
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isolation of acute myeloid leukemia blasts from blood using a microfluidic device†
Alexandra
Teixeira
 abc,
Maria
Sousa-Silva
ad,
Alexandre
Chícharo
a,
Kevin
Oliveira
a,
André
Moura
e,
Adriana
Carneiro
afg,
Paulina
Piairo
a,
Hugo
Águas
e,
Belém
Sampaio-Marques
bc,
Isabel
Castro
bc,
José
Mariz
h,
Paula
Ludovico
bc,
Sara
Abalde-Cela
a and
Lorena
Diéguez
*a
abc,
Maria
Sousa-Silva
ad,
Alexandre
Chícharo
a,
Kevin
Oliveira
a,
André
Moura
e,
Adriana
Carneiro
afg,
Paulina
Piairo
a,
Hugo
Águas
e,
Belém
Sampaio-Marques
bc,
Isabel
Castro
bc,
José
Mariz
h,
Paula
Ludovico
bc,
Sara
Abalde-Cela
a and
Lorena
Diéguez
*a
aInternational Iberian Nanotechnology Laboratory (INL), Avda. Mestre José Veiga, 4715-310 Braga, Portugal. E-mail: lorena.dieguez@inl.int
bLife and Health Sciences Research Institute (ICVS), Escola de Medicina, Universidade do Minho, Campus Gualtar, 4710-057 Braga, Portugal
cICVS/3B's – PT Government Associate Laboratory, Braga/Guimarães, Portugal
dRUBYnanomed LDA, Praça Conde de Agrolongo, 4700-312 Braga, Portugal
eCENIMAT|i3N, Department of Materials Science, NOVA School of Science and Technology, Campus de Caparica, NOVA University of Lisbon and CEMOP/UNINOVA, 2829-516 Caparica, Portugal
fIPO Experimental Pathology and Therapeutics Group, Research Center of IPO Porto (CI-IPOP)/RISE@CI-IPOP (Health Research Network), Portuguese Oncology Institute of Porto (IPO Porto), Porto Comprehensive Cancer Center (Porto.CCC), 4200-072 Porto, Portugal
gInstituto de Ciências Biomédicas Abel Salazar (ICBAS), Universidade do Porto, Porto, Portugal
hDepartment of Oncohematology, Portuguese Institute of Oncology Francisco Gentil Porto, Portugal
First published on 13th April 2024
Abstract
Acute myeloid leukemia (AML) is the most common form of acute leukemia in adults and associated with poor prognosis. Unfortunately, most of the patients that achieve clinical complete remission after the treatment will ultimately relapse due to the persistence of minimal residual disease (MRD), that is not measurable using conventional technologies in the clinic. Microfluidics is a potential tool to improve the diagnosis by providing early detection of MRD. Herein, different designs of microfluidic devices were developed to promote lateral and vertical mixing of cells in microchannels to increase the contact area of the cells of interest with the inner surface of the device. Possible interactions between the cells and the surface were studied using fluid simulations. For the isolation of leukemic blasts, a positive selection strategy was used, targeting the cells of interest using a panel of specific biomarkers expressed in immature and aberrant blasts. Finally, once the optimisation was complete, the best conditions were used to process patient samples for downstream analysis and benchmarking, including phenotypic and genetic characterisation. The potential of these microfluidic devices to isolate and detect AML blasts may be exploited for the monitoring of AML patients at different stages of the disease.
1. Introduction
Acute myeloid leukemia (AML) is a malignant neoplastic disease of hematopoietic stem cells and the most common form of acute leukemia in adults.1 AML is characterised by abnormal accumulation of myeloid progenitors in the bone marrow (BM), blood, and other tissues, impairing the production of normal blood cells. The conventional diagnosis of this hematological malignancy is performed by a BM biopsy to identify the presence of leukemic cells (or blasts).The most common form of therapy used in AML patients is chemotherapy with approximately 80% of cases achieving complete remission (CR). CR of the disease is considered when less than 5% of leukemic cells are found in the BM biopsy and blood counts are normal.2,3 Unfortunately, the majority of AML patients (∼50%) will ultimately relapse3,4 due to the persistent of some undetectable leukemic cells that will reinitiate the disease. This condition is named as minimal or, more appropriately, measurable residual disease (MRD).5,6 In MRD conditions, the number of leukemic cells, called leukemic stem cells (LSCs), is down to 1 in 104–106 total leukocytes, frequencies lower than routine measurement by morphology or cytogenetics are capable to detect.3,7,8 Hence the isolation and detection of these blast cells remain one of the main challenges for MRD assessment in the clinic, highlighting the need of new techniques. In fact, next generation sequencing (NGS) has been used in some cases and has demonstrated high benefits in terms of diagnosis, prognosis, accurate risk stratification, and precision therapy in AML.9–12 Nevertheless, the implementation of NGS in clinical diagnostics laboratories has been slow, mainly due to the associated costs and the requirement of specialised technicians.13
Recently, microfluidics has been one of the main technologies applied for the isolation and detection of rare cells. Microfluidic devices have been successfully used for size-based,14–17 immune-based,18,19 or chromatographic-based separation,20 for cell encapsulation,21–23 and also microfluidic flow cytometry.24–28 Furthermore, microfluidic devices also offer high efficiency for processing complex fluids with minimal damage to the cells contained in these fluids, even the most sensitive.29–32 For example, Ribeiro-Samy et al. demonstrated the isolation of circulating tumor cells (CTCs) from whole blood using pillar structures to separate the cells of interest from the other blood cells, based on size and deformability.15
Another strategy that is commonly used to isolate rare cells is through the biological properties of the cells, like the expression of specific markers. Immune-based isolation is typically achieved by the immunomagnetic approach using functionalised magnetic particles, but it can also be done by the immobilisation of specific antibodies that will recognize the cells of interest on the surface of microfluidic devices.33 Interestingly, in most microfluidic devices the cells follow streamlines with minimal molecular diffusion through the flow channels since they are under laminar and uniaxial flow conditions.34,35 The design of microfluidic devices can be tuned to increase the mixing and the contact of cells against the surface to enable the efficient isolation of cancer cells. For example, Stroock et al. proposed a structured channel ceiling with a staggered herringbone working as a chaotic mixer, increasing the contact of the cells with the surface.36 This concept was later used for the first time by Stott and coworkers for the isolation of CTCs,37 followed by examples from other researchers applied to different cancer types, including AML, by introducing modifications to the microfluidic device.38–40 In the AML context, a size-based strategy for blast isolation is not optimal given that blast cells present similar sizes to the blood cells. Thus, the biological properties of blasts should be explored or their isolation by microfluidics devices.
Herein, we present the development of a microfluidic device to isolate and concentrate AML blasts more efficiently. This device is composed by two structures, micropillars and herringbones, and the surfaces of these structures functionalised with specific antibodies to recognise AML cells. The strategy developed herein involves different geometries and structures, alone or in combination, in order to disrupt the flow lines, provide lateral and vertical mixing, and maximize number of contacts and contact duration between the target cells and the antibody-coated walls themselves. After defining the strategy that results in the best capture efficiency, the system was tested with clinical samples using BM and peripheral blood (PB) from AML patients. The developed device has the potential to be applied to AML as well as to promote a more further and individual analysis.
2. Results and discussion
2.1. Design and fabrication of microfluidic devices for the isolation of AML cells
Devices with different geometries were designed to find the best combination for the isolation of AML blasts (Fig. 1). First, the device dimensions were adjusted to the standard dimensions of a microscope holder, being the dimensions of a conventional glass slide used 75 mm × 25 mm (Fig. 1A). Then, taking into account principles for lateral and vertical mixing, four different molds with different geometries were designed and fabricated. Micropillar devices were designed with dimensions, triangular arrangement and a shift every three rows similar to the work described by Nagrath et al.41 This arrangement was considered one of the most efficient to isolate cancer cells (such as CTCs),41 but a similar approach was never tested in AML. Indeed, the application of microfluidic devices for the enrichment of AML blasts has not yet been widely explored. This is due to the fact that blasts enrichment is even more challenging than CTC isolation, since expression and morphology of leukemic blasts is very similar to that of healthy blood cells.42 Thus, three different micropillar designs were considered according to the insertion of spacings between consecutive row: one with a gap every three rows, another with a gap every four rows, and the last one without any gap (Fig. 1B). This difference among the three designs was implemented to study the dynamic differences of the motion of cells through the devices, which would have a direct impact on the isolation efficiency and also to test if removing some lines helps in the cell counting and does not significantly affect cell enrichment. Finally, a herringbone design was also produced, based on herringbone structures previously described in the literature,37 to study the potential of this structure alone or in combination with the pillar-based isolation approach (Fig. 1C). | ||
| Fig. 1 Schematic representation of the different microfluidic devices designed using AutoCAD. Dimensions of microfluidic devices (A): device total surface: 75 mm of length and 25 mm of width (i); isolation area: 70 mm of length and 11 mm of width (ii) and diameter of the pillars and interpillar space: 50 μm (iii). AutoCAD designs of the geometry of pillars (B): micropillars device design (i); zoom in of a small section containing the pre-filters and isolation area of the different designs: gap every 3 lines, gap every 4 lines and no gap (from left to right); (ii) different spaces between rows of the pillars in the different designs and demonstration of the arrangement of the devices, pillars are arranged in an equilateral triangular array shape with a shift after every 3 rows, and including a gap every 3 lines (iii), a gap every 4 lines (iv) and no gap (v). Herringbone design, the respective zoom in and dimensions of the structure, from the top to the bottom (C). Depth of the masters was 50 μm. | ||
2.2. Simulation of the behavior of the microfluidic devices
COMSOL Multiphysics was used to perform numerical simulations. Microfluidics and Particle Tracing modules were used, simulating a three-dimensional model and reduced versions of the designs.The designs of the devices were used in COMSOL to simulate the process of cells transiting through microfluidic devices to demonstrate the optimal design and combination (micropillars or herringbone, alone, or in combination). In order to observe which pattern presents the best theoretical isolation efficiency, a series of simulations were conducted between all the different designs at different input flow rates. Fig. 2A shows the results of the simulations in the microfluidic device for the no gap design combined either with a glass slide (Fig. 2A(i)) or with a herringbone structure (Fig. 2A(ii)). The simulations of the other designs are included in the ESI (Fig. S1†).
 | ||
| Fig. 2 Simulation results. Performance of the micropillar device without spacing between rows (A(i)), when combined with a herringbone (A(ii)). Theoretical capture efficiency per one section of the device and extrapolated for all the area of the device, for all the designs (B). | ||
The simulation results showed the designs (no gap only and no gap combined with herringbone) and flow rates (2, 20 and 40 μL min−1) that resulted in the highest number of particle-to-wall interactions, consequently allowing a higher chance of having captured cells.
Moreover, the predicted capture efficiency in one section of the device and also the extrapolation for the total area were calculated (Fig. 2B). According to the data obtained from the simulations, the best option was the device with no gap at 20 μL min−1 with 37.67% in one section and 98.58% in the extrapolation for the whole device (ESI, Table S1†). Furthermore, it was found that when pillars are combined with herringbone a slight decrease in the predicted capture efficiency occurred. It is important to note that results and the high values of capture efficiency obtained by the simulations were performed under perfect conditions. These optimal conditions were defined by no interference from other cells than the particle/cell of interest and every time the particle touches the surface it is supposed to remain in the isolation area. Following these results and considering the best conditions that resulted from the simulations, experimental optimisations using microfluidic chips were performed to compare theoretical and experimental results.
2.3. Optimisation of the isolation of AML cells, using microfluidic devices
Additionally, these two different functionalisation strategies were replicated in silicon wafers and analysed by X-ray photoelectron spectroscopy (XPS) – Fig. S2.† The XPS results demonstrated that both functionalisation protocols undergo modifications in the surface. However, considering the merge of XPS results before and after antibody immobilisation, demonstrated in the Fig. S2,† functionalisation B presented the highest N 1s difference (Fig. S2B(vii)†) when compared with functionalisation A (Fig. S2A(vii)†), being characteristic of the presence of the antibody in the surface.48–50 Thus, the functionalisation that provides a better immobilisation of the antibody on the surface, according with XPS results, is the functionalisation B.
Then, to determine the capacity of the developed microfluidic devices to isolate AML cells spiked in peripheral blood mononuclear cell (PBMC) suspension, different parameters and combinations were tested and the capture efficiencies evaluated. The first parameter selected for testing in the microfluidic devices was the different functionalisation protocols, and to confirm whether functionalisation B resulted in a better capture efficiency. Thus, the two functionalisations A and B were tested, maintaining the same flow rate at 20 μL min−1 and the design of gap every 4 lines bonded to a glass slide for both. The results observed in Fig. 3C showed the highest average of capture efficiency of 36.96% for functionalisation B, while functionalisation A showed a more humble 17.25%. These results demonstrate that functionalisation B work better, corroborating the XPS results. Once the best functionalisation protocol was selected, the next parameter to briefly observe if the flow rate affects the capture efficiency. For this purpose, two flow rates were tested, 20 μL min−1 and 40 μL min−1 in the microfluidic device with a gap every 4 lines combined with glass slide (Fig. 3D). At 20 μL min−1 the average capture efficiency was 36.96%, while at 40 μL min−1 was 34.31%, without significative differences (Fig. 3D). In this sense and to have a higher throughput, the flow rate selected for further experiments was 40 μL min−1 and no significant differences were found.
 | ||
| Fig. 3 Schematic representation of the different combinations and respective brightfield images of the devices images (A and B): micropillar and herringbone devices bonded against a glass slide (PDMS/glass) (A), and micropillars bonded against a herringbone replica (PDMS/PDMS combination) (B). Capture efficiency (%) of target cells in devices combining PDMS and glass and functionalised following protocol A and B at a flow rate of 20 μL min−1 (C) and functionalised with best functionalisation protocol B when using different flow rates (20 μL min−1 and 40 μL min−1) and (D). Efficiency of the different designs and combinations of microfluidic devices (E and F). All the designs of devices were tested with the functionalisation B and flow rate at 40 μL min−1, using combinations PDMS/glass (E) and PDMS (pillars)/PDMS (herringbone) (F). For all these experiments were used KG-1 cells. The results in (C), and (D) are presented as mean ± SD of 3 independent biological replicates. Student's t test was applied to compare the funtionalisation A and B at 20 μL min−1 (C) as well as the different flow rates 20 μL min−1 and 40 μL min−1, using the same functionalisation B (D). *p < 0.05. The results in E and F are presented as mean ± SD of 3 independent biological replicates. One-way ANOVA and Tukey's post hoc test were used to compare the 4 different microfluidic devices, bonded into a glass slide, at 40 μL min−1, as well as the different devices, bonded into a herringbone. *p < 0.05. | ||
Then, the combination of micropillars and herringbone (Fig. 3F), which combines vertical and lateral mixing can increase the contact between the cells and the antibody-coated surfaces promoting an increase in the capture efficiency.
Importantly, this combination of pillars and herringbone was already described for the isolation of Hep3B liver tumor cells, but with a different micropillar design and using a different functionalisation strategy (3-mercaptopropyl trimethoxysilane and GMBS (N-γ-maleimidobutyryl-oxysuccinimide ester)). The reported cell capture efficiency was outstanding (∼80%) compared with other reports.51 However, applying this system to AML blasts can be challenging and give different results. Commonly, successful microfluidic strategies used for CTCs, and that worked well, are not applicable to AML blasts. For these reasons, the different pillars designs were bonded against the herringbone, and used to process the cell suspension and to assess the capture efficiency. The results presented in the Fig. 3F demonstrated that adding the herringbone to the micropillars increased the capture efficiency for all the pillar designs tested. However, no significant differences were observed among the efficiencies obtained using the three different combinations. It is important to note that the anticipated enhanced effect resulting from the combination of pillars and herringbone structures was not observed. This can be explained by the fact that antibody–antigen recognition is a slow process. It is conceivable that using lower flow rates than those used could reveal a synergistic effect from the double mixing and leading to higher capture efficiencies. However, such optimization could be at the expense of yield – a critical parameter in a clinical context. Besides that, the dimensions of the herringbone could also be adapted to smaller target cells. Still, the combination of the herringbone with the micropillar device with gaps every 4 lines presented the highest capture efficiency at 55.17%, followed by gap every 3 lines with 49.01% and no gap 44.87% (Fig. 3F).
According to the results obtained, the combination of gap every 4 lines pillars with herringbone was selected as the best strategy/model to use. Taking into account the concentrations tested, and for the optimised conditions, this device should be able to detect blasts in concentrations as little as 2 AML blasts in 106 PBMCs.
 | ||
| Fig. 4 Flow rates and control experiments of the microfluidic devices. KG-1 cells were tested using the best combination (gap every 4 lines of pillars bonded into a herringbone) and using different flow rates: 20 μL min−1, 40 μL min−1 and 60 μL min−1 (A). The controls experiments with KG-1 and HL-60 cells and using gap every 4 lines combined with herringbone (using KG-1 cells), negative controls: using microfluidic devices without antibody in the surface (using KG-1 cells) and testing a different cell line (HL-60 cells) without expression of CD34 and only antibody without previous functionalisation were also done (B). Flow cytometry analysis for the expression of CD34 in the two different AML cell lines (C). CD34 positivity was significantly different and higher in the KG-1 cells. The results presented as mean ± SD of 3 independent biological replicates. For the flow rates and controls an one-way ANOVA and Tukey's post hoc test were used to compare the different flow rates in the same type of device (gap every 4 lines), as well as for the different controls using the same device and flow rate. For the analysis of flow cytometry results a Student's t test was applied to compare the expression of CD34 in KG-1 and HL-60 cells. *p < 0.05. | ||
Next, some control experiments were performed to evaluate the specificity of the devices. For this purpose, devices with no antibody immobilised on the surface were tested with HL-60 cells, which present low expression of CD34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 52 (Fig. 4B).
52 (Fig. 4B).
The results shown in Fig. 4B demonstrated a decrease in the capture efficiency in the two negative controls, showing similar values of 37.89% in the case of the HL-60 cells, and 32.50% when no antibody was immobilised on the surface. The results obtained in the absence of the antibody showed that the vertical and lateral mixing alone can have an impact in the capture of the cells, although this capture would be potentially weak, and the cells should detach upon the application of stronger rinsing processes. Similarly, HL-60 cells were retained within devices, mostly due to non-specific binding, suggesting that they can be washed away if we tune the rinsing step. However, flow cytometry (FC) analysis (Fig. 4C) demonstrated that HL-60 cells present some residual expression of CD34, more precisely 5.80%, while another AML cell line, KG-1 cells show almost 100%.
Additionally, to observe the interference of the functionalisation in the immobilisation of the antibody and consequently in the cell isolation, control experiment where the functionalisation was not performed and only the antibody was passed through the device was analysed (data not shown). This experimental control demonstrated the possibility of having some cells isolated in the device (29.60%).
To better understand these results, a microcontact printing experiment was performed in order to evaluate the capacity of antibodies to bind to the surface just by physisorption (Fig. S3†).
The microcontact printing results showed that the antibody attaches on the surface of the glass slides, namely to those without previous functionalisation, demonstrating the possibility of the antibody to bind to the surface of microfluidic devices, even in a non-organised way, and recognise some cells of interest. Furthermore, to target a wider variety of AML blasts, an experiment was carried out immobilising a combination of antibodies. This experiment also served to test the specificity of the devices. Briefly, two antibodies (CD33 and CD34) and two different cells (KG-1 and Jurkat) with different expressions of the markers were used. KG-1 cells are positive for both and Jurkat negative for both. In Fig. S4,† it was observed that the highest capture efficiency is observed for KG-1 when functionalising the devices with the cocktail of antibodies, with 57.43%, since both biomarkers are expressed in this cell line. Despite Jurkat cells are negative for both biomarkers, the capture efficiency observed was higher than expected, which is explained by a high non-specific binding of the cells to the functionalised surface. As a conclusion, in the optimised conditions, the devices demonstrated a reasonable capacity to isolate the cells of interest. However, the real capture efficiency might be even higher as the method used to assess capture efficiency, accounted for cells captured in the main isolation area only but, since the whole device was functionalised from inlet to outlet, it is very likely that many more cells are captured outside this area, and are not being counted. In addition, it was possible to verify that, in the optimal conditions, the number of cells captured decreases from top to bottom of the isolation area, suggesting that cell capture is specific, and that most cells have been captured by the end of the isolation area. Finally, it is important to mention that the objective of these devices is not to enumerate the captured cells, but to enrich and concentrate the possible blasts contained in the sample, with the final goal to increase the sensitivity of downstream molecular analysis, particularly important in the MRD context.
With the objective to identify aberrant markers in the isolated AML cells, expression of CD7 and CD56 was tested by FC using fluorescently labeled antibodies (Fig. S5A†). Expression was also assessed by ICC in well plates (Fig. S5B†), and compared against in situ ICC in microfluidic devices (Fig. 5).
 | ||
| Fig. 5 Representative fluorescence microscopic images obtained from immunocytochemistry (ICC) assays of AML cells within the microfluidic devices. Phenotypic characterisation using CD7 and CD56 markers in AML cells, (A) KG-1 and (B) HL-60 cells trapped inside the microfluidic device combining pillars and herringbone. (C) Zoom in of (i) KG-1 and (ii) HL-60 cells. All cells were stained for NucBlue™ (1:10), and CD7-FITC and CD56-R667 (1:50), in blue, green and red, respectively. The bar represents 100 μm. | ||
Two different AML cell lines were used: KG-1 (positive for both markers) and HL-60 (negative for both). The FC results, Fig. S5A,† confirmed the expected expression values of the different biomarkers on KG-1 cells (60.8% CD7+CD56−, 3.4% CD7−CD56+, and 32.8% CD7+CD56+) and on HL-60 cells, where both markers were close to 0%. Similarly, ICC tests in well-plate demonstrated that KG-1 cells were positive for both biomarkers, as expected, although CD56 was only expressed in a small number of cells (Fig. S6B(i)†), while HL-60 cells were negative for both (Fig. S6B(ii)†). The same results were observed in the microfluidic devices (Fig. 5). These last results demonstrated that immunophenotyping of AML cells was possible in situ directly inside the microfluidic devices. This is a tremendous improvement, since, up to date, the identification of these biomarkers in isolated AML cells, it is usually done by recovering the cells,52 which can lead to danification or loss of cells.
2.4. Testing using clinical AML samples
Samples were processed using the optimised conditions and identification of aberrant markers (CD7 and CD56) was performed by in situ ICC. Results were compared against the gold standard technique, FC. As it can be seen in Fig. 6, the results obtained by both techniques were similar and showed a low number of cells expressing these aberrant markers, below or about 1%.
 | ||
| Fig. 6 Enumeration of AML blasts with expression of aberrant markers. PBMCs isolated from BM and PB samples from AML patients were stained with CD7 and CD56 antibodies. (A) Determination of the percentage of CD7+ and CD56+ blasts, or both, by FC. (B) Representative enumeration of leukemic blasts positive for aberrant markers (CD7 or CD56, or both) by ICC on the microfluidic device, in some specific areas, not in all isolation area. | ||
The selected NGS panel (described in Methods) was chosen not only to find the known mutations but also to identify new ones that could potentially contribute to disease progression and serve as valuable disease biomarkers. NGS analysis considered only the variants that presented an allele frequency (VAF) higher than 5%, and revealed 17 variants in the two patients studied (Table S2†). Classification using the American College of Medical Genetics and Genomics (ACMG) score revealed four pathogenic and likely pathogenic variants (TIER I), which are predictive, prognostic or diagnostic variants.
Analysis of patient 1 showed variants in FLT3, IDH2, NPM1 and SRSF2 genes, all associated with AML. Alterations in KIT were also observed but without known clinical relevance. Concerning patient 2, pathogenic variants were detected in TP53, and other genes not yet associated with AML, which can become new targets for study.
Importantly, according to the data obtained, PB appears to be just as effective as BM samples for the detection of mutational variants in AML patients, as already described in the literature.53
3. Experimental
3.1. Microfluidic device design and fabrication
Two types of microfluidic devices were designed to isolate blasts from acute myeloid leukemia patients, consisting of patterns based on micropillars or herringbones. For the first type (pillars design), thousands of micropillars with diameter 100 μm and spaced 50 μm were geometrically distributed across the isolation area. The gap size, geometry, and aspect ratio were carefully chosen to allow cells to flow through and mix horizontally to enhance their contact with the surface of the device. These devices present a set of pre-filters with 120 μm gaps to prevent large clumps or debris from clogging the setup. On the herringbone devices, a chevron pattern was designed using line widths and spacings of 50 μm.The microfluidic masters were designed in 2D AutoCAD software (Autodesk) and fabricated on a 200 mm silicon wafer. Fabrication of silicon master mold was performed using a SiO2 hard mask for the silicon dry etching process. For this purpose, a 1 μm thick plasma enhanced chemical vapor deposition (PECVD) SiO2 layer is firstly deposited on a single-side polished (1 0 0) 200 mm Si wafer using a CVD system (MPX from SPTS). The wafer (P/Boron, 〈100〉, Siegert Wafer, Germany) was exposed to hexamethyldisilazane (HDMS, Sigma Aldrich) vapour prime to improve the adhesion of the photoresist to the sample obtained by spin coating of 1.2 μm of AZP4110 (Microchemicals GmbH, Germany) on a SÜSS MicroTec optical track (SÜSS MicroTec AG, Germany). Direct write laser (DWL 2000 Heidelberg Instruments) was used to pattern the photoresist for the top geometry microfluidic channels. Following the post bake, the exposed photoresist was developed with AZ400K (Microchemicals GmbH), and the wafer was rinsed with deionized water and dried. Etching of SiO2 was completed on a reactive ion etching (RIE) tool (APS from SPTS) with a C4F8 etching chemistry followed by removal of the photoresist, striped with an oxygen plasma (PVA GIGAbatch 360 M from Tepla). The silicon wafer was then etched by a dry etching process performed on an inductively coupled plasma (ICP) – RIE tool (Pegasus from SPTS), using a SF6/C4F8 plasma, to transfer the SiO2 mask features to the bulk silicon. The top remaining SiO2 mask was removed on APS from SPTS. Trench depth was measured using an optical profilometer (KLA – Tencor P-16 Surface Profiler) until the desired depth of 50 μm was reached and finally, the master was characterised using Scanning Electron Microscopy (Quanta SEM, FEI, USA). Before master replication, the wafer was hydrophobized performing an overnight vapor-phase treatment with trichloro(1H,1H,2H,2H-perfluorooctyl)silane (Sigma Aldrich), in a desiccator at room temperature.
3.2. Simulations
COMSOL Multiphysics® (COMSOL AB, Sweden) version 5.6 was used to perform the numerical simulations in a 32 GB RAM computer. Along with the core software, the Microfluidics and Particle Tracing modules were used. Reduced versions of the various three-dimensional models were used. Each model was cropped in such a way that a repeating unit cell could be considered. First all the micropillar designs were simulated alone, and later in combination with the herringbone. A one-way coupling approach was followed, meaning that the fluid flow calculations were performed first, and the particles were released afterwards without influencing the flow. To make this happen, a stationary study was performed to determine the fluid dynamics inside the microfluidic channels as the flow is only pressure-driven. A laminar flow is assumed as the Reynolds number is much lower than 100 due to the channels’ small dimensions and velocities.54 Thus, incompressible Navier–Stokes's equations (eqn (1) and (2)) were solved.55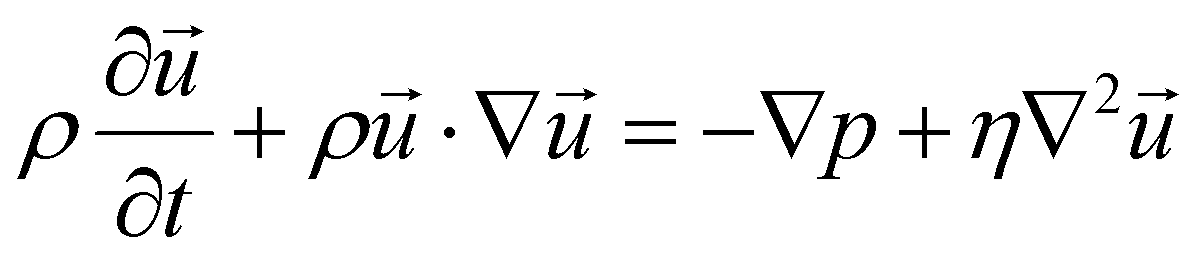 | (1) |
 | (2) |
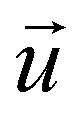 the fluid velocity, p is the pressure and η is the fluid's dynamic viscosity.
the fluid velocity, p is the pressure and η is the fluid's dynamic viscosity.
The working fluid considered was water. The fluid entering the computational model was a fully developed flow at a flow rate of 0.125, 1.25 and 2.5 μL min−1, considering that this model represents 1/16 of the experimental platform (in parallel) where 2, 20 and 40 μL min−1 would be applied. The outlet had a prescribed absolute pressure of 1 atm. The lateral boundaries were set with a periodic boundary condition in case of the simple designs, while symmetry was used on the designs using the herringbone structure, as this structure would not match perfectly laterally. The remaining boundaries (top, bottom, and pillar walls) were prescribed with a no slip wall condition.
After this first study, a second one was performed to determine the position of the cells and their velocity. Their mass was considered to be 4 ng and their diameter 12.5 μm.56 The particles were dragged by the surrounding fluid, applying to them a Stokes force and their initial velocity was given by the fluid velocity. 300 particles were released at the same time. They were initially distributed proportionally to the magnitude of the fluid velocity across the inlet boundary. As soon as the particles crossed the outlet boundary, they disappeared from the model. Similarly, to the fluid flow study, a periodic condition was prescribed to the lateral walls of the simple designs while the symmetry condition was used on the herringbone designs.
To make the meshing process easier, the final geometry was finalised by forming an assembly, which could possibly lead to some fluid loss due to a mesh mismatch. However, these losses were never observed to be higher than 1% when comparing the inflow with the outflow.
The stationary study results were fed into the time dependent study, which was carried out from 0 s to 10 min in time steps of 0.5 s. The simulation studies took under 2 h each to compute.
3.3. PDMS replicas fabrication and bonding of the devices
For the fabrication of PDMS replicas, a mixture of PDMS prepolymer and cross-linker (ratio 10:1, w/w, SYLGARD™ 184 Silicone Elastomer, Dow Chemical) was poured on top of the master, degassed, and then cured for 2 h at 65 °C. The cured replica was cut and peeled from the master, and holes for tubing were made with a biopsy punch (diameter 1.25 mm, Kai Medical). Lastly, for the preparation of the microfluidic devices, clean glass slides (25 × 75 mm, Thermo Fisher Scientific) and PDMS replicas were treated with oxygen plasma (PDC-002-CE, Harrick Plasma) for 30 s. Afterwards, the exposed surfaces were brought together for irreversible bonding and fluorinated ethylene propylene tubing (FEP tubing, IDEX) was inserted in inlets and outlets to proceed with functionalisations and immobilisation of the antibody in all the surface of the devices.
3.4. Functionalisation of devices
Upon bonding, the microfluidic devices were connected to a syringe pump and filled with ethanol at 100 μL min−1 to enhance wettability. Then, two different protocols were used, protocol A and B.First, for functionalisation A, after stabilisation in ethanol, 2% (3-aminopropyl)triethoxysilane (APTES, Sigma Aldrich) in ethanol was flown into the device for 30 minutes and rinsed with ethanol. Then the buffer was changed to MilliQ water and stabilised for 10 minutes before injecting 1% glutaraldehyde (GA, Sigma Aldrich) in water for 30 min and rinsed with water. After that, filtered 10 mM phosphate buffered saline (PBS, Sigma Aldrich), was flowed through the device and equilibrated for 10 minutes before introducing 22 μg of CD34 (Biolegend) in 150 μL of PBS that was left to react overnight at 4 °C. The unreacted antibody was then rinsed with PBS and the surface blocked with 2% Bovine Serum Albumin (BSA, Sigma Aldrich) in PBS.
For functionalisation B, after wetting the devices with ethanol, they were rinsed with MilliQ water. Then, a solution of 1% carboxyethylsilanetriol (CTS, Gelest) and 10% acetic acid in water was passed through the devices for 6 minutes and incubated for 10 minutes and then washed with PBS. After that, a solution of 2% 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC, Sigma Aldrich) and 3% N-hydroxysuccinimide (NHS, Sigma Aldrich) in PBS was flown into the device for 6 minutes. This EDC/NHS solution was used to activate the carboxylic groups, and an incubation time of 10 min was used, followed by a washing step using PBS. Finally, for the immobilisation of the antibody, the same amount of antibody used in functionalisation A was added to the devices and incubated overnight at 4 °C. On the day after, the activation step (2% EDC/3% NHS solution) was repeated and the surface was blocked with methoxypolyethylene glycol amine (PEG, Merck), before rinsing the device with PBS. The surface functionalisation was characterised by X-ray photoelectron spectroscopy (XPS).
3.5. Cell culture
Acute myeloid leukemia cell lines, KG-1 and HL-60, were obtained from the German Collection of Microorganisms and Cell cultures (DSMZ – Deutsche Sammlung von Mikroorganismen und Zellkulturen—German). Cells were maintained in culture in RPMI 1640 medium (Merck), supplemented with 10% heat-inactivated FBS and 1% antibiotic–antimycotic solution (Invitrogen) in a humidified, 37 °C, 5% CO2 atmosphere. When reaching confluence, cells were counted using a hemacytometer, centrifuged and labelled with 1:50 NucBlue™ (Invitrogen) for 30 minutes in the incubator. Stained cells were later used in the spiking experiments.
3.6. Spiking experiments
PB samples (15 mL) from healthy donors were collected in EDTA-coated tubes, and PBMCs were isolated directly using Ficoll-Histopaque-10771 (Histopaque®, Sigma Aldrich) gradient centrifugation, according to the manufacturer's instructions. Briefly, anticoagulated blood was diluted with an equal volume of PBS (Gibco), pH 7.4, containing 2% fetal bovine serum (FBS, Gibco). Then, diluted blood was layered over the Histopaque and samples were centrifuged at 400g for 30 min at room temperature in a swing-out rotor with no brakes. The PBMC interface was carefully removed by pipetting, and washed twice with PBS–2% FBS by centrifugation at 300g for 10 min. The PBMC pellet was re-suspended in PBS and the cells counted using a hemocytometer.Then, 200 AML cultured cells stained with NucBlue™ were used as target and spiked in 0.5 mL of PBS containing 3 × 106 of PBMCs previously isolated from healthy donors, to mimic an AML patient sample. Simultaneous to spiking the cells in PBS, the same amount of cells (200 cells) was added to a well plate that was used as a control.
Spiked samples were injected at different flow rates in the various functionalised microfluidic devices using a syringe pump (New Era Pump Systems, Inc.). Trapped cells were rinsed with 2% Bovine Serum Albumin (BSA, Sigma Aldrich) in PBS, fixed with 4% paraformaldehyde (PFA, Sigma Aldrich) for 20 min at room temperature (RT), and finally washed with PBS. Following sample processing, a fluorescence microscope (Ti-E, Nikon) was used to image the cells captured in the isolation area, using a 20× objective.
The full setup is depicted in the Scheme 1.
 | ||
| Scheme 1 Schematic representation of the sample processing in the microfluidic devices. | ||
To assess the isolation efficiency, the number of stained cells captured in the device was divided by the number of spiked cells initially inserted in the device, for that was used the number of cells inside the well plate, as in the eqn (3). Experiments were done in triplicate.
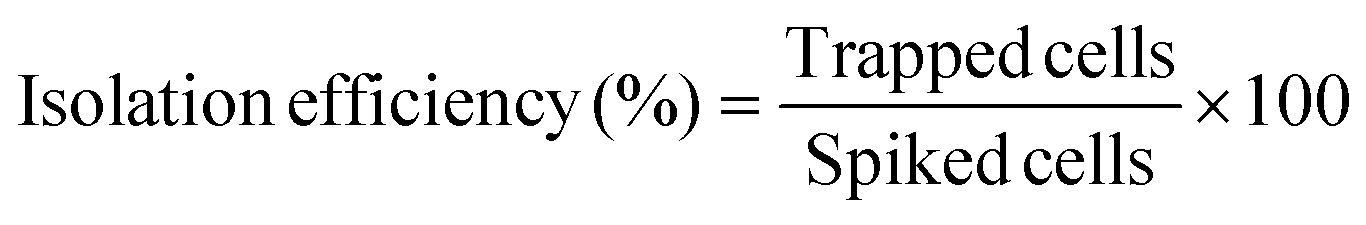 | (3) |
3.7. Cells preparation for immunocytochemistry (ICC)
KG-1 and HL-60 cells were used to test the ICC using the chosen aberrant markers, CD7 and CD56. For that, 100000 cells were resuspended in 0.5 mL of PBS and seeded onto a well plate.
First the cells were fluorescently labeled with 1:50 CD7 (Vio® Bright FITC, Miltenyi) and 1:50 CD56 (Vio® Bright R667, Miltenyi) for 1 h, and then washed with PBS. After that, the cells were fixed with 4% PFA (Sigma Aldrich), for 20 min, washed with PBS, and permeabilised with 0.25% Triton X-100 solution (Sigma Aldrich) for 10 min, and washed with PBS. Subsequently, cells were incubated with 1:10 NucBlue™ (Invitrogen) during 1 h, to stain the cell nucleus. After the incubation period, cells were washed with PBS. All the incubations were done at RT.
Staining experiments were replicated inside the microfluidic devices. For those, the same number of cells were injected at 40 μL min−1 in the functionalised microfluidic devices using a syringe pump (New Era Pump Systems, Inc.). Trapped cells were rinsed with 2% BSA (Sigma Aldrich) in PBS, prior to following the ICC protocol.
3.8. AML patient samples
Clinical samples for this study were provided by the Instituto Português de Oncologia do Porto (IPO) – Porto, after patients provided written informed consent. This study was approved, in accordance with the Declaration of Helsinki, by the ethics committees of ICVS (SECVS 010/2015, 104/2017-1) and IPO-Porto (CES.142A/014).3.9. Clinical sample processing
PB and BM samples (3 mL) were collected at diagnosis in EDTA-coated tubes from two patients with AML. PBMCs were isolated directly using Ficoll-Histopaque-1077 (Histopaque®, Sigma Aldrich) gradient centrifugation, according to the manufacturer's instructions. The PBMC pellet was re-suspended in 1 mL of human serum, and kept at −80 °C until use. Thawed samples were distributed for the different tests: 400 μL for direct DNA extraction, 200 μL for FC and 400 μL were run through the device at 40 μL min−1 (Scheme 2). For FC, cells were stained with conjugated CD7 and CD56 antibodies and run directly. Samples for NGS, proceeded directly for genomic DNA (gDNA) extraction using the NZY Blood gDNA Isolation kit (NZYTech, Lisbon, Portugal) based on the manufacturer's instructions. The gDNA extracted was stored at 4 °C or −20 °C. The final concentration of gDNA extracted from each sample was measured using Qubit. Finally, for microfluidic testing, samples were run through the devices, stained with CD7 and CD56 and then phenotypical analysis was done using fluorescence microscopy. | ||
| Scheme 2 Schematic representation of the distribution of the clinical samples for the different tests. | ||
3.10. Next-generation sequencing
After gDNA extraction, the obtained gDNA was used to perform NGS using a myeloid gene panel. NGS analysis was performed as an outsourced service at GENOMED – DIAGNÓSTICOS DE MEDICINA MOLECULAR, S.A. and using a panel denominated ‘Painel de 30 genes mielóides por NGS’. More precisely, the myeloid gene panel targeted 30 genes: ABL1, ASXL1, BRAF, CALR, CBL, CEBPA, CSF3R, DNMT3A, ETV6, EZH2, FLT3, HRAS, IDH1, IDH2, JAK2, KIT, KRAS, MPL, NPM1, NRAS, PTPN11, RUNX1, SETBP1, SF3B1, SRSF2, TET2, TP53, U2AF1, WT1, ZRSR2. Amplicon sequencing libraries were prepared from 100 ng of DNA per sample.3.11. X-ray photoelectron spectroscopy (XPS)
Silicon wafers were prepared following all the steps described in the different functionalisation protocols. Before the functionalisation, silicon wafer pieces were cleaned with ethanol absolute and dried with a nitrogen gun. Afterwards, the silicon wafer pieces were cleaned with a plasma cleaner for 30 seconds to remove any organic contaminants, and to activate the surface. Prior to the functionalisation, the pieces were rinsed with ethanol. Each functionalisation step was reproduced by immersing the silicon piece on a vial containing the corresponding solution for incubation, and rinsing between steps, according to the functionalisation protocol. The antibody was immobilised overnight at 4 °C and then washed. Before XPS analysis (ESCALAB 250Xi, Thermo Scientific), samples were kept in a sealed container to avoid contamination.3.12. Microcontact printing (μCP)
Glass slides were prepared using the all steps described in the functionalisation protocol B, but replacing the anti-CD34 antibody by an anti-pan cytokeratin FITC antibody (Sigma Aldrich; F3418). A PDMS stamp containing a micropillar pattern was treated with the plasma cleaner and incubated with 22 μg ml−1 pan-FITC-conjugated cytokeratin for 20 min at room temperature. The excess solution was removed from the stamp using a washing step in PBS, and the stamp was air-dried for 1 min. The stamp was then placed, patterned side down, onto the glass slide previously functionalised, and appropriate pressure was applied on top of the stamp. This ensured contact between stamp and glass slide to allow the transference of the antibody. Two different times for the contact glass–PDMS stamp were used, 1 and 2 minutes. Bare glass slides were tested as negative controls where no functionalisation was applied, and just clean glass slides were brought in contact with the antibody using the stamp. Finally, the immobilisation of the antibodies onto the glass slides was evaluated using the fluorescence microscope.3.13. Flow cytometry analysis
KG-1 and HL-60 cells at a concentration of 0.25 × 106 cells per mL were used per each condition. Briefly, cells were centrifuged and incubated with 1:50 of CD33, CD34 and CD117 antibodies (Biolegend) for 90 minutes at room temperature. Then, cells were again centrifuged followed by the incubation with 1:1000 secondary antibody (Goat Anti-Mouse IgG, Abcam) for 1 h at room temperature in the dark. For the aberrant markers (CD7 and CD56), KG-1 and HL-60 cells were used at a concentration of 0.5 × 106 cells per mL. Cells were incubated with 1:50 of conjugated 1:50 anti-CD7 and 1:50 anti-CD56 antibodies for 1 h 30 minutes at room temperature in the dark. After the incubations, a washing step with PBS was performed and after another centrifugation 200 μL microlitres of FACS buffer was added to each sample. Samples were run in a FACS LSRII flow cytometer (BD Biosciences®). FACS Diva was used as the acquisition software. Analysis of the results was performed using the FlowJo 7.6 (Tree Star®) software. At least, 3 independent biological replicates were performed.
3.14. Statistical analysis
All data are reported as the mean ± standard error of the mean (SEM). Statistical analysis was performed using the Student's t test to compare the functionalisation A and B using the same device, and also applied to compare the functionalisations, but using the same flow rate and device. The one-way ANOVA and Tukey's post hoc tests were used to compare the performance of the different microfluidic devices, the influence of the different flow rates, and the difference with control experiments, when all the other parameters were kept constant. A p-value lower than 0.05 was assumed to denote a significant difference.4. Conclusions
In conclusion, this work demonstrated the ability of our optimised microfluidic device for the efficient enrichment of AML blasts from clinical samples. For this, a microfluidic device composed by a combination of micropillars and a herringbone, with an anti-CD34 antibody immobilised onto the surface was selected to recognise and capture immature blasts, characteristic of AML. The functionalisation using CTS and the processing of the sample at 40 μL min−1 yielded the best results, achieving approximately 55% capture efficiency. Despite other methods, combining different geometries, functionalisation strategies and antibodies, have reported higher capture efficiencies,40,52,57,58 most of them did not demonstrate combined phenotypic and molecular analysis of the isolated cells.52,57,58 Our device allows phenotypic analysis of AML blasts in situ and high sensitive downstream molecular analysis. This approach enables the fast identification of blasts for assessment of MRD, as well as mutation analysis towards subtyping and therapy selection, in both PB and BM samples.In the future these devices could be used in the clinical setting to concentrate very residual amounts of blasts contained in the samples to increase the sensitivity of downstream analysis.
The application of this technology to AML patients is crucial as it can allow earlier diagnosis of the disease, which is an important and decisive step to achieve complete remission, since promotes the application of therapy (and more targeted) at an earlier stage of the disease. Additionally, microfluidics can be used as a complementary technique for patient monitoring, analysing the presence of AML blasts in BM or even in PB, during treatment, to evaluate therapeutic response, assess the presence of MRD and, in the long term, to evaluate disease relapse in a non-invasive way.
Author contributions
S. A.-C., B. S.-M., P. L. and L. D. contributed to conceptualisation and ideation, S. A.-C. and L. D. designed the experimental strategy, A. T., M. S.-S., contributed to the experimental execution of the work and data interpretation, A. T., A. Ca. and P. P. contributed to cell culture; A. T., K. O., S. A.-C. and L. D. contributed in the design of the devices; A. Ch. and K. O. contributed in the fabrication of the masters; A. M. and H. Á. contributed in the simulations; I. C. and J. M. contributed in the recruitment of patients and collecting of clinical samples; A. T., M. S.-S., contributed to the original draft preparation, original draft revision S. A.-C. and L. D., and all authors contributed to the final revision of the manuscript before submission. All authors have read and agreed to the final version of the manuscript.Conflicts of interest
Authors declare no conflict of interest.Acknowledgements
This work was supported by “Innovative Microfluidic Platform for Analysis of myeloid Leukemia blasts” project (PTDC/EMD-EMD/30782/2017) co-funded by FCT and the European Regional Development Fund (ERDF) through COMPETE2020; by the Foundation for Science and Technology (FCT) projects UIDB/50026/2020 and UIDP/50026/2020; and by the project NORTE-01-0145-FEDER-000055, supported by Norte Portugal Regional Operational Programme (NORTE 2020), under the PORTUGAL 2020 Partnership Agreement, through the European Regional Development Fund (ERDF). This project also received funding of the project health from Portugal (C630926586-00465198), supported by Component C5 – Capitalisation and Business Innovation, under the Portuguese Resilience and Recovery Plan, through the NextGenerationEU Fund. A. M. and H. A. acknowledge FCT – Fundação para a Ciência e a Tecnologia concerning the projects LA/P/0037/2020, UIDP/50025/2020 and UIDB/50025/2020 of the Associate Laboratory Institute of Nanostructures, Nanomodelling and Nanofabrication – i3N, and also under project dPCR4FreeDNA of the same research unit – PTDC/BTM-SAL/31201/2017. A. T. acknowledges the FCT studentship SFRH/BD/148091/2019. A. C. acknowledges the FCT scholarship 2021.05593.BD. B. S.-M. acknowledges funding by FCT, grant number DL 57/2016. The authors kindly thank Dr Oleksandr Bondarchuk for his support in the XPS analysis. This work was carried out in part through the use of the INL User Facilities.References
- B. Deschler and M. Lübbert, Cancer, 2006, 107, 2099–2107 CrossRef PubMed.
- G. J. Schuurhuis and G. Ossenkoppele, Expert Rev. Hematol., 2010, 3, 1–5 CrossRef PubMed.
- A. Teixeira, L. Carreira, S. Abalde-Cela, B. Sampaio-Marques, A. C. Areias, P. Ludovico and L. Diéguez, Cancers, 2023, 15, 1362 CrossRef CAS PubMed.
- N. R. Ramos, C. C. Mo, J. E. Karp and C. S. Hourigan, J. Clin. Med., 2015, 4, 665–695 CrossRef CAS PubMed.
- M. T. Voso, T. Ottone, S. Lavorgna, A. Venditti, L. Maurillo, F. Lo-Coco and F. Buccisano, Front. Oncol., 2019, 9, 655 CrossRef PubMed.
- F. Ravandi, R. B. Walter and S. D. Freeman, Blood Adv., 2018, 2, 1356–1366 CrossRef CAS PubMed.
- C. Dix, T.-H. Lo, G. Clark and E. Abadir, J. Clin. Med., 2020, 9, 1714 CrossRef PubMed.
- G. J. Schuurhuis, M. Heuser, S. Freeman, M.-C. Béné, F. Buccisano, J. Cloos, D. Grimwade, T. Haferlach, R. K. Hills, C. S. Hourigan, J. L. Jorgensen, W. Kern, F. Lacombe, L. Maurillo, C. Preudhomme, B. A. van der Reijden, C. Thiede, A. Venditti, P. Vyas, B. L. Wood, R. B. Walter, K. Döhner, G. J. Roboz and G. J. Ossenkoppele, Blood, 2018, 131, 1275–1291 CrossRef CAS PubMed.
- M.-E. Percival, C. Lai, E. Estey and C. S. Hourigan, Blood Rev., 2017, 31, 185–192 CrossRef PubMed.
- E. Paietta, Hematology Am. Soc. Hematol. Educ. Program, 2012, 2012, 35–42 CrossRef PubMed.
- T. Haferlach, Hematol. Rep., 2020, 12, 8957 CrossRef CAS PubMed.
- R. L. Levine and P. J. M. Valk, Haematologica, 2019, 104, 868–871 CrossRef CAS PubMed.
- R. R. Singh, R. Luthra, M. J. Routbort, K. P. Patel and L. J. Medeiros, Expert Rev. Precis. Med. Drug Dev., 2016, 1, 109–120 CrossRef.
- Y. Chen, P. Li, P.-H. Huang, Y. Xie, J. D. Mai, L. Wang, N.-T. Nguyen and T. J. Huang, Lab Chip, 2014, 14, 626–645 RSC.
- S. Ribeiro-Samy, M. I. Oliveira, T. Pereira-Veiga, L. Muinelo-Romay, S. Carvalho, J. Gaspar, P. P. Freitas, R. López-López, C. Costa and L. Diéguez, Sci. Rep., 2019, 9, 8032 CrossRef PubMed.
- C. Lopes, P. Piairo, A. Chícharo, S. Abalde-Cela, L. R. Pires, P. Corredeira, P. Alves, L. Muinelo-Romay, L. Costa and L. Diéguez, Cancers, 2021, 13, 4446 CrossRef CAS PubMed.
- A. Carneiro, P. Piairo, A. Teixeira, D. Ferreira, S. Cotton, C. Rodrigues, A. Chícharo, S. Abalde-Cela, L. L. Santos, L. Lima and L. Diéguez, Cells, 2022, 11, 376 CrossRef CAS PubMed.
- L. Diéguez, M. A. Winter, K. J. Pocock, K. E. Bremmell and B. Thierry, Analyst, 2015, 140, 3565–3572 RSC.
- L. Diéguez, M. Winter, S. Molan, P. Monis, B. King and B. Thierry, J. Biol. Eng., 2018, 12, 4 CrossRef PubMed.
- P. Li, Z. S. Stratton, M. Dao, J. Ritz and T. J. Huang, Lab Chip, 2013, 13, 602–609 RSC.
- J. F. Edd, D. Di Carlo, K. J. Humphry, S. Köster, D. Irimia, D. A. Weitz and M. Toner, Lab Chip, 2008, 8, 1262–1264 RSC.
- E. W. M. Kemna, R. M. Schoeman, F. Wolbers, I. Vermes, D. A. Weitz and A. van den Berg, Lab Chip, 2012, 12, 2881–2887 RSC.
- S. Da Ling, Y. Geng, A. Chen, Y. Du and J. Xu, Biomicrofluidics, 2020, 14, 61508 CrossRef PubMed.
- A. B. Shrirao, Z. Fritz, E. M. Novik, G. M. Yarmush, R. S. Schloss, J. D. Zahn and M. L. Yarmush, Technology, 2018, 6, 1–23 CrossRef PubMed.
- X. Li, B. Fan, L. Liu, D. Chen, S. Cao, D. Men, J. Wang and J. Chen, Sci. Rep., 2018, 8, 14229 CrossRef PubMed.
- A. Chícharo, M. Martins, L. C. Barnsley, A. Taouallah, J. Fernandes, B. F. B. Silva, S. Cardoso, L. Diéguez, B. Espiña and P. P. Freitas, Lab Chip, 2018, 18, 2593–2603 RSC.
- A. Chícharo, D. M. Caetano, S. Cardoso and P. Freitas, Microfluidics and Biosensors in Cancer Research: Applications in Cancer Modeling and Theranostics, Springer, 2022, pp. 413–444 Search PubMed.
- A. Teixeira, A. Carneiro, P. Piairo, M. Xavier, A. Ainla, C. Lopes, M. Sousa-Silva, A. Dias, A. S. Martins and C. Rodrigues, Microfluidics and Biosensors in Cancer Research: Applications in Cancer Modeling and Theranostics, Springer, 2022, pp. 553–590 Search PubMed.
- U. Dharmasiri, S. Balamurugan, A. A. Adams, P. I. Okagbare, A. Obubuafo and S. A. Soper, Electrophoresis, 2009, 30, 3289–3300 CrossRef CAS PubMed.
- J. A. Phillips, Y. Xu, Z. Xia, Z. H. Fan and W. Tan, Anal. Chem., 2009, 81, 1033–1039 CrossRef CAS PubMed.
- Y. Dong, A. M. Skelley, K. D. Merdek, K. M. Sprott, C. Jiang, W. E. Pierceall, J. Lin, M. Stocum, W. P. Carney and D. A. Smirnov, J. Mol. Diagn., 2013, 15, 149–157 CrossRef PubMed.
- U. Dharmasiri, S. K. Njoroge, M. A. Witek, M. G. Adebiyi, J. W. Kamande, M. L. Hupert, F. Barany and S. A. Soper, Anal. Chem., 2011, 83, 2301–2309 CrossRef CAS PubMed.
- L. Descamps, D. Le Roy and A.-L. Deman, Int. J. Mol. Sci., 2022, 23, 1981 CrossRef CAS PubMed.
- S. L. Stott, C.-H. Hsu, D. I. Tsukrov, M. Yu, D. T. Miyamoto, B. A. Waltman, S. M. Rothenberg, A. M. Shah, M. E. Smas, G. K. Korir, F. P. Floyd, A. J. Gilman, J. B. Lord, D. Winokur, S. Springer, D. Irimia, S. Nagrath, L. V. Sequist, R. J. Lee, K. J. Isselbacher, S. Maheswaran, D. A. Haber and M. Toner, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18392 LP–18397 LP CrossRef PubMed.
- J. Marschewski, S. Jung, P. Ruch, N. Prasad, S. Mazzotti, B. Michel and D. Poulikakos, Lab Chip, 2015, 15, 1923–1933 RSC.
- A. D. Stroock, S. K. W. Dertinger, A. Ajdari, I. Mezić, H. A. Stone and G. M. Whitesides, Science, 2002, 295, 647 LP–651 LP CrossRef PubMed.
- S. L. Stott, C.-H. Hsu, D. I. Tsukrov, M. Yu, D. T. Miyamoto, B. A. Waltman, S. M. Rothenberg, A. M. Shah, M. E. Smas, G. K. Korir, F. P. Floyd, A. J. Gilman, J. B. Lord, D. Winokur, S. Springer, D. Irimia, S. Nagrath, L. V. Sequist, R. J. Lee, K. J. Isselbacher, S. Maheswaran, D. A. Haber and M. Toner, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18392–18397 CrossRef CAS PubMed.
- S. Wang, K. Liu, J. Liu, Z. T. Yu, X. Xu, L. Zhao, T. Lee, E. K. Lee, J. Reiss and Y. Lee, Angew. Chem., 2011, 123, 3140–3144 CrossRef.
- S. Wang, A. Thomas, E. Lee, S. Yang, X. Cheng and Y. Liu, Analyst, 2016, 141, 2228–2237 RSC.
- Q. Lai, J. Song, J. Zha, H. Zheng, M. Deng, Y. Liu, W. Lin, Z. Zhu, H. Zhang, B. Xu and C. Yang, Biosens. Bioelectron., 2023, 219, 114803 CrossRef CAS PubMed.
- S. Nagrath, L. V. Sequist, S. Maheswaran, D. W. Bell, D. Irimia, L. Ulkus, M. R. Smith, E. L. Kwak, S. Digumarthy, A. Muzikansky, P. Ryan, U. J. Balis, R. G. Tompkins, D. A. Haber and M. Toner, Nature, 2007, 450, 1235–1239 CrossRef CAS PubMed.
- V. Weeda, S. G. C. Mestrum and M. P. G. Leers, Int. J. Mol. Sci., 2022, 23, 10529 CrossRef CAS PubMed.
- M. Yüce and H. Kurt, RSC Adv., 2017, 7, 49386–49403 RSC.
- M.-H. Park, E. Reátegui, W. Li, S. N. Tessier, K. H. K. Wong, A. E. Jensen, V. Thapar, D. Ting, M. Toner, S. L. Stott and P. T. Hammond, J. Am. Chem. Soc., 2017, 139, 2741–2749 CrossRef CAS PubMed.
- S. W. Shaner, J. K. Allen, M. Felderman, E. T. Pasko, C. D. Wimer, N. D. P. Cosford, S. Kassegne and P. Teriete, AIP Adv., 2019, 9, 65313 CrossRef.
- J. H. Myung and S. Hong, Lab Chip, 2015, 15, 4500–4511 RSC.
- W. Zeijlemaker, A. Kelder, J. Cloos and G. J. Schuurhuis, Curr. Protoc. Cytom., 2019, 91, e66 CrossRef PubMed.
- R. A. Zangmeister, J. Pharm. Sci., 2012, 101, 1639–1644 CrossRef CAS PubMed.
- J. E. Baio, F. Cheng, D. M. Ratner, P. S. Stayton and D. G. Castner, J. Biomed. Mater. Res., Part A, 2011, 97A, 1–7 CrossRef CAS PubMed.
- B. Thierry, M. Kurkuri, J. Y. Shi, L. E. M. P. Lwin and D. Palms, Biomicrofluidics, 2010, 4, 32205 CrossRef PubMed.
- P. Xue, Y. Wu, J. Guo and Y. Kang, Biomed. Microdevices, 2015, 17, 39 CrossRef PubMed.
- J. M. Jackson, J. B. Taylor, M. A. Witek, S. A. Hunsucker, J. P. Waugh, Y. Fedoriw, T. C. Shea, S. A. Soper and P. M. Armistead, Analyst, 2016, 141, 640–651 RSC.
- R. Weinkauff, E. H. Estey, P. Starostik, K. Hayes, Y. O. Huh, C. Hirsch-Ginsber, M. Andreeff, M. Keating, H. M. Kantarjjan and E. J. Freireich, Am. J. Clin. Pathol., 1999, 111, 733–740 CrossRef CAS PubMed.
- M. Itomlenskis, P. S. Fodor and M. Kaufman, Proc. COMSOL Conf., Boston, 2008.
- P. Xue, L. Zhang, J. Guo, Z. Xu and Y. Kang, Microfluid. Nanofluid., 2016, 20, 1–9 CrossRef CAS.
- J. Prinyakupt and C. Pluempitiwiriyawej, Biomed. Eng. Online, 2015, 14, 1–19 CrossRef PubMed.
- B. L. Khoo, M. Shang, C. H. Ng, C. T. Lim, W. J. Chng and J. Han, npj Precis. Oncol., 2019, 3, 30 CrossRef PubMed.
- V. J. Lyons and D. Pappas, Analyst, 2019, 144, 3369–3380 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4an00158c |
| This journal is © The Royal Society of Chemistry 2024 |