
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A microfluidic-based quantitative analysis system for the multiplexed genetic diagnosis of human viral infections using colorimetric loop-mediated isothermal amplification†
Daigo
Natsuhara
 *a,
Akira
Miyajima
a,
Tomoya
Bussho
a,
Shunya
Okamoto
a,
Moeto
Nagai
ab,
Masaru
Ihira
c and
Takayuki
Shibata
*a
*a,
Akira
Miyajima
a,
Tomoya
Bussho
a,
Shunya
Okamoto
a,
Moeto
Nagai
ab,
Masaru
Ihira
c and
Takayuki
Shibata
*a
aDepartment of Mechanical Engineering, Toyohashi University of Technology, Aichi 441-8580, Japan. E-mail: d-natsuhara@mems.me.tut.ac.jp; shibata@me.tut.ac.jp
bInstitute for Research on Next-generation Semiconductor and Sensing Science (IRES2), Toyohashi University of Technology, Aichi 441-8580, Japan
cFaculty of Clinical Science for Biological Monitoring, Fujita Health University, Aichi 470-1192, Japan
First published on 26th April 2024
Abstract
In this study, a microfluidic-based system utilizing colorimetric loop-mediated isothermal amplification (LAMP) is introduced for the quantitative analysis of nucleic acid targets. This system offers a user-friendly and cost-effective platform for the multiplexed genetic diagnosis of various infectious diseases across multiple samples. It includes time-lapse imaging equipment for capturing images of the microfluidic device during the LAMP assay and a hue-based quantitative analysis software to analyze the LAMP reaction, streamlining diagnostic procedures. An electric pipette was used to simplify the loading of samples and LAMP reagents into the device, allowing easy operation even by untrained individuals. The hue-based analysis software employs efficient image processing and post-processing techniques to calculate DNA amplification curves based on color changes in multiple reaction chambers. This software automates several tasks, such as identifying reaction chamber areas from time-lapse images, quantifying color information within each chamber, correcting baselines of DNA amplification curves, fitting experimental data to theoretical curves, and determining the threshold time for each curve. To validate the developed system, conventional off-chip LAMP assays were conducted with a 25 μL reaction mixture in 0.2 mL polymerase chain reaction (PCR) tubes using a real-time turbidimeter. The results indicated that the threshold time obtained using the colorimetric LAMP assay in the developed system is comparable to that obtained with real-time turbidity measurements in PCR tubes, demonstrating the system's capability for quantitative analysis of target nucleic acids, including those from human herpesviruses.
Introduction
Viral infections have become a significant global concern, affecting human health, safety, and well-being due to increased morbidity and mortality rates.1,2 Therefore, advancing genetic diagnostic technologies is crucial to protect individual well-being. These advancements are key for early virus detection, minimizing the risk of severe symptoms, and preventing the spread of infectious diseases. Loop-mediated isothermal amplification (LAMP) offers a promising approach for the diagnosis of infectious viral diseases, enabling rapid virus detection, typically within 30–60 min, at a constant temperature (60–65 °C) using four–six specially designed LAMP primers and Bst DNA polymerase with high strand-displacement activity.3–7 Notably, LAMP stands out for its simplicity because it does not require sophisticated thermal cycler instruments commonly used in the conventional polymerase chain reaction (PCR). Instead, LAMP can be performed using readily available equipment such as a heating block or water bath.8 The accessibility and efficiency of LAMP for viral detection make it a valuable tool in the fight against infectious diseases, offering a promising pathway for enhancing public health and safety worldwide.The versatility and effectiveness of the LAMP method have made it widely used for diagnosing various infectious diseases, including severe acute respiratory syndrome (SARS) coronavirus 2 (SARS-CoV-2),9 influenza,10 and human immunodeficiency virus.11 It provides the significant benefit of straightforward detection techniques for LAMP amplification products, such as direct colorimetric detection.12–14 For example, phenol red, a pH-sensitive colorimetric indicator, changes from red to yellow owing to proton generation during the LAMP reaction.15 Hydroxynaphthol blue (HNB) is another indicator that changes color in response to the reduction in free Mg2+ ion concentration, a result of magnesium pyrophosphate formation during DNA amplification.16 However, current standard methods for DNA quantification in LAMP assays typically involve turbidity measurement or fluorescence detection using a fluorescent intercalator such as SYBR Green.
However, these methods require expensive sensors and precise light sources for each tube, prompting research into affordable quantitative analyses through colorimetric indicator changes during LAMP assays. For example, Papadakis et al.17 introduced a real-time colorimetric LAMP approach for detecting SARS-CoV-2 RNA and Influenza A DNA, using phenol red and HNB. This system comprises a heater for LAMP assays, light-emitting diode (LED) lights, and a camera to monitor color shifts, enabling DNA-content quantification in samples through temporal analysis of color changes. Similarly, González-González et al.18 demonstrated a cost-effective, quantitative method for SARS-CoV-2 RNA detection from patient samples by assessing phenol red color shifts during LAMP. Priye et al.19 proposed the image processing algorithm to quantify DNA amplification by LAMP reaction. They demonstrated that the smartphone application automatically detected the circular area in the PCR tube and quantitatively analyzed the fluorescence signal during LAMP assays. However, the presence of unexpected air bubbles generated in the PCR tube or uneven fluorescence intensity in the image has the potential to obstruct the identification of the circular area, leading to analysis failure.
Quantitative colorimetric LAMP assays hold significant promise for diagnostics, especially in resource-constrained settings. However, challenges remain in delivering point-of-care testing effectively. A key strategy in managing a viral pandemic involves diagnosing multiple viruses simultaneously. However, this requires preparing and testing numerous sample/reagent mixtures individually for each virus, a labor-intensive process demanding substantial reagent volumes (approximately 10–25 μL) per test. To overcome these issues, lab-on-a-chip technologies have been developed for multiplexed LAMP assays within microfluidic devices, allowing for the concurrent detection of several infectious diseases.20–25 Previous studies have demonstrated the feasibility of multiplexed LAMP assays and the quantitative analyses of nucleic acid quantities in microfluidic devices using colorimetric indicators. Yin et al.26 reported a real-time colorimetric LAMP method for quantifying human papillomavirus 16 (HPV 16) DNA within a microfluidic device using Eriochrome Black T (EBT) as an indicator, achieving quantitative detection with a limit of fewer than 100 copies per reaction, on par with traditional real-time fluorescent LAMP assays’ detection limits. Furthermore, Yin et al.27 reported a real-time colorimetric LAMP detection using a microfluidic device capable of distributing sample/reagent across multiple chambers, successfully identifying nucleic acids from SARS-CoV-2 and human enteric pathogens in wastewater for immediate diagnosis.
Image processing required for deriving DNA amplification curves for nucleic acid quantification involves manually selecting specific areas within reaction chambers. Additionally, air bubbles in these chambers can interfere with color analysis, leading to imprecise DNA amplification curves. Nguyen et al.28 performed colorimetric detection of Escherichia coli W within 50 min using a portable LAMP system integrated with a microfluidic device and a smartphone, demonstrating quantitative detection ranging from 1.32 × 102 to 1.32 × 107 copies per chamber using EBT. However, adjusting image analysis parameters to identify reaction chambers and air bubbles necessitates a well-trained operator. These advancements promise to significantly improve point-of-care testing efficiency and accessibility for infectious diseases, especially in settings with limited resources. However, a fully automated method for image analysis remains undeveloped.
In our previous studies,29–32 we devised a versatile microfluidic device for multiplexed detection of target nucleic acids via the LAMP method, applicable in various domains such as crop disease detection,29 rapid toxic plant identification for emergency care,30 infectious disease diagnosis,31 and food allergen detection.32 This microfluidic device facilitates sequential dispensing of sample/reagent mixtures into an array of reaction chambers in a single operation, potentially serving as a platform for swift and straightforward sample-to-answer diagnostics. We showcased the device's capability to simultaneously detect multiple viral infections, including SARS-CoV-2, SARS, seasonal influenza A, and pandemic influenza A (H1N1) 2009, in under 30 min using a colorimetric reverse transcription LAMP assay.31 To ensure food safety and allergen prevention, we demonstrated the concurrent detection of DNA from wheat, buckwheat, and peanuts as plant food allergens using colorimetric LAMP assays within 60 min.32 Notably, in these studies, color shifts in each reaction chamber were evaluated to determine positive reactions, offering only qualitative endpoint results without quantitative nucleic acid analysis in the samples.
In this study, a microfluidics-based system was developed for the quantitative analysis of viral infectious diseases across multiple test samples. This system integrates time-lapse imaging equipment to capture images during the LAMP reaction and employs real-time hue-based quantitative data analysis software. This software is designed to calculate DNA amplification curves corresponding to color changes in multiple reaction chambers. An automatic thresholding method is proposed in the image-processing phase to accurately identify and extract data related to microchamber areas. In the postprocessing phase, a highly accurate function for curve fitting is introduced, enabling the derivation of theoretical DNA amplification curves. This developed system not only automates the generation of DNA amplification curves but also facilitates the quantitative assessment of target nucleic acids from human herpesviruses in samples. This advancement marks a significant stride toward precise and automated infectious disease diagnosis and aids in monitoring the efficacy of antiviral drugs in patients. Furthermore, the image analysis method proposed in this study can benefit research using a conventional colorimetric LAMP assay, which is typically considered as a binary or semi-quantitative nucleic acid amplification test, by enabling it to serve as a quantitative nucleic-acid detection technique.
Experimental
Fabrication process of the multiplexed diagnostic device
Fig. 1a shows the fabricated polydimethylsiloxane (PDMS)-based microfluidic device for multiplexed genetic diagnosis of viral infections in multiple samples. The device comprises two main sections: a mixing region and a dispensing region, which double as reaction and detection areas. Five microchambers are positioned in a circular arrangement within two separate microfluidic networks. The microfluidic device has a diameter of approximately 35 mm, with the rectangular microchannels measuring approximately 202 μm in width and 109 μm in height.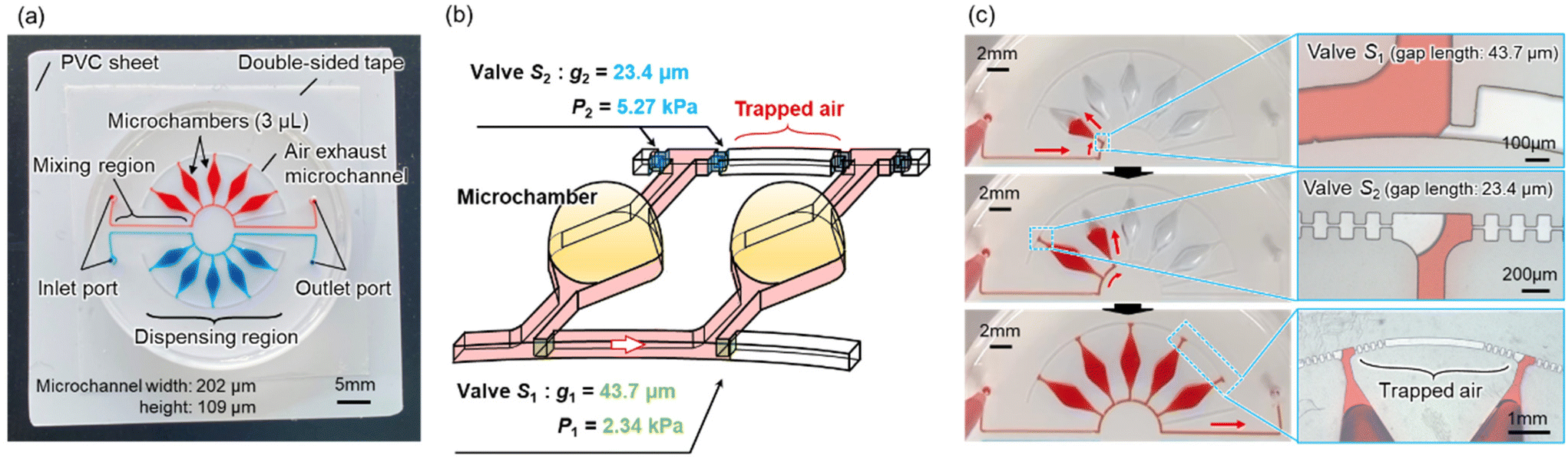 | ||
| Fig. 1 Schematic of the microfluidic diagnostic device allowing sequential liquid dispensing for a multiplexed genetic diagnosis in two samples. (a) A fabricated PDMS microfluidic device consisting of an array of five reaction microchambers (volume of each, ≈3 μL) arranged in a circle in two individual microfluidic networks filled with colored water. (b) Detailed design of the reaction microchamber integrated with a pair of passive stop valves. (c) Experimental result showing sequential liquid dispensing into the microchambers using an electric pipette. | ||
The fabrication process employed is summarized as follows: initially, a negative thick photoresist (SU-8 3050; MicroChem, Newton, MA, USA) was patterned on a 4-inch single-crystal silicon wafer (e-Prize, Yokohama, Japan) to form a mold using a single-step photolithography technique. Subsequently, to construct deep microchamber structures (up to 1 mm deep) suitable for colorimetric LAMP assays, pieces of wax (Ferris File-A-Wax, Freeman Manufacturing & Supply, Avon, OH, USA) were positioned at the center of each SU-8 chamber pattern. A reflow process was then carried out by heating the mold on a hotplate at 120 °C for 15 min (EC1200-N; AS ONE, Osaka, Japan). The SU-8 master mold, featuring semi-elliptical wax structures, was replicated using PDMS (Silpot 184; Dow Corning Toray, Tokyo, Japan) and cured on a hot plate at 80 °C for 40 min. After detaching the PDMS from the SU-8 master mold, circular holes (1.0 mm in diameter) for the inlet and outlet ports were created in the PDMS device using a biopsy punch tool (Kai Industries, Gifu, Japan). The microchambers and microchannels on the PDMS surface were finally sealed with a white polyvinyl chloride (PVC) sheet (EB-235; Hikari, Osaka, Japan) utilizing silicone-based adhesive double-sided tape (No. 5303 W; Nitto Denko, Osaka, Japan). The detailed fabrication process of the microfluidic device is elaborated in our prior study.32
Sequential liquid dispensing using an electric pipette
In our previous studies,31,32 we proposed a theory for the sequential dispensing of liquids into multiple reaction microchambers by managing the burst pressures of passive stop valves (S1 and S2) integrated within each chamber (Fig. 1b). As shown in Fig. 1c, the dispensing procedure operates as follows: initially, the liquid flow halts at the temporary stop valve S1 (with a burst pressure of P1 = 2.34 kPa) and redirects towards the microchamber. Once the microchamber fills, the flow ceases at the permanent stop valve S2 (with a burst pressure of P2 = 5.27 kPa), aiding in air exhaustion from the microchambers. The liquid proceeds to the second microchamber, surpassing valve S1 because P1 < P2. The burst pressure of valve S2 significantly increases after air entrapment between the S2 valves of adjacent microchambers is alleviated. This process is replicated to sequentially fill all microchambers.32 According to our theory,31,32 the optimal flow rate for filling a series of five microchambers is estimated at approximately 200 μL min−1. Detailed explanations on the calculation of each passive stop valve's burst pressure and flow control theory, offering guidelines for the device's design for sequential liquid dispensing, are provided in the ESI (Fig. S1†).For operational simplicity, a custom electric pipette (pipetty 250 μL; Icomes Lab, Iwate, Japan) was utilized to introduce the DNA sample and LAMP reagent mix into the microfluidic device, diverging from the syringe pump used in earlier studies.31,32 This pipette, modified from its original 200 μL configuration to fit a commercially available 50 μL pipette tip (Finntip #9400373; Thermo Fisher Scientific, Tokyo, Japan), allowed for quicker mixture introduction. The dispensing flow rate with the electric pipette escalates with the filling ratio, the volume of the dispensed liquid relative to the pipette tip's capacity. As shown in Fig. S2,† the flow rate correlates with the filling ratio across varying pipette tip volumes, enabling the sequential dispensing of colored water into five microchambers within 12 s (Video S1†). Utilizing of an electric pipette simplifies sample introduction, minimizes contamination risk, and shortens sample handling time, facilitating easy device operation by individuals without specialized training.
Operating procedure for the colorimetric LAMP assay in the microfluidic device
The method for conducting a multiplexed colorimetric LAMP assay within a microfluidic device for the simultaneous genetic diagnosis of infectious diseases from multiple samples is outlined as follows: initially, 0.5 μL of specific primer sets, designed to amplify targeted viral nucleic acids, were pre-spotted and dried at the center of each reaction chamber (each with a volume of approximately 3 μL) using a hot plate set at 80 °C for 3 min. The microchambers and microchannels on the PDMS surface were subsequently sealed using a white PVC sheet adhered with silicone-based double-sided tape. A mixture comprising the DNA sample and LAMP reagents, totaling 32 μL, was introduced into the PDMS microfluidic device at a flow rate of approximately 35 μL min−1via an electric pipette. This process ensures effective mixing of the sample and reagents as they traverse through the planar asymmetric contraction-and-expansion micromixer33 in the mixing region, before being dispensed into the chambers. Once the microchambers were filled, the inlet and outlet ports were sealed with silicone-based double-sided tape, and the device was secured using a homemade clamp (Fig. 2a and b). The assembly was then placed in a hot-water bath (TB-1N; AS ONE, Osaka, Japan) to facilitate the amplification of targeted nucleic acids via the LAMP reaction under stable isothermal conditions at 63 °C for 60 min (Fig. 2c). The operating procedure is illustrated in Fig. S3.†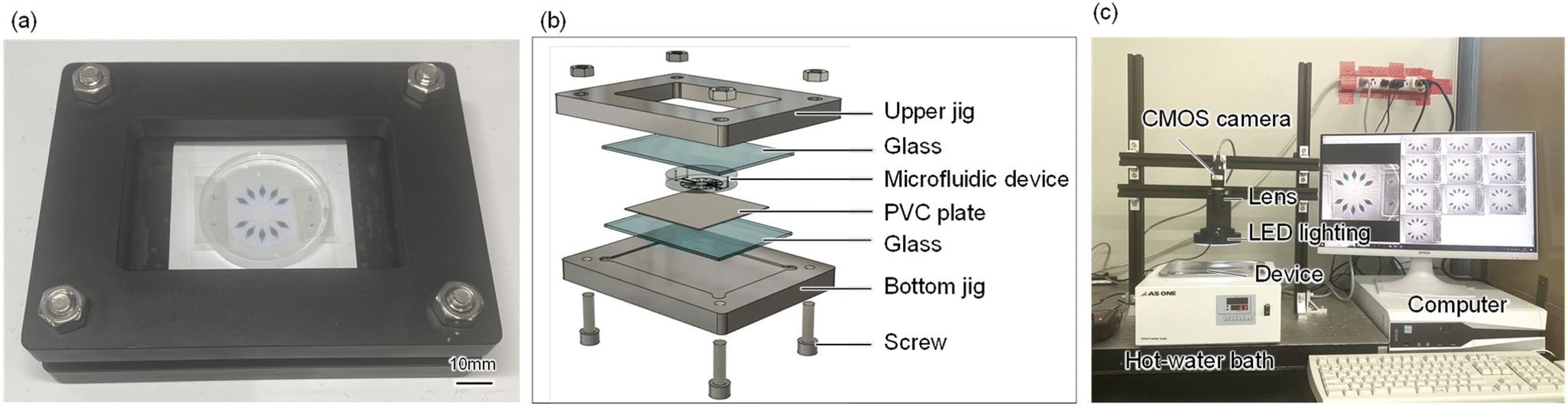 | ||
| Fig. 2 Schematic of the microfluidic device mechanically clamped with a jig and the homemade time-lapse imaging equipment. (a) Photograph of the device clamped with a jig. (b) Configuration of the microfluidic device and the jig. (c) Photograph of the time-lapse imaging equipment. | ||
In this study, LAMP primer sets for the detection of herpes simplex virus 1 (HSV-1) targeted the US4 region (data not yet published; Fujita Health University, Japan), while those for HSV 2 aimed at the gG region.34 Plasmid DNA samples containing the target sequences of HSV-1 and HSV-2 were used as templates in LAMP assays. HNB from FUJIFILM Wako Pure Chemical, Osaka, Japan, was utilized as a colorimetric indicator to signal LAMP reactions, with a positive reaction indicated by a color shift from violet to sky blue because pH-sensitive colorimetric indicators (i.e., phenol red, neutral red, and cresol red) have potential to be affected by acid clinical samples such as saliva.35 Each reaction chamber's final HNB concentration was adjusted to 150 μM within the blend of sample and LAMP reagents. The LAMP reactions employed the Loopamp® DNA amplification kit from Eiken Chemical, Tokyo, Japan, which includes a 2× reaction mix and thermostable Bst polymerase. A mixture totaling 37.5 μL, comprising the DNA sample and LAMP reagents, was prepared for colorimetric LAMP assays in the microfluidic device (Table S1†). For comparative purposes, traditional off-chip LAMP assays were conducted using a 25 μL reaction mixture (Table S2†) in 0.2 mL PCR tubes (SnapStrip®II PCR Tubes; Scientific Specialties, CA, USA) with a real-time turbidimeter (LoopampEXIA; Eiken Chemical, Tokyo, Japan).
Automated quantitative analysis for the detection of multiple nucleic acid targets
As shown in Fig. 2c, the device's time-lapse images were captured during the LAMP assays using custom-built imaging apparatus. This setup included a complementary metal–oxide semiconductor camera (STC-MCS163U3 V; Omron Sentech, Kanagawa, Japan) with a fixed focal lens (M117FM35; Tamron, Saitama, Japan) and an LED ring light (LED-R48; Arms System, Tokyo, Japan) to ensure even lighting. Images of the device were taken every 30 s using a Python macro program, and the colorimetric LAMP assay proceeded for 60 min.Following the introduction of a DNA sample and reagent mixture, the time-dependent color alteration of each microchamber was automatically analyzed using custom software from a series of time-lapse images of the device. This study conducted quantitative evaluations of color differences between positive (sky blue) and negative (violet) reactions in each microchamber, employing the Commission on Illumination (CIE) L*a*b* color space (CIELAB). Defined by the CIE in 1976, CIELAB is a widely utilized three-dimensional color space.36 In this context, the hue angle change (Δh) in the CIELAB color space served as an indicator of gene amplification reaction progression.
Results and discussion
Multiplexed colorimetric LAMP assay in the microfluidic diagnostic device
Fig. 3 presents a typical experimental result for DNA amplification of HSV-1 and HSV-2 within the reaction chambers connected to each microfluidic network. The time-lapse imaging equipment captured clear images. Prior to introducing a DNA sample and reagent mixture, two specific primer sets of 0.5 μL, designed to amplify two distinct types of specific DNA targets, were pre-spotted and dried in each reaction chamber (no. 1a, 3a, 1b, and 3b for HSV-1; no. 2a, 4a, 2b, and 4b for HSV-2), while chambers 5 and 10 had no primers pre-spotted. The HSV-1 DNA sample, mixed with LAMP reagents (DNA concentration: 106 copies per μL), was introduced into the five reaction chambers on the upper side, and the mixture containing the HSV-2 DNA sample (106 copies per μL) was introduced into the five reaction chambers on the lower side. The LAMP assay was conducted at 63 °C for 60 min in a hot water bath. As anticipated, a positive reaction, indicated by a color shift of HNB in the LAMP reaction solution from violet to sky blue, was observed only in chambers 1a and 3a among the upper five microchambers after 20 min of running the LAMP assay, signifying the detection of HSV-1 specific DNA. After 30 min, HSV-2 specific DNA was detected in chambers 2b and 4b among the lower five microchambers.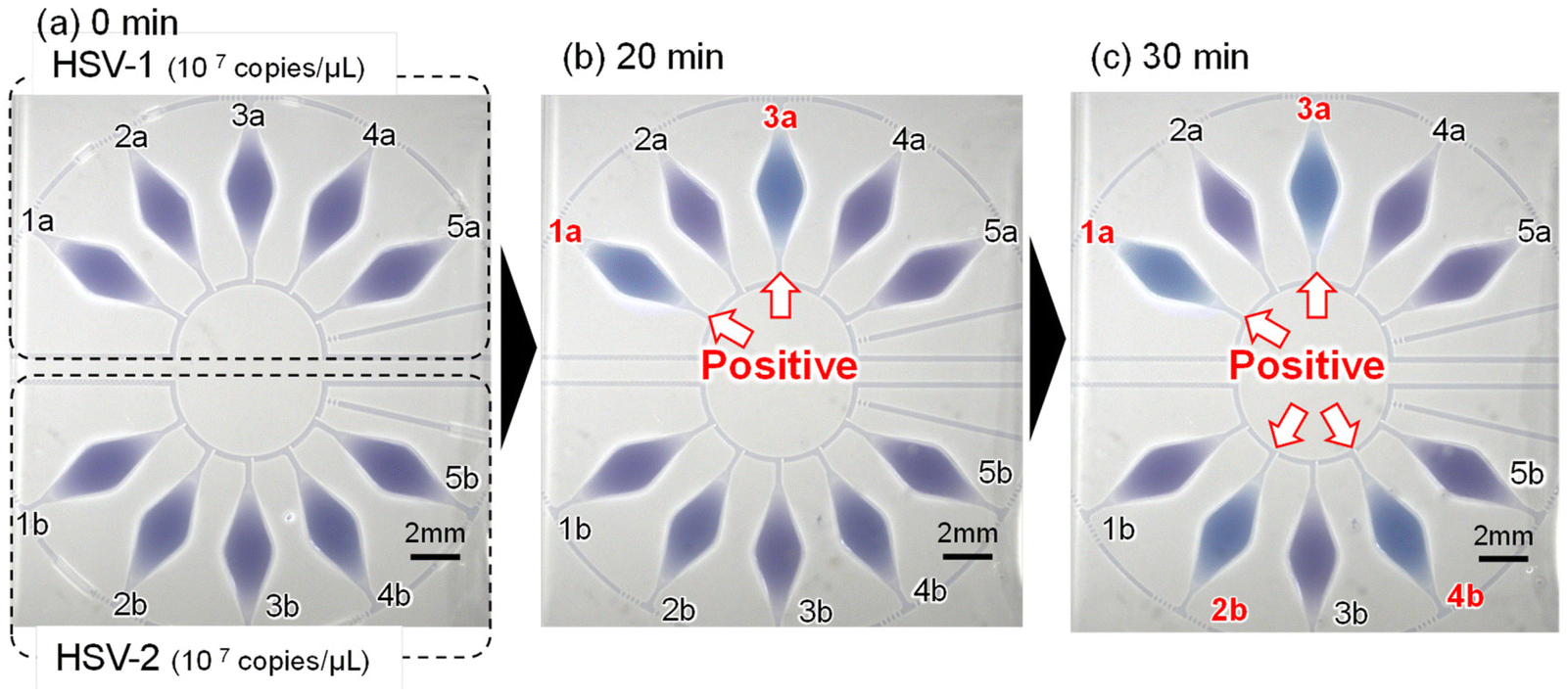 | ||
| Fig. 3 Time-lapse images demonstrating the LAMP assay for the detection of HSV-1 and HSV-2 DNA (DNA concentration of 106 copies per μL). Photographs showing the LAMP assay (a) before running (0 min), and after running for (b) 20 min and (c) 30 min. | ||
Development of an automated hue-based quantitative analysis software
In our previous studies,31,32 endpoint analyses were performed to estimate color differences between positive and negative reactions by measuring the hue angle h in the CIELAB color space as follows: | (1) |
However, to quantitatively assess the concentration of targeted viral DNA in a sample, it is imperative to derive a DNA amplification curve reflecting the increment in DNA product quantity over time. To this end, automated analysis software was developed to capture the color evolution within each reaction chamber, keyed to hue angle (Δh) shifts in the CIELAB color space during the LAMP reaction. This software, scripted in Python, runs on Visual Studio Code.
The initial phase involves autonomously identifying the areas of reaction chambers from time-lapse device imagery captured by custom imaging equipment (Fig. 2c). This stage utilizes a binarization technique, commonly applied to highlight areas of interest from grayscale images surpassing a designated threshold.37Fig. 4a illustrates the 10 microchambers identified through this binarization, marked by green circles. It's critical to choose an optimal threshold to accurately delineate reaction chamber information. Fig. 4b graphs the standard deviation in areas of the ten reaction chambers against the threshold values employed. Here, the standard deviation in estimated chamber areas reaches a minimum at a threshold of 80, indicating consistent chamber region extraction. The threshold yielding the lowest standard deviation in the initial image was adopted for all subsequent time-lapse images, curtailing analysis duration. Applying this determined threshold stabilized reaction chamber area measurements across LAMP experiments, countering the effects of inconsistent thresholding which could lead to significant discrepancies in chamber area assessments (Fig. S4†). This method effectively minimizes diagnostic errors potentially introduced by external variables like lighting discrepancies or minor fabrication-induced chamber shape variances. This binarization technique also allows for the removal of unexpected air bubbles generated within the reaction chambers during LAMP assays (Fig. S5†).
 | ||
| Fig. 4 Schematic illustrating the method for the automatic recognition of regions in reaction chambers based on binary image processing. (a) Green circular areas represent the 10 microchambers that were determined through the binarization process. (b) Standard deviations of areas (in terms of the number of pixels) identified for 10 microchambers as a function of the threshold values used in the binarization process. | ||
Next, the color data (red, green, blue values) within each reaction chamber was derived from a sequence of time-lapse device images captured at 30-second intervals. This step involved computing changes in the a* and b* values within the CIELAB color space, as outlined in prior research. Fig. 5a presents an analysis example where the time-dependent color shift within each reaction chamber is charted on the a*–b* color plane over a 60 min colorimetric LAMP assay duration. Here, data points corresponding to positive reactions in chambers 1a, 3a, 2b, and 4b moved towards the lower-left quadrant (indicating a color transition from violet to sky blue), signaling gene amplification progression. Conversely, the data point for a negative reaction in chamber 2a stayed stationary.
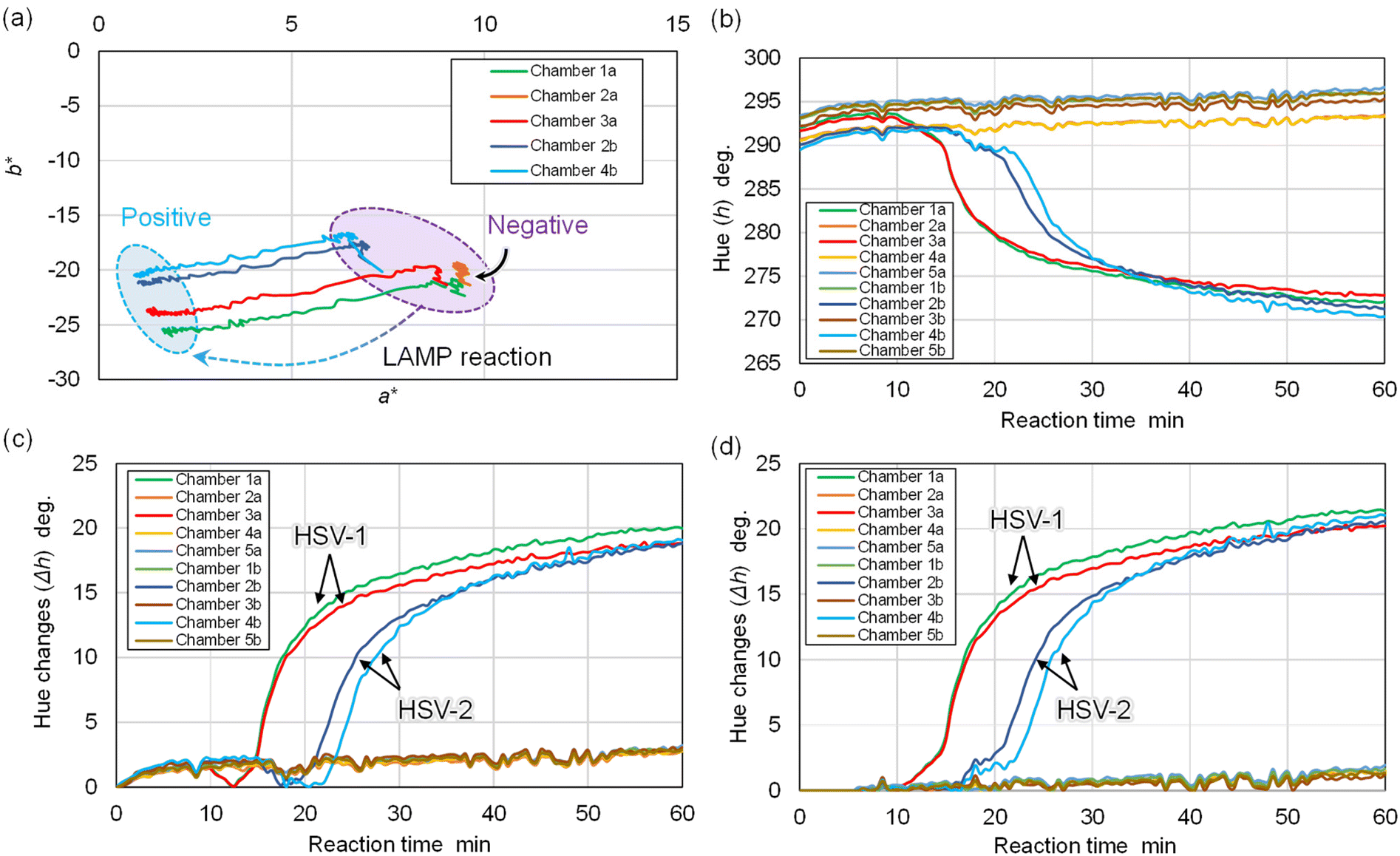 | ||
| Fig. 5 Method for automatically generating DNA amplification curves from a set of time-lapse images. (a) Time-dependent color changes in each reaction chamber in the a*–b* chromatic plane during the LAMP reaction for 60 min. (b) Color change in hue angles (h) as a function of the LAMP reaction time. (c) Change in the difference in hue angles (Δh) without considering the baseline correlation. (d) Change in the difference in hue angles (Δh) by considering the baseline correlation, which resulted in the baseline remaining at zero until 5.5 min. | ||
As per eqn (1), the hue angle (h) induced color shifts were plotted against LAMP reaction time (Fig. 5b). Initial value discrepancies at 0 min for each data point likely stem from minor lighting condition variations, attributed to LED lighting device inhomogeneity and/or water surface fluctuations. Following this, the hue angle difference (Δh) was calculated from previously determined hue angles (h) to generate DNA amplification curves (Fig. 5c). However, during the LAMP reaction's initial phase (around 5.5 min), minimal hue angle change was observed. This phenomenon could result from minor pH alterations in LAMP reagents due to temperature increase upon device immersion in hot water.
In the process of baseline correction for DNA amplification curves, the following approach was adopted: initially, the standard deviation of Δh, denoted as SD0, was computed using the first five images (spanning 0–2.0 min). Next, SD0.5 was calculated from the subsequent five images (covering 0.5–2.5 min). This calculation procedure was repeated to pinpoint the moment when SDn (n = 0.5m; where m represents zero and positive integers) dropped below 0.3 times the original SD0 value. In this experiment, SD5.5 (ranging between 5.5–7.5 min) met this criterion. Consequently, the Δh value at 5.5 min was adjusted to zero, and hue angles before this time were similarly corrected to zero, ensuring the baseline remained at zero up until 5.5 min (as illustrated in Fig. 5d). This correction negated the minor hue angle increment observed at the reaction's onset due to initial temperature changes. Thus, the proprietary software was configured to automatically derive the DNA amplification curves following the aforementioned algorithm. As depicted in Fig. 5d, the DNA amplification curves for HSV-1 in chambers 1a and 3a escalated to a Δh of 15° within 25 min. Meanwhile, the HSV-2 samples in chambers 2b and 4b reached a Δh of 15° within 35 min. In contrast, chambers 5a and 5b, which showed negative reactions, exhibited negligible hue angle shifts (<2°), indicating no cross-contamination.
Optimization of the curve fitting method
For the fourth phase, experimental data underwent curve fitting. To facilitate quantitative LAMP analysis, accurately determining the threshold time (Tt),38 which is calculated as the peak value of the second derivative of each DNA amplification curve, must be precisely determined.39–41 However, fluctuations on the water's surface introduced noise into the curves, complicating accurate Tt determination. Therefore, a curve-fitting technique was employed to accurately calculate Tt. In real-time PCR analyses, experimentally derived DNA amplification curves are typically fitted to a sigmoidal function as follows:42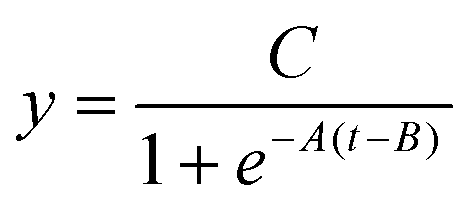 | (2) |
Fig. 6a presents the results of applying a sigmoid function to fit the experimental data. However, the fit was not precise, especially in the early, steep part of the DNA amplification curve. The mean absolute error (MAE) was calculated to be 0.79° (average for n = 4) using the equation
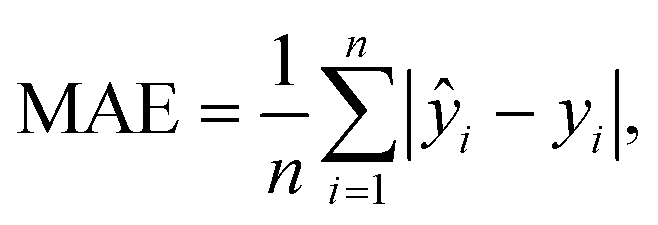 | (3) |
 and yi are the fitting and experimental data, respectively, at each reaction time.
and yi are the fitting and experimental data, respectively, at each reaction time.
 | ||
| Fig. 6 DNA amplification curves fitted with (a) a conventional sigmoidal function and (b) a modified sigmoidal function. | ||
The shape of the experimental curves closely resembled the amplification profiles observed in conventional LAMP reactions based on turbidity measurements (Fig. S6†). This similarity arises because the color shift in HNB is triggered by a reduction in free Mg2+ ion concentration due to the formation of insoluble magnesium pyrophosphate, which increases turbidity as DNA amplification progresses.43 To achieve more accurate curve fitting, we proposed a new function, hereafter referred to as the modified sigmoidal function, for use in the curve-fitting process as follows:
 | (4) |
Fig. 6b compares the experimental findings with theoretical curves derived using the modified sigmoidal function. The accuracy of the fit was notably enhanced, yielding an MAE of 0.19° (for n = 4). The final step involved automatically determining the moment when the second derivative of the fitted curve peaked, defining this point as the Tt value.
Quantitative analysis of human herpesviruses
To assess the effectiveness of the developed hue-based quantitative analysis system, LAMP assays were conducted in microfluidic devices (with a 3 μL reaction volume) at 63 °C for 60 min across a range of DNA concentrations from 102 to 107 copies per μL for HSV-1 samples and 104 to 108 copies per μL for HSV-2 samples. The procedure involved introducing two samples of identical DNA concentration into a single device; this was repeated for each DNA concentration level. The LAMP primer sets were pre-applied in the reaction chambers as shown in Fig. 3, assigning four reaction chambers to the targeted nucleic acids (n = 4 for each DNA concentration). The Tt value from a single LAMP assay was averaged across the four positive reaction chambers. If DNA detection failed in any chamber, it was deemed below the limit of detection (LOD), and a Tt value was not calculated. For comparison, conventional off-chip LAMP assays (n = 3 for each DNA concentration) utilizing real-time turbidity measurements were performed in PCR tubes (25 μL reaction volume) as shown in Fig. S6.† A LAMP assay with a sample lacking template DNA served as a negative control, as indicated in ESI Fig. S7,† which resulted in no LAMP reactions in any microchambers.Fig. 7a displays the DNA amplification curves for varying DNA concentrations of HSV-1. The sample with the highest DNA concentration (107 copies per μL) showed a significant increase in Δh at the shortest reaction time, whereas lower DNA concentrations resulted in longer reaction times. Fig. 7b presents a standard curve for HSV-1 specific DNA, plotting the calculated Tt values (represented by solid circles) against the DNA concentration in the sample (n = 4). A pronounced negative correlation (R2 = 0.970) was found for the detection of specific DNA within the microfluidic devices.
 | ||
| Fig. 7 Experimental results showing the quantitative analysis of human herpesviruses using the developed microfluidic-based system. (a) DNA amplification curves of HSV-1 samples (102–107 copies per μL) fitted with a modified sigmoidal function. (b) Relationship between HSV-1 viral DNA concentration and Tt in the microfluidic device compared with the Tt values obtained from real-time turbidity measurements in PCR tubes. (c) DNA amplification curves of HSV-2 samples (105–108 copies per μL) fitted with a modified sigmoidal function. (d) Relationship between HSV-2 viral DNA concentration and Tt in the microfluidic device compared with the Tt values obtained from real-time turbidity measurements in PCR tubes. | ||
Additionally, the Tt values determined using the microfluidic devices were in good agreement with those from conventional turbidity-based LAMP assays (indicated by solid triangles), demonstrating comparable LOD. Specifically, the LOD for HSV-1, set at 102 copies per μL, matched that of traditional off-chip LAMP assays. Regarding HSV-2 detection within a sample range of 104–108 copies per μL, DNA amplification curves and the corresponding standard curve are depicted in Fig. 7c and d. Despite similar Tt values between device-based and turbidity measurements, the LOD for HSV-2 was found to be an order of magnitude lower (105 copies per μL) in the device, attributed to unsuccessful DNA detection at a concentration of 104 copies per μL in two out of four reaction chambers. This outcome implies that the LOD is influenced by the absolute DNA quantity in the sample. To potentially enhance the LOD, future studies could explore strategies to prevent the adsorption of target DNA and/or enzymes on the PDMS surface.
The equivalence of Tt values obtained in the microfluidic devices (with a 3 μL reaction volume) to those from conventional turbidity measurements in PCR tubes (25 μL reaction volume) underscores the comparability of HNB-based colorimetric detection to turbidity assessment timings. Curry et al.'s research44 on the impact of solution volume changes on cycle threshold (Ct) values in PCR methods highlighted that Ct values are more dependent on the target DNA concentration than on the solution's total target DNA amount. This study also examined how varying the total LAMP mixture volumes (12.5, 25.0, and 50.0 μL) affected the Tt value in PCR tubes for HSV-1 samples at a DNA concentration of 107 copies per μL (Fig. S8†), confirming that DNA concentration dictates the Tt value, independent of the sample's total DNA amount. Consequently, the microfluidic device's LAMP assay, combined with the newly developed automated hue-based quantitative analysis system, achieves an analytical performance on par with that of real-time turbidity measurements in PCR tubes, offering advantages for high-throughput, multi-sample, and multi-target genetic diagnostics in a single procedure. Acquiring gene amplification curves enhances the accuracy and reliability of diagnostic tests by allowing precise differentiation between positive and negative results based on the curve's shape, thus reducing uncertainties inherent in endpoint visual color evaluations. Moreover, this approach offers the advantage of decreasing the time required for diagnostics compared to traditional endpoint methods. Fig. 8 illustrates the temporal variation of the Tt values. In this system, the color image analysis captures device images at 0.5 min intervals. The fitting curve, derived from changes in the hue angle (Δh) from the start of the gene amplification reaction to a certain elapsed time (tx), facilitates the calculation of Tt values from each curve. These values are plotted against tx in Fig. 8a and b. Early in the LAMP reaction (tx < 15–25 min), Tt values fluctuate significantly but stabilize to a constant value as the reaction progresses. Tables S3 and S4† present a comparison of Tt values derived from the complete dataset of hue angle change (Δh) after a 60 min reaction and the elapsed time (tx) at which Tt values stabilize. The criterion for Tt value stabilization was set at a standard deviation (SDx) of less than 0.2 across five Tt values, including data from tx and previous time points. For instance, SD20.0 at tx = 20.0 min is calculated from five Tt values between 18.0 and 20.0 min. Fig. S9† details the temporal variation of SDx up to 60 min. The findings indicate that positive results can be confirmed after a certain period (tx), approximately twice (= tx/Tt) the actual Tt value. Additionally, a strong negative correlation (R2 = 0.978) between the true Tt values and tx/Tt is observed (Fig. 8c). Future research will focus on real-time gene amplification curve acquisition during LAMP assays to expedite diagnostics through timely Tt value changes.
 | ||
| Fig. 8 Experimental result showing the temporal variation of the Tt values of (a) HSV-1 (102–107 copies per μL) and (b) HSV-2 (105–108 copies per μL) up to 60 min. (c) Relationship between true Tt values and their ratio to the time required to confirm a positive result (tx/Tt). | ||
Conclusions
To provide a swift and user-friendly platform for the multiplexed genetic diagnosis of various samples, this study introduced a microfluidics-based quantitative analysis system employing the colorimetric LAMP technique. The developed PDMS-based microfluidic device facilitates the concurrent diagnosis of up to five distinct viral infections across two samples. Utilizing an electric pipette for the introduction of DNA samples and LAMP reagents into the device enhances usability. The system incorporates time-lapse imaging equipment for capturing images during LAMP assays and software for hue-based analysis to evaluate hue variations within the reaction chambers. The system's capacity to automate the creation of DNA amplification curves and accurately quantify DNA concentrations in samples, specifically for HSV-1 and HSV-2, was demonstrated against respective standard curves. The LOD for the device proved comparable for HSV-1 and notably lower for HSV-2 than the LODs of traditional turbidity-based LAMP assays performed in PCR tubes, indicating that LOD is influenced by the total amount of target nucleic acids in the sample. Moreover, Tt values obtained from microfluidic-based LAMP assays were found to be similar to those from conventional off-chip assays, underscoring that Tt values are determined by DNA concentration rather than reaction volume.Future research will focus on the real-time analysis of time-lapse images during LAMP assays to facilitate the direct visualization of all postprocessing stages. This encompasses the acquisition of gene amplification curves, their alignment with theoretical models, and the calculation of Tt values. Furthermore, this will be integrated into a portable microfluidic-based diagnostic system using a smartphone and a microcomputer for portability, enabling on-site diagnosis. Additionally, incorporating a DNA extraction process into the microfluidic device is essential for standardized LAMP assays. The system offers the flexibility to tailor nucleic acid targets (DNA/RNA) by varying the primer types or combinations pre-spotted in the reaction chambers, making it a highly adaptable tool for the onsite multiplexed genetic detection of a wide range of infectious agents (viruses, bacteria, fungi, and parasites) in humans, animals, and plants, as well as foodborne pathogens, allergens, and illegal substances, eliminating the necessity for extensive and multiple manual procedures.
Author contributions
Daigo Natsuhara: Methodology, investigation, writing—original draft preparation; Akira Miyajima: methodology, investigation; Tomoya Bussho: methodology, investigation; Shunya Okamoto: writing—review and editing, supervision; Moeto Nagai: writing—review and editing, supervision; Masaru Ihira: methodology, writing—review and editing, supervision, funding acquisition; Takayuki Shibata: conceptualization, writing—review and editing, supervision, project administration, funding acquisition. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was partially supported by “Knowledge Hub Aichi”, Priority Research Project from Aichi Prefectural Government, and JSPS KAKENHI Grant Number JP22KJ1627. We would like to thank Editage (https://www.editage.com) for English language editing.References
- B. Hu, H. Guo, P. Zhou and Z.-L. Shi, Nat. Rev. Microbiol., 2021, 19, 141–154 CrossRef CAS PubMed.
- P. Palese, Nat. Med., 2004, 10, S82–S87 CrossRef CAS PubMed.
- T. Notomi, H. Okayama, H. Masubuchi, T. Yonekawa, K. Watanabe, N. Amino and T. Hase, Nucleic Acids Res., 2000, 28, E63 CrossRef CAS PubMed.
- Y. Mori and T. Notomi, J. Infect. Chemother., 2009, 15, 62–69 CrossRef CAS PubMed.
- N. Tomita, Y. Mori, H. Kanda and T. Notomi, Nat. Protoc., 2008, 3, 877–882 CrossRef CAS PubMed.
- Y. Mori, H. Kanda and T. Notomi, J. Infect. Chemother., 2003, 19, 404–411 CrossRef PubMed.
- T. Notomi, Y. Mori, N. Tomita and H. Kanda, J. Microbiol., 2015, 53, 1–5 CrossRef CAS PubMed.
- M. Parida, S. Sannarangaiah, P. K. Dash, P. V. L. Rao and K. Morita, Rev. Med. Virol., 2008, 18, 407–421 CrossRef CAS PubMed.
- C. Yan, J. Cui, L. Huang, B. Du, L. Chen, G. Xue, S. Li, W. Zhang, L. Zhao, Y. Sun, H. Yao, N. Li, H. Zhao, Y. Feng, S. Liu, Q. Zhang, D. Liu and J. Yuan, Clin. Microbiol. Infect., 2020, 26, 773–779 CrossRef CAS PubMed.
- L. L. M. Poon, C. S. W. Leung, K. H. Chan, J. H. C. Lee, K. Y. Yuen, Y. Guan and J. S. M. Peiris, J. Clin. Microbiol., 2005, 43, 427–430 CrossRef CAS PubMed.
- N. Hosaka, N. Ndembi, A. Ishizuka, S. Kageyama, K. Numazaki and H. Ichimura, J. Virol. Methods, 2009, 157, 195–199 CrossRef CAS PubMed.
- Y. L. Lau, M. Y. Lai, B. T. Teoh, J. Abd-Jamil, J. Johari, S. S. Sam, K. K. Tan and S. AbuBakar, PLoS One, 2015, 10, e0138694 CrossRef PubMed.
- K. Nawattanapaiboon, E. Pasomsub, P. Prombun, A. Wongbunmak, A. Jenjitwanich, P. Mahasupachai, P. Vetcho, C. Chayrach, N. Manatjaroenlap, C. Samphaongern, T. Watthanachockchai, P. Leedorkmai, S. Manopwisedjaroen, R. Akkarawongsapat, A. Thitithanyanont, M. Phanchana, W. Panbangred, S. Chauvatcharin and T. Srikhirin, Analyst, 2021, 146, 471–477 RSC.
- G. S. Park, K. Ku, S. H. Beak, S. J. Kim, S. I. Kim, B. T. Kim and J. S. Maeng, J. Mol. Diagn., 2020, 22, 729–735 CrossRef CAS PubMed.
- A. T. Scott, T. R. Layne, K. C. O'Connell, N. A. Tanner and J. P. Landers, Anal. Chem., 2020, 92, 13343–13353 CrossRef CAS PubMed.
- M. Goto, E. Honda, A. Ogura, A. Nomoto and K. Hanaki, BioTechniques, 2009, 46, 167–172 CrossRef CAS PubMed.
- G. Papadakis, A. K. Pantazis, N. Fikas, S. Chatziioannidou, V. Tsiakalou, K. Michaelidou, V. Pogka, M. Megariti, M. Vardaki, K. Giarentis, J. Heaney, E. Nastouli, T. Karamitros and A. Mentis, Sci. Rep., 2022, 12, 3775 CrossRef CAS PubMed.
- E. González-González, I. M. Lara-Mayorga, I. P. Rodríguez-Sánchez, Y. S. Zhang, S. O. Martínez-Chapa, G. T. Santiago and M. M. Alvarez, Anal. Methods, 2021, 13, 169–178 RSC.
- A. Priye, C. S. Ball and R. J. Meagher, Anal. Chem., 2018, 90, 12385–12389 CrossRef CAS PubMed.
- H. Zhang, Y. Xu, Z. Fohlerova, H. Chang, C. Iliescu and P. Neuzil, TrAC, Trends Anal. Chem., 2019, 113, 44–53 CrossRef CAS PubMed.
- D. Yuan, J. Kong, X. Li, X. Fang and Q. Chen, Sci. Rep., 2018, 8, 8682 CrossRef PubMed.
- X. Fang, H. Chen, S. Yu, X. Jiang and J. Kong, Anal. Chem., 2011, 83, 690–695 CrossRef CAS PubMed.
- V. M. Phan, S. W. Kang, Y. H. Kim, M. Y. Lee, H. V. Nguyen, Y. L. Jeon, W. I. Lee and T. S. Seo, Sens. Actuators, B, 2023, 390, 133962 CrossRef CAS.
- W. Yu, Y. Chen, Z. Wang, L. Qiao, R. Xie, J. Zhang, S. Bian, H. Li, Y. Zhang and A. Chen, Food Chem., 2021, 351, 129348 CrossRef CAS PubMed.
- S. J. Oh, B. H. Park, J. H. Jung, G. Choi, D. C. Lee, D. H. Kim and T. S. Seo, Biosens. Bioelectron., 2016, 75, 293–300 CrossRef CAS PubMed.
- K. Yin, V. Pandian, K. Kadimisetty, X. Zhang, C. Ruiz, K. Cooper and C. Liu, Sci. Rep., 2020, 10, 9009 CrossRef CAS PubMed.
- K. Yin, X. Ding, Z. Xu, Z. Li, X. Wang, H. Zhao, C. Otis, B. Li and C. Liu, Sens. Actuators, B, 2021, 344, 130242 CrossRef CAS PubMed.
- H. Q. Nguyen, V. D. Nguyen, H. V. Nguyen and T. S. Seo, Sci. Rep., 2020, 10, 15123 CrossRef CAS PubMed.
- D. Natsuhara, K. Takishita, K. Tanaka, A. Kage, R. Suzuki, Y. Mizukami, N. Saka, M. Nagai and T. Shibata, Micromachines, 2020, 11, 540 CrossRef PubMed.
- S. Misawa, D. Natsuhara, Y. Kiba, T. Yamamuro, R. Suzuki, T. Shibata and M. Kitamura, Forensic Toxicol., 2021, 39, 259–265 CrossRef CAS.
- D. Natsuhara, R. Saito, H. Aonuma, T. Sakurai, S. Okamoto, M. Nagai, H. Kanuka and T. Shibata, Lab Chip, 2021, 21, 4779–4790 RSC.
- D. Natsuhara, S. Misawa, R. Saito, K. Shirai, S. Okamoto, M. Nagai, M. Kitamura and T. Shibata, Sci. Rep., 2022, 12, 12852 CrossRef CAS PubMed.
- D. Natsuhara, R. Saito, S. Okamoto, M. Nagai and T. Shibata, Micromachines, 2022, 13, 1386 CrossRef PubMed.
- Y. Enomoto, T. Yoshikawa, M. Ihira, S. Akimoto, F. Miyake, C. Usui, S. Suga, K. Suzuki, T. Kawana, Y. Nishiyama and Y. Asano, J. Clin. Microbiol., 2005, 43, 921–955 CrossRef PubMed.
- C. Uribe-Alvarez, Q. Lam, D. A. Baldwin and J. Chernoff, PLoS One, 2021, 16, e0250202 CrossRef CAS PubMed.
- E. Saldaña, R. Siche, W. Castro, R. Huamán and R. Quevedo, LWT – Food Sci. Technol., 2014, 59, 1220–1226 CrossRef.
- O. I. Singh, T. Sinam, O. James and T. R. Singh, Int. J. Comput. Appl., 2012, 51(6), 5–10 Search PubMed.
- X. Zhang, S. B. Loweb and J. J. Gooding, Biosens. Bioelectron., 2014, 61, 491–499 CrossRef CAS PubMed.
- D. P. Manage, L. Chui and L. M. Pilarski, Microfluid. Nanofluid., 2013, 14, 731–741 CrossRef CAS.
- R. Lenarčič, D. Morisset, M. Pirc, P. Llop, M. Ravnikar and T. Dreo, PLoS One, 2014, 9, e96027 CrossRef PubMed.
- Q. J. Zhou, L. Wang, J. Chen, R. N. Wang, Y. H. Shi, C. H. Li, D. M. Zhang, X. J. Yan and Y. J. Zhang, J. Microbiol. Methods, 2014, 104, 26–35 CrossRef CAS PubMed.
- R. G. Rutledge, Nucleic Acids Res., 2004, 32, e178 CrossRef CAS PubMed.
- Y. Mori, K. Nagamine, N. Tomita and T. Notomi, Biochem. Biophys. Res. Commun., 2001, 289, 150–154 CrossRef CAS PubMed.
- J. D. Curry, C. McHale and M. T. Smith, Mol. Biol. Today, 2002, 3, 79–84 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4an00215f |
| This journal is © The Royal Society of Chemistry 2024 |