. A. Toth, Z. Kuklenyik, J. I. Jones, B. A. Parks, M. S. Gardner, D. M. Schieltz, J. C. Rees, M. L. Andrews, L. G. McWilliams, J. L. Pirkle and J. R. Barr, On-column trypsin digestion coupled with LC-MS/MS for quantification of apolipoproteins, J. Proteomics, 2017, 150, 258–267 CrossRef CAS PubMed.
- I. van den Broek, K. Sobhani and J. E. Van Eyk, Advances in quantifying apolipoproteins using LC-MS/MS technology: implications for the clinic, Expert Rev. Proteomics, 2017, 14, 869–880 CrossRef CAS PubMed.
- J. R. Whiteaker, L. Zhao, L. Anderson and A. G. Paulovich, An automated and multiplexed method for high throughput peptide immunoaffinity enrichment and multiple reaction monitoring mass spectrometry-based quantification of protein biomarkers, Mol. Cell. Proteomics, 2010, 9, 184–196 CrossRef CAS.
- J. Woessmann, V. Petrosius, N. Üresin, D. Kotol, P. Aragon-Fernandez, A. Hober, U. Auf dem Keller, F. Edfors and E. M. Schoof, Assessing the Role of Trypsin in Quantitative Plasma and Single-Cell Proteomics toward Clinical Application, Anal. Chem., 2023, 95, 13649–13658 CrossRef CAS.
- F. Li, C. M. Schmerberg and Q. C. Ji, Accelerated tryptic digestion of proteins in plasma for absolute quantitation using a protein internal standard by liquid chromatography/tandem mass spectrometry, Rapid Commun. Mass Spectrom., 2009, 23, 729–732 CrossRef CAS PubMed.
- C. A. Toth, Z. Kuklenyik, J. I. Jones, B. A. Parks, M. S. Gardner, D. M. Schieltz, J. C. Rees, M. L. Andrews, L. G. McWilliams, J. L. Pirkle and J. R. Barr, On-column trypsin digestion coupled with LC-MS/MS for quantification of apolipoproteins, J. Proteomics, 2017, 150, 258–267 CrossRef CAS.
- U. Zaman, F. M. Richter, R. Hofele, K. Kramer, T. Sachsenberg, O. Kohlbacher, C. Lenz and H. Urlaub, Dithiothreitol (DTT) Acts as a Specific, UV-inducible Cross-linker in Elucidation of Protein-RNA Interactions, Mol. Cell. Proteomics, 2015, 14, 3196–3210 CrossRef CAS.
- J. van den Akker, E. VanBavel, R. van Geel, H. L. Matlung, B. Guvenc Tuna, G. M. Janssen, P. A. van Veelen, W. C. Boelens, J. G. De Mey and E. N. Bakker, The redox state of transglutaminase 2 controls arterial remodeling, PLoS One, 2011, 6, e23067 CrossRef CAS.
- F. Yang, D. Li, R. Kufer, L. Cadang, J. Zhang, L. Dai, J. Guo, S. Wohlrab, M. Greenwood-Goodwin, A. Shen, D. Duan, H. Li and I. H. Yuk, Versatile LC-MS-Based Workflow with Robust 0.1 ppm Sensitivity for Identifying Residual HCPs in Biotherapeutic Products, Anal. Chem., 2022, 94, 723–731 CrossRef CAS PubMed.
- P. Singla, O. Singh, S. Chabba, V. K. Aswal and R. K. Mahajan, Sodium deoxycholate mediated enhanced solubilization and stability of hydrophobic drug Clozapine in pluronic micelles, Spectrochim. Acta, Part A, 2018, 191, 143–154 CrossRef CAS PubMed.
- K. Nakajima, Y. Tokita and A. Tanaka, Hypothesis II: The majority of VLDL-apoB48 remnants in postprandial plasma are derived from the liver, not from the intestine, Clin. Chim. Acta, 2019, 490, 12–16 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1–S4 and Fig. S1–S5. See DOI: https://doi.org/10.1039/d4an00221k |
| This journal is © The Royal Society of Chemistry 2024 |